
Abstract
The study of molecular valence electron dynamics and their coupling with nuclear motion is one of the frontiers of ultrafast physics and ultrafast chemistry. With time-resolved strong-field ion momentum spectroscopy, we study electron valence and nucleus wavepacket evolution on a femtosecond timescale. Two orientation-dependent bond-breaks of N2O molecules from the same electronic state are studied, and the influence of orbital hybridization and polarization effect during molecular breaking is analyzed based on the measured time-resolved asymmetric Pzsum distributions, allowing a visual representation of electron localization during the dissociation of molecules into ions and atoms. Comparison of experimental and theoretical results on orientation-dependent dissociation dynamics allows us to understand how nuclear motions evolve during fragmentation and to control ultrafast molecular reactions.
Similar content being viewed by others

Introduction
Understanding the process of electron rearrangement during molecular breakage is a crucial area of interest in molecular physics and photochemistry because it initiates the subsequent movements of the nucleus, which ultimately determines the outcome of molecular reactions1,2,3,4,5. Tracing the ultrafast dynamics of electrons and nuclei after photoexcitation or photoionization has been pursued for decades. The development of ultrafast spectroscopy based on time-resolved photoelectron momenta, strong-field dissociation ion momenta, and high-order harmonic generation (HHG) enabled real-time observation of molecular processes at femto- and attosecond time scales5,6,7,8,9. Attosecond spectroscopies can probe purely electronic dynamics in atomic and molecular systems and track the electron-nucleus coupling in real time9,10,11,12. These studies will help us better understand electron dynamics and nuclear evolution on unstable potential energy surfaces (PESs), as well as help us control chemical reactions in the future7,8,9,10,11,12,13,14.
The electron distribution profiles of atoms and molecules can be captured by utilizing tunneling ionization in strong laser fields since the angular distribution of strong-field ionization (SFI) is determined by the shape of electron orbitals and the polarization of samples along the laser fields15,16,17. It shows great success in imaging the orbital shape of the molecular ground state and excitation state, like the centrosymmetric molecules H2, N2, O2, CO2, and C4H618,19,20,21,22, and non-centrosymmetric molecules HCl, CO, OCS, CH3X (X = Br, I), and C6H5CN9,23,24,25,26,27,28. The orientation-dependent ionization yields are also determined by the asymmetric responses of molecular polarization to strong laser fields24,25,27,29,30. Furthermore, the autoionization of CO molecules can be studied31, and the transient valence charge localization during the dissociative ionization of HCl molecules can be captured by strong-field ionization spectroscopy32. Using the SFI-pump and SFI-probe scheme, the valence electron wavepackets of atomic coherent states can be tracked with femtosecond-resolution ion and electron momentum spectroscopy33,34,35. This method has also been used to track the electron rearrangement during the dissociation of Br29 and the excitation of NO molecules36.
In this study, we examine electron localization during the dissociation of N2O molecules utilizing time-resolved strong-field ion momentum spectroscopy and track the evolution of asymmetric valence electron wavepackets at two orientation-dependent dissociation channels. We investigate the electronic and nuclear dynamics starting from the same electronic state \({\widetilde{B}}^{2}\Pi\). Time-resolved kinetic energy release (KER) enables us to determine the motion of nuclei, whereas time-dependent Pzsum reveals the localization of electrons. The electron localization is attributed to the coupling between electronic states at short inter-nuclear distances, while polarization interactions between dissociated ions and atoms dominate at longer inter-nuclear distances.
Results and discussion
Ultrafast electron localization
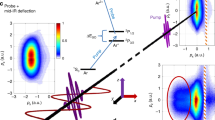
Molecular dissociation from an excited state can lead to multiple different products, followed by the localization of electrons to fragments. When N2O+ is excited to the \({\widetilde{B}}^{2}\Pi\) state, it can follow two different dissociation paths, resulting in either NO++N or NN++O products37,38,39. Our measurement involves ionizing the N2O molecules to the \({\widetilde{B}}^{2}\Pi\) state using a linear polarized pumping laser and subsequently ionizing them to dication states using an elliptical polarized probing laser. The probing laser propagates along the x-axis in the COLTRIMS with an ellipticity of 0.7. The minor axis of the laser field is along the z-direction, and the main axis is along the y-direction (see Supplementary Note 1 and Supplementary Fig. 1). Due to the angular streaking of the rotating laser fields, the electron is primarily emitted along the major axis (y-axis) but ends up with a final momentum along the minor axis (z-axis)40 (see Supplementary Note 1 and Supplementary Fig. 2). Under the momentum conservation between ions and electrons after ionization and dissociation, the total momentum of NO+ and N+ ions in the z-direction (Pzsum) can be expressed as: Pzsum = Pz\({}_{N{O}^{+}}\) + Pz\({}_{{N}^{+}}\) = −(Pze1 + Pze2). The equation shows that the Pzsum distributions can serve as a monitor for the ionization dynamics, as it is equivalent to the negative total momentum in the z-direction (Pze1 and Pze2) of the two electrons participating in the ionization process. The Pzsum distribution has been demonstrated very successfully in the study of the sequential ionization of atoms41, the multiorbital ionization, the enhanced ionization, the transient valence charge localization, and the strong-field autoionization of molecules9,24,31,32,42. Therefore, the ionization dynamics of dissociating molecules can be inferred from the time-dependent Pzsum distributions, as shown in Fig. 1. To eliminate the effect of direct double ionization, the signal is chosen by setting the KER in the range of 0.5–5.0 eV for the NO++N+ channel and 0.3–4.6 eV for the NN++O+ channel. To consider the redistribution of electrons in various dissociation channels, we select \({{{{{{{{{\rm{Py}}}}}}}}}_{N}}^{+}\) and \({{{{{{{{{\rm{Py}}}}}}}}}_{O}}^{+}\) in positive and negative directions (Py>0 or <0) for two channels and analyze the time-dependent Pzsum distributions. This analysis reveals three characteristic features, as demonstrated in Fig. 1a, b, and Fig. 1d, e for two dissociation channels.

a and b are from the NO++N+ channel, where \({{{{{{{{{\rm{Py}}}}}}}}}_{N}}^{+}\) is selected to be greater and less than 0, respectively. The time-dependent asymmetry of Pzsum distributions for this channel is presented with blue and red lines in (c). d and e show the measured time-dependent Pzsum for the NN++O+ channel when \({{{{{{{{{\rm{Py}}}}}}}}}_{O}}^{+}\) is selected to be greater and less than 0, respectively. f The measured time-dependent asymmetry of Pzsum distributions for the NN++O+ channel are presented with blue and red lines. The vertical black dashed lines indicate the delay time at 0 and 100 fs. Error bars denote statistical mean error.
Asymmetric distributions for both channels are observed in the negative delay (probe pulse before the pump pulse), where an electron is first ionized from the N2O molecule by the elliptically polarized laser, followed by the release of a second electron to form a dication by the linearly polarized laser. The asymmetry arises from the SFI of the valence orbital of N2O molecules, which reflects the asymmetric distribution of electron orbitals in the neutral molecules43. When we set Py along the positive and negative directions, the asymmetric distribution for the same channel is reversed, indicating the orientation-dependent ionization of non-centrosymmetric molecules44. Additionally, in the short positive delay regimes, there is a surprising observation that the Pzsum distribution transitions from a single peak to a symmetric bimodal distribution as the delay time increases, as indicated by the black dashed line in Fig. 1a. The combined laser fields contribute to the rapid variations around zero delay time, resulting from the overlapping of two pulses. Another regime occurs at a long positive delay time, where Pzsum exhibits a symmetric distribution with two peaks. In this regime, the linearly polarized laser ionizes the molecule and triggers dissociation into an ion and a neutral atom. Then, the second electron is released from the atom by the elliptically polarized laser, resulting in a symmetric bimodal distribution.
We define the time-dependent asymmetry of Pzsum distributions for two channels by
and the results are given in Fig. 1c, f. In the negative delay, the signs of Asy for the two channels marked by the red and blue lines are reversed. This occurs because \({{{{{{{{{\rm{Py}}}}}}}}}_{N}}^{+}\) and \({{{{{{{{{\rm{Py}}}}}}}}}_{O}}^{+}\) point in opposite directions when the same oriented molecule breaks, owing to the SFI of non-centrosymmetric molecules. By comparing the measured Asy with previous studies and considering the higher ionization yield from the HOMO-1 orbital compared to the HOMO-2 orbital39,43, it has been determined that the \({\widetilde{B}}^{2}\Pi\) state is most likely populated through SFI of one electron from the HOMO-1 orbital to the \({\widetilde{A}}^{2}{\Sigma }^{+}\) state, followed by excitation within the pump pulse. Given the less asymmetry in the orbital distribution of HOMO-2 compared to HOMO-1, it is reasonable to anticipate that the asymmetry will be reduced when an electron is emitted from the HOMO-2 orbital. Consequently, we can conclude that the contribution of HOMO-2 can be disregarded in the population of the \({\widetilde{B}}^{2}\Pi\) state.The Pzsum distributions and Asy plots in the positive delay indicate the electron and nucleus evolution after being populated in the \({\widetilde{B}}^{2}\Pi\) state. To better understand these observations, we calculate the corresponding three-dimensional potential energy surfaces (PESs) for N2O+ dissociation using the complete active space self-consistent(CASSCF) field method45,46 and the orbitals associated with two dissociation channels, as depicted in Fig. 2 (see “Numerical method” for details).

The two-dimensional PESs of N2O+ illustrate two dissociation paths leading to NO++N and NN++O, indicated by arrows. The electronic configuration of the \({\widetilde{B}}^{2}\Pi\) state is (1σ)2(2σ)2(3σ)2(4σ)2(5σ)2(6σ)2(1π)3(7σ)2(2π)4. The radial coupling interactions at conical intersections cause electronic transitions from 2π to 8σ for the NO++N channel and from 7σ to 3π for the NN++O channel.
The asymmetry in the positive delay is much weaker than that in the negative delay, as shown in Fig. 1, because, in the positive delay, both nuclei and electrons have redistribution, which is reflected by the orbital evolution in Fig. 2. In addition, it is worth noting that the sign of Asy changes when the directions of the N and O ions are selected to be the same, i.e., \({{{{{{{{{\rm{Py}}}}}}}}}_{N}}^{+}\) >0 or \({{{{{{{{{\rm{Py}}}}}}}}}_{O}}^{+}\) >0, for delays smaller than 100 fs, as indicated by the black dash lines in Fig. 1c, f. This behavior is similar to what was observed for the negative delay, suggesting that the valence electron still retains its molecular orbital distribution when the bond distance is short. Thus, the fast variation of Asy due to electron localization during molecular dissociation is captured in the short delay time. The sign of Asy remains unchanged when N and O ions point in the same direction for two channels in the longer positive delay (>100 fs). For example, both the values for blue lines are positive and red lines are negative, and they decrease and approach zero with increasing delay time. In this case, the molecules are dissociated into a molecular ion (NO+ or NN+) and an atom (N or O) in the positive delay. As shown in Fig. 2, the valence electron is localized to the atom when the bond distance (R) is larger than 7 a.u. Additionally, Asy also depends on the dipole moment, which always points from the ion to the atom after dissociation. Therefore, when N and O point in the same direction for both channels, the dipole moment changes to the same direction.
Ultrafast nuclear dynamics
In order to provide further insight into the dynamics of electron-nucleus coupling during the dissociation of N2O+, we present the time-dependent KER spectra obtained from ionizing N2O+ to the dication states for the two channels in Fig. 3. The spectra obtained from the dissociation of N2O2+ to NO++N+ and NN++O+ split into two branches, as illustrated in Fig. 3a, c. One of the branches with the unchanged KER during the scanning comes from the direct Coulomb explosion (CE) of N2O2+, either from the pump or the probe laser. The other branch exhibits a gradual decrease in KER, reaching asymptotic energies of 0.6 eV for the NO++N+ channel and 0.4 eV for the NN++O+ channel at a delay time of 6 ps. In this branch, the molecules are populated to the \({\widetilde{B}}^{2}\Pi\) state, which can rapidly couple to the 2Δ state through conical intersections, as shown in Fig. 2. Consequently, the ion dissociates along the 2Δ PES, and the electron configuration changes during this dissociation process. The coupling between the related states in the two channels also leads to different rising times for molecular dissociation (see Supplementary Note 2 and Supplementary Fig. 3).

The time-dependent KER spectra of the Coulomb explosion channels, both measured and calculated, are shown in a and b for the NO++N+ channel and c and d for the NN++O+ channel. To facilitate comparison between the experimental and theoretical spectra, the black dashed curves overlaid on the measured spectra correspond to the simulation spectra.
In the simulation, the time-dependent KER decreases as the delay time increases, with the NO++N+ channel asymptotic energy reaching 1.1 eV and the NN++O+ channel reaching 0.5 eV at a delay time of 6 ps, as shown in Fig. 3b, d. The semiclassical trajectory simulations are performed based on the calculated PESs (see “Numerical method” and Supplementary Note 3), and the fast dissociation trajectories of two channels are shown in Supplementary Fig. 4. These simulations included the excitation of the \({\widetilde{B}}^{2}\Pi\) state and its coupling to other states using the Landau–Zener surface hopping method47,48,49. The theoretically calculated time-dependent KER traces agree well with the measured ones, as indicated by the black dashed line in Fig. 3. Both the experiments and simulations demonstrate that the KER decreases rapidly in the first ps and remains nearly constant in the subsequent 5 ps. The KER (t) = E0 + Ecou(t), where the Coulomb energy Ecou(t) = 1/R(t) is determined by the molecular structure at the arrival of the probe laser, R(t) is the distance between the two fragments, and E0 is the initial KER from the dissociation of the \({\widetilde{B}}^{2}\Pi\) state. The KER reaches the dissociation energy for both channels at around 1 ps, suggesting that the ultrafast dissociation of both channels is complete within the first ps. The discrepancy between the experiment and theory in the energy difference is due to the neglect of the vibrational and rotational energy of the NO+ and NN+ fragments in the simulation, where only the ground states are included.
R-dependent molecular orbital hybridization and polarization effect
To further analyze the electron localization dynamics during a molecular cation turns into a cation with a neutral atom, we present the Pzsum distributions and corresponding Asy for two channels at different inter-nuclear distances in Fig. 4a, b. The CE approximation is used to calculate the inter-nuclear distances. The inset of Fig. 4a shows the orientation-dependent PysumPzsum distributions for the NO++N+ channel, which exhibit significant asymmetric distributions at RNN = 11 a.u. (200 fs), but become symmetric at RNN = 140 a.u. (1500 fs). As the inter-nuclear distances increase, the R-dependent asymmetries for both channels weaken, and the Asy decreases to around zero when R reaches 40 a.u. or 15.5 a.u. for the NO++N or NN++O channel, respectively. To clarify the experimental results, we utilized two theoretical methods: modified tunneling calculation for electronic dynamics and semiclassical trajectory simulations for nuclear dynamics. These methods are applied to explain the dissociation of the N2O+ molecule into NO++N and NN++O channels, which the results of the semiclassical trajectory simulations for nuclear dynamics agree well with the experimental observations, demonstrating that the dissociation of both channels starts from the \({\widetilde{B}}^{2}\Pi\) state. In the meanwhile, the simulated trajectories give the inter-nuclear distances between fragments at a certain delay time (see Supplementary Fig. 4). The electronic transition from 2π to 8σ in the NO++N channel and from 7σ to 3π in the NN++O channel is induced by the radial coupling as shown in Fig. 2. The R-dependent asymmetry parameters for both channels are calculated using the modified tunneling approach and are depicted by the blue and red lines in Fig. 4a, b. The obtained Asy traces show that both channels exhibit characteristic behavior where an asymmetry exists at short R and becomes zero as R increases. However, the simulation fails to capture the rapid rise observed for the NO++N channel before 200 fs, as illustrated in Fig. 1c. During this regime, orbital hybridization dominates the dissociation process, as shown in Fig. 2, and the advanced time-dependent quantum chemistry calculations are required to replicate the observed phenomenon.

a and b show the asymmetry parameters (Asy) obtained from measurement (blue and red circles) and calculation (blue and red lines), respectively, as a function of elongation of either the N-NO+ (RNN) or NN+-O (RNO) bond distance. The inset of (a) presents the measured PysumPzsum distributions at 200 and 1500 fs from NO++N+. c and d show the calculated difference dipole moment (Δμ) between N2O2+ and N2O+ when the N-NO+ or NN+-O bonds are breaking, starting from the \({\widetilde{B}}^{2}\Pi\) state. Error bars denote statistical mean error.
The R-dependent modified tunneling theory is used to calculate the ionization yields from oriented N2O molecules, with details provided in “Methods”. The theory accounts for the ac-Stark shifts of the ionization energies for both dissociating N2O+ and N2O2+, taking into consideration the contribution of the R-dependent dipole moments, \({\mu }^{{N}_{2}{O}^{2+}}\)(R) and \({\mu }^{{N}_{2}{O}^{+}}\)(R). The difference in permanent dipole moments between N2O2+ and N2O+ (Δμ = \({\mu }^{{N}_{2}{O}^{2+}}\)(R) − \({\mu }^{{N}_{2}{O}^{+}}\)(R)) is calculated for different inter-nuclear distances of molecules in the two channels at the cc-pVTZ level using the CI method45,46. These results are presented in Fig. 4c, d. Based on the R-dependent modified tunneling theory, the asymmetry in the electron distribution is closely linked to the difference in permanent dipole moment, Δμ, between N2O2+ and N2O+ at varying bond distances. At short bond distances, the valence electrons retain similar behavior to the molecular orbitals, resulting in a significant asymmetric distribution upon ionization. As the inter-nuclear separation increases, N2O+ dissociates into a molecular ion and an atom, and the probe laser only releases electrons from the atoms with Δμ tending to zero, as depicted in Fig. 4c, d. As a result, the atom’s SFI lacks asymmetry.
Two physical mechanisms contribute to the changing of dipole moment during the dissociation of N2O+ molecules. The first mechanism is molecular orbital hybridization, which dominates the contribution when R ≤ 10 a.u., causing electrons to redistribute between molecular ions (NN+ or NO+) and atoms (O or N). The difference in dipole moments for the two channels is similar when dominated by molecular orbital hybridization, as illustrated in Fig. 4c, d. This mechanism becomes weaker as R increases and disappears when R exceeds 10 a.u. The second mechanism is polarization, which occurs when the molecular ions (NN+ or NO+) polarize the nearby atoms (O or N), causing an axial dipole moment. This effect is long-range and can influence dipole moment up to dozens of a.u. Moreover, for R > 10 a.u., the polarization effect on the N atom is stronger than that on the O atom, resulting in a stronger dipole moment that decreases more slowly as R increases. The primary factor contributing to the difference in dipole moment between the N and O atoms is the excited states populated after the dissociation. In the NO++N channel, the N atom is excited to the 2D0 state, which is more easily polarized than the ground state 3P of the O atom in the NN+ and O dissociation channel. Despite the small dipole moment (<10−3 a.u.) caused by polarization at R > 20 a.u. in the NO++N channel, the measured Pzsum still exhibits significant asymmetry and agrees well with theoretical calculations. These findings highlight the ability of time-dependent strong-field ion momentum spectroscopy to capture the coupling between electron and nucleus during molecular dissociation.
Conclusions
We studied the orientation-dependent ultrafast bond-breakage of N2O molecules using time-resolved ion momentum spectroscopy based on strong-field tunneling ionization. Through the femtosecond-resolved measurements and semiclassical trajectory calculations, the nuclear dynamics of N-NO and NN-O dissociation channels from the same \({\widetilde{B}}^{2}\Pi\) state are tracked by time-dependent KER spectra. The electron localization of these two dissociation channels during the bond breaking has been revealed by the time- and orientation-dependent Pzsum distributions, which illustrates the entire view when the molecules are split into two separate fragments during bond breaking. During the process of bond breaking, we can follow the evolution of the differences of molecular dipole moments between charged states and their couplings to atomic motions at femtosecond time scales. The results of this study can be used to better understand nuclei and electron wavepackets evolution during molecular dissociation and to control chemical reactions.
Methods
Experimental setup
In the experiment, N2O molecules are ionized by a pair of femtosecond laser pulses (800 nm, ∽35 fs), and the resulting molecular dissociation dynamics are probed by measuring the ions’ coincident momenta. The measurements are conducted in a COLTRIMS50,51, and the fragments ions are projected to a delay-line detector by a weak, homogeneous electric field (16.9 V/cm) for mass-to-charge ratio and three-dimensional momentum distribution analysis. A pump-probe setup is employed to measure the ultrafast dynamics of the molecules, where a linearly polarized pump laser (1.0 × 1014 W/cm2) initially ionizes the molecules to the cation state, followed by the elliptically polarized probe laser (ε ∽ 0.7, 3.5 × 1014 W/cm2) further ionizes the cation to dication state. The pump and probe beams are integrated into a Mach–Zehnder interferometer, and the time zero is determined by measuring the time-dependent N2O+ signal with a cross-correlation profile of 90 fs FWHM from Gaussian fitting. Further details on the experimental setup can be found in the supplementary material and our previous publications39,47,52.
Numerical method
In theory, we employ the complete active space self-consistent field method to calculate the three-dimensional PESs for N2O+ dissociation by using Molpro program45,46. This method combines an SCF computation with full configuration interaction using a subset of orbitals (known as the active space). Figure 2 shows the PESs for elongating the N-NO or NN-O bond distance at a bending geometric configuration (ΘNNO = 165°) to describe N2O+ fragmentation. To perform the simulation of the two-body fragmentation of the two dissociated channels, we utilized a home-built program that employed the semiclassical Landau–Zener surface hopping method48. This program was evaluated in our previous works47,49. The simulation begins with initial sampling in the \({\widetilde{B}}^{2}\Pi\) state, and the ground vibration state for neutral N2O sets the initial positions of RN−NO and RNN−O. Additionally, the Wigner function determines the initial momentum. As the particles approach the avoidable point, transfer rates are calculated according to the Landau–Zener theory to achieve the transition between different electronic states. The decay dynamics of the N2O+ include two competing pathways:
N-NO channel:
NN-O channel:
Fragmentation occurs in both channels as a result of orbital overlapping and the conical intersection between the \({\widetilde{B}}^{2}\Pi\) state and 2Δ states. Statistical collection of dissociation information from both channels allows for the generation of final time-resolved KER spectra. This is accomplished by projecting the real-time nuclear geometry of molecules during the evolutions among PESs to the dication states, where the KER is accumulated from the Coulomb energies between charged species.
To explain the electron localization during the dissociation, we model the ionization process by R-dependent modified tunneling theory. The model is based on the static tunneling rate calculation53,54 and takes the Stark shifts of both dissociating N2O+ and N2O2+ energies into account. The Stark shifts lead to an effective ionization potential27,29,55, I\({}_{p}^{eff}\)(θ,t), used in the tunneling model, given by:
here, \({\mu }^{{N}_{2}{O}^{2+}}\)(t) and \({\mu }^{{N}_{2}{O}^{+}}\)(t) are the permanent dipole moment of N2O2+ and N2O+ obtained at a different inter-nuclear distance (R) of molecules from two channels; the relation between t and R is simulated from the nuclear dynamics above. Ip0(t) is the time-dependent ionization potential of N2O+ without the external fields; the polar angle between the dipole axis and the elliptically polarized probe laser’s instantaneous direction is represented by θ. The molecular parameters used for calculating ionization yields are obtained through Molpro software calculations.
Furthermore, we simulated the motion of electrons ionized by the probe laser. In this simulation, the interactions between ionized electrons and ions are set as Coulomb interactions. So, the initial positions of the electron before laser ionization satisfy the following equations
and the tunneling position of the electron follows
where, \({I}_{p}^{eff}(\theta ,t)\) is the effective ionization potential calculated by equation Eq. (6), and E(t) is the intensity of the laser field. \({{{{{{{{\boldsymbol{r}}}}}}}}}_{e}^{b}\) and \({{{{{{{{\boldsymbol{r}}}}}}}}}_{e}^{a}\) are the positions of electrons before and after tunneling ionization, respectively. rNO/NN and rN/O are the positions of molecular fragments. After tunneling ionization, the electron moves under the influence of the laser field and Coulomb field. Finally, we can obtain the concerned orientation-dependent Pzsum.
Data availability
All data supporting the findings of this study are available from the corresponding author upon a reasonable request.
Code availability
The codes used to benchmark the experimental results are available upon reasonable request.
References
Blanchet, V., Zgierski, M. Z., Seideman, T. & Stolow, A. Discerning vibronic molecular dynamics using time-resolved photoelectron spectroscopy. Nature 401, 52–54 (1999).
Bisgaard, C. Z. et al. Time-resolved molecular frame dynamics of fixed-in-space CS2 molecules. Science 323, 1464–1468 (2009).
Sansone, G. et al. Electron localization following attosecond molecular photoionization. Nature 465, 763–766 (2010).
Haessler, S. et al. Attosecond imaging of molecular electronic wavepackets. Nat. Phys. 6, 200–206 (2010).
Hockett, P., Bisgaard, C. Z., Clarkin, O. J. & Stolow, A. Time-resolved imaging of purely valence-electron dynamics during a chemical reaction. Nat. Phys. 7, 612–615 (2011).
Goulielmakis, E. et al. Real-time observation of valence electron motion. Nature 466, 739–743 (2010).
Calegari, F. et al. Ultrafast electron dynamics in phenylalanine initiated by attosecond pulses. Science 346, 336–339 (2014).
Kraus, P. M. et al. Measurement and laser control of attosecond charge migration in ionized iodoacetylene. Science 350, 790–795 (2015).
Li, W. et al. Visualizing electron rearrangement in space and time during the transition from a molecule to atoms. PNAS 107, 20219–20222 (2010).
Wörner, H. J., Bertrand, J. B., Kartashov, D. V., Corkum, P. B. & Villeneuve, D. M. Following a chemical reaction using high-harmonic interferometry. Nature 466, 604–607 (2010).
Cattaneo, L. et al. Attosecond coupled electron and nuclear dynamics in dissociative ionization of H2. Nat. Phys. 14, 733–738 (2018).
Vos, J. et al. Orientation-dependent stereo Wigner time delay and electron localization in a small molecule. Science 360, 1326–1330 (2018).
Galbraith, M. et al. Few-femtosecond passage of conical intersections in the benzene cation. Nat. Commun. 8, 1018 (2017).
Karamatskos, E. T. et al. Time-resolving the UV-initiated photodissociation dynamics of OCS. Faraday Discuss. 228, 413 (2021).
Tong, X. M., Zhao, Z. X. & Lin, C. D. Theory of molecular tunneling ionization. Phys. Rev. A 66, 033402 (2002).
Litvinyuk, I. V. et al. Alignment-dependent strong field ionization of molecules. Phys. Rev. Lett. 90, 233003 (2003).
Meckel, M. et al. Laser-induced electron tunneling and diffraction. Science 320, 1478–1482 (2009).
Staudte, A. et al. Angular tunneling ionization probability of fixed-in-space H2 molecules in intense laser pulses. Phys. Rev. Lett. 102, 033004 (2009).
Alnaser, A. S. et al. Effects of molecular structure on ion disintegration patterns in ionization of O2 and N2 by short laser pulses. Phys. Rev. Lett. 93, 113003 (2004).
Pavičić, D., Lee, K. F., Rayner, D. M., Corkum, P. B. & Villeneuve, D. M. Direct measurement of the angular dependence of ionization for N2, O2, and CO2 in intense laser fields. Phys. Rev. Lett. 98, 243001 (2007).
Mikosch, J. et al. Channel- and angle-resolved above threshold ionization in the molecular frame. Phys. Rev. Lett. 110, 023004 (2013).
Schell, F. et al. Molecular orbital imprint in laser-driven electron recollision. Sci. Adv. 4, eaap8148 (2018).
Akagi, H. et al. Laser tunnel ionization from multiple orbitals in HCl. Science 325, 1364–1367 (2009).
Wu, J. et al. Multiorbital tunneling ionization of the CO molecule. Phys. Rev. Lett. 108, 183001 (2012).
Zhang, B., Yuan, J. & Zhao, Z. Dynamic core polarization in strong-field ionization of CO molecules. Phys. Rev. Lett. 108, 183001 (2012).
Luo, S. et al. Multiorbital effects in strong-field ionization and dissociation of aligned polar molecules CH3I and CH3Br. Phys. Rev. A 96, 063415 (2017).
Holmegaard, L. et al. Photoelectron angular distributions from strong-field ionization of oriented molecules. Nat. Phys. 6, 428–432 (2010).
Trabattoni, A. et al. Setting the photoelectron clock through molecular alignment. Nat. Commun. 11, 2546 (2020).
Dimitrovski, D., Martiny, C. P. J. & Madsen, L. B. Strong-field ionization of polar molecules: Stark-shift-corrected strong-field approximation. Phys. Rev. A 82, 053404 (2010).
Tolstikhin, O. I., Morishita, T. & Madsen, L. B. Theory of tunneling ionization of molecules: weak-field asymptotics including dipole effects. Phys. Rev. A 84, 053423 (2011).
Luo, S. et al. Revealing molecular strong field autoionization dynamics. Phys. Rev. Lett. 126, 103202 (2021).
Ma, J. et al. Transient valence charge localization in strong-field dissociative ionization of HCl molecules. Phys. Rev. Lett. 127, 183201 (2021).
Fleischer, A. et al. Probing angular correlations in sequential double ionization. Phys. Rev. Lett. 107, 113003 (2011).
Walt, S. G. et al. Dynamics of valence-shell electrons and nuclei probed by strong-field holography and rescattering. Nat. Commun. 8, 15651 (2017).
Kübel, M. et al. Spatiotemporal imaging of valence electron motion. Nat. Commun. 10, 1042 (2019).
Endo, T. et al. Imaging electronic excitation of CO by ultrafast laser tunneling ionization. Phys. Rev. Lett. 116, 163002 (2016).
Lebech, M., Houver, J. C. & Dowek, D. Dissociative photoionization of N2O in the region of the N2O+ (B2Π) state studied by ion-electron velocity vector correlation. J. Chem. Phys. 120, 8226 (2004).
Chambaud, G., Gritli, H., Rosmus, P., Werner, H. J. & Knowles, P. J. The ion-molecule reaction O+(4S) + N2(X1Σ+) → NO+ (X1Σ+, v\({}^{{\prime} }\)) + N(4S) and the predissociation of the A2Σ+ and B2Π states of N2O+. Mol. Phys. 98, 1793–1802 (2000).
Zhao, X. et al. Ultrafast dissociation dynamics of singly and doubly ionized N2O in strong laser fields. Phys. Rev. A 101, 013416 (2020).
Eckle, P. et al. Attosecond angular streaking. Nat. Phys. 4, 565–570 (2008).
Pfeiffer, A. N., Cirelli, C., Smolarski, M., Dörner, R. & Keller, U. Timing the release in sequential double ionization. Nat. Phys. 7, 428–433 (2011).
Wu, J. et al. Probing the tunnelling site of electrons in strong field enhanced ionization of molecules. Nat. Commun. 3, 1113 (2012).
Afaneh, F. & Schmidt-Böcking, H. Imaging of strong field dissociative single and double ionization channels of N2O. Int. J. Mod. Phys. B 31, 1750215 (2017).
Li, X. et al. Multiorbital and excitation effects on dissociative double ionization of CO molecules in strong circularly polarized laser fields. Phys. Rev. A 100, 013415 (2019).
Knowles, P. J. & Werner, H. J. An efficient second-order MC SCF method for long configuration expansions. Chem. Phys. Lett. 115, 259 (1985).
Werner, H. J., Knowles, P. J., Knizia, G., Manby, F. R. & Schütz, M. Molpro: a general-purpose quantum chemistry program package. WIREs Comput. Mol. Sci. 2, 242 (2012).
Zhao, X. et al. Tracking the nuclear movement of the carbonyl sulfide cation after strong-field ionization by time-resolved Coulomb-explosion imaging. Phys. Rev. A 103, 053103 (2021).
Belyaev, A. K., Domcke, W., Lasser, C. & Trigila, G. Nonadiabatic nuclear dynamics of the ammonia cation studied by surface hopping classical trajectory calculations. J. Chem. Phys. 142, 104307 (2015).
Hu, X. Q. et al. Breakdown of the Coulomb-explosion imaging technique induced by the ultrafast rotation of fragments. Phys. Rev. A 101, 012707 (2020).
Dörner, R. et al. Cold target recoil ion momentum spectroscopy: a ‘momentum microscope’ to view atomic collision dynamics. Phys. Rep. 330, 95–192 (2000).
Ullrich, J. et al. Recoil-ion and electron momentum spectroscopy: reaction-microscopes. Rep. Prog. Phys. 66, 1463 (2003).
Yu, X. et al. Femtosecond time-resolved neighbor roles in the fragmentation dynamics of molecules in a dimer. Phys. Rev. Lett. 129, 023001 (2022).
Ammosov, M. V., Delone, N. B. & Krainov, V. P. Tunnel ionization of complex atoms and of atomic ions in an alternating electromagnetic field. Sov. Phys. JETP 64, 1191 (1986).
Bisgaard, C. Z. & Madsen, L. B. Tunneling ionization of atoms. Am. J. Phys. 72, 294 (2004).
Dimitrovski, D. et al. Ionization of oriented carbonyl sulfide molecules by intense circularly polarized laser pulses. Phys. Rev. A 83, 023405 (2011).
Acknowledgements
This work was supported by the National Basic Research Program of China (No. 2019YFA0307700) and the National Natural Science Foundation of China (Grants No.12134005, No.12004135, No.11934004, No.92250306, No.12074143 and No.12104063).
Author information
Authors and Affiliations
Contributions
S.L., X.H., and D.D. conceived and supervised the project. X.Zhao, X.Y., D.R., M.L., X.Zhang, X.L., P.M., D.Z., C.W., S.L., and D.D. performed the experiments, T.X., X.H., Q.W., Y.W., and J.W. carried out the simulations. X.Zhao, X.H., S.L., and D.D. wrote the manuscript. All authors discussed the results and commented on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Physics thanks Jochen Küpper and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhao, X., Xu, T., Yu, X. et al. Probing electron localization during molecular dissociation by femtosecond strong-field ion momentum spectroscopy. Commun Phys 6, 124 (2023). https://doi.org/10.1038/s42005-023-01248-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42005-023-01248-3
- Springer Nature Limited