
Abstract
Aptamers are oligonucleotides with antibody-like binding function, selected from large combinatorial libraries. In this study, we modified a DNA aptamer library with N-hydroxysuccinimide esters, enabling covalent conjugation with cognate proteins. We selected for the ability to bind to mouse monoclonal antibodies, resulting in the isolation of two distinct covalent binding motifs. The covalent aptamers are specific for the Fc region of mouse monoclonal IgG1 and are cross-reactive with mouse IgG2a and other IgGs. Investigation into the covalent conjugation of the aptamers revealed a dependence on micromolar concentrations of Cu2+ ions which can be explained by residual catalyst remaining after modification of the aptamer library. The aptamers were successfully used as adapters in the formation of antibody-oligonucleotide conjugates (AOCs) for use in detection of HIV protein p24 and super-resolution imaging of actin. This work introduces a new method for the site-specific modification of native monoclonal antibodies and may be useful in applications requiring AOCs or other antibody conjugates.
Similar content being viewed by others

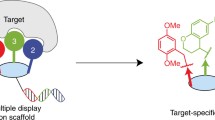

Introduction
Owing to their exquisite recognition function, antibodies (Abs) are used throughout biotechnology and medicine. Antibody-oligonucleotide conjugates (AOCs) enable technologies that combine antibody-based recognition of proteins with DNA hybridization, amplification, quantification and sequencing (reviewed by Dovgan et al.1). For example, proximity-based assays using AOCs have been developed for the sensitive detection and quantification of proteins, protein-protein interactions, and post-translational modifications2,3. Multiplexed monitoring of protein expression in single cells has been performed in conjunction with single cell transcriptome profiling with the use of AOCs4,5. DNA-PAINT is a microscopy technique that uses transient hybridization of short fluorophore-coupled oligonucleotides to AOCs to achieve sub-diffraction limit resolution6.
These aforementioned advances in protein monitoring are tempered by the inherent difficulty of generating AOCs. A number of non-specific conjugation methods have been developed, typically targeting surface lysine residues on antibodies1,7,8,9,10. Non-specific functionalization, resulting in oligodeoxynucleotide (ODN) appendages near the variable regions, can cause loss of function, and over-functionalization can result in instability. Therefore, there is continued interest in developing technologies enabling site-specific AOC synthesis. Genetic incorporation of cysteines or non-natural amino acids for specific covalent conjugation11,12 is limited to antibodies for which laborious genetic manipulation, protein expression and purification is feasible. Chemical modification of glycan moieties on the Fc fragment has recently been demonstrated for AOC synthesis13. Chemoenzymatic DNA conjugation targeting antibody glycans with a commercial kit has also been shown14. Recently, two groups published similar methods of AOC synthesis using Fc-binding protein A or protein G to site-specifically attach DNA to antibodies15,16. While this method is effective, it requires specialized variants of protein A or protein G containing the non-natural amino acid 4-benzoylphenylalanine and multiple conjugation steps, including photocrosslinking. Another study attempted to take advantage of a purported metal binding site17 on the antibody Fc region for the isolation of AOCs18. In this study, researchers were able to localize the conjugation reaction by annealing the amine-reactive ODN to an anchor DNA strand that was functionalized with a copper-binding ligand, nitriloacetic acid. While the researchers did achieve regio-specificity in the ligation reaction, recovery was low (10% yield) and Fab-conjugation was still detected18.
Other targeting agents may enable site-specific attachment of ODNs to Abs. For example, aptamers are short pieces of DNA or RNA selected from large random libraries and bind to target biomolecules with high affinity and specificity in a process known as SELEX19,20. They represent an attractive platform for generating AOCs because they are composed of nucleic acid and can potentially be synthesized along with the ODN to be attached (Fig. 1). In the first example of aptamer-based AOC synthesis, Skovsgaard et al. used a previously described human IgG1-binding aptamer for attachment of ODN with preference for the IgG heavy chain using reductive amination chemistry21. This approach- the post-SELEX addition of covalent reactivity to an aptamer, can result in a wide range of reactivities and specificities, depending on the localization of the covalently reactive group to a compatible functional group on the protein and the accessibility of the covalently reactive group to other parts of the target protein or other proteins present in the reaction.

Different parts of a typical IgG antibody and DNA conjugate are drawn and labeled. In this framework, a covalent aptamer would function as an adapter in connecting an ODN to the antibody Fc. The short red lines represent a specific residue that can react with the aptamer to form a covalent bond.
Various nucleobase chemical modifications have been developed for conjugating DNA or RNA with proteins (please refer to refs. 22,23,24,25,26 and review27). These chemistries and strategies may have utility in aptamer-based AOC synthesis by allowing the reactive group to be within the aptamer sequence and close to the antibody residue to be conjugated. For example, the reductive amination reaction used by Skovsgaard et al. for AOC synthesis utilized an aldehyde-modified thymidine base21. Copper-catalyzed azide-alkyne cycloaddition (CuAAC) or “click” chemistry has been used extensively for the modification of DNA (review by Fantoni et al.28), and specifically for the attachment of covalently reactive groups to DNA or RNA base analogs allowing for protein conjugation24,25,26.
Selection of aptamers from libraries with increased chemical diversity has been a focus in the aptamer field in the past decade29,30. A key requirement is that the chemical modifications accommodate copying and amplification of the library in SELEX (Systemic Evolution of Ligands by Exponential enrichment), the iterative process by with aptamers are selected. To enable greater diversity in library modification, display technologies termed SELMA (SELection with Modified Aptamers) have been developed where an unmodified double-stranded DNA copy is physically attached to the modified DNA or RNA library31,32,33,34,35,36. Recently, RNA-SELMA was used for addition of N-hydroxysuccinimide (NHS) esters, which react with primary amines to form amide bonds, to RNA aptamer libraries, thereby introducing the potential for covalent binding37. Upon selection against immobilized streptavidin, the library became enriched with covalent aptamers (CAs) specific for streptavidin. In this report, we choose antibodies as a target for an NHS ester-modified DNA library. We show that CAs selected from a random library can be used for the generation of AOCs that are suitable for proximity assays and DNA-PAINT.
Results
Selection of mouse IgG1-binding aptamers from an NHS ester-modified random library
Given their potential application in AOC synthesis, the possibility of isolating antibody-binding covalent DNA aptamers was explored using a DNA-SELMA display system similar to one used previously31. A summary of the library and selection scheme is in Fig. 2. A major difference between the libraries is the regeneration method, where enzymatic ligation was used in the new library, allowing for a photocleavable linker that can be used to elute the unmodified dsDNA library from the CA/IgG complex by irradiation with 350 nm light (Fig. 2). This elution strategy should result in a lower background recovery because of the retention of non-specifically bound library on the IgG-binding surface (e.g. protein A/G beads). The base composition of the library was controlled, allowing for 10% ethynyldeoxyuridine (EdU) incorporation (30% each of adenosine, cytosine, guanosine) in the random region (Fig. 2, step 1), resulting in 2-3 modifications per construct on average. The rationale for this proportion of EdU is two-fold. First, an increase in modification sites leads to potentially increased heterogeneity in the library, depending on both the CuAAC efficiency and sulfo-HSAB purity (NHS esters degrade over time). Second, chemical oligonucleotide synthesis is both simpler and more economical when fewer EdU nucleotides are incorporated into any given oligonucleotide. SELMA was initiated with a library of ~1013 different sequences. After 4 rounds of selection using a mouse monoclonal IgG1 anti-streptavidin antibody as bait and streptavidin magnetic beads for recovery, enrichment of the library was apparent based on the lowering in PCR cycle number required for efficient amplification of the library from 19 to 15 (Fig. 2, step 5). At this point, the target was changed to a different mouse IgG1 monoclonal Ab and protein A/G magnetic beads for recovery to select for binding to conserved parts of the antibody subclass. In rounds 5 and 6, amplification of the eluted library was achieved at 12 PCR cycles.

Step 1) Enzymatic incorporation of commercially available 5-ethynyl-2’-deoxyuridine triphosphate (EdUTP; replacing thymidine triphosphate) along with natural dATP, dCTP, and dGTP followed by buffer exchange and annealing of the displacement primer. Step 2) Polymerase-based displacement of the EdU-containing strand. Step 3) CuAAC modification with sulfo-HSAB. Step 4) Exposure of Mouse IgG1 monoclonal Ab (100 nM) to the library. Step 5) Magnetic bead capture of IgG1-aptamer conjugates followed by washing, elution with 350 nm light and PCR amplification of eluted conjugates. Step 6) Isolation of the non-biotinylated PCR strand and annealing of the regeneration oligonucleotide (P = 5’ phosphate). Step 7) Ligation of the two oligonucleotides to complete a single round of selection. The EdU nucleoside, its CuAAC product and the conjugate with a lysine in the target antibody are identified with and their corresponding chemical structures shown in a blue dashed box. Note that rare conjugation-proficient constructs as shown are a small fraction of the library from which they are selected.
Enriched aptamers are covalently reactive with mouse IgG1 and bind to the Fc fragment
We used gel shift assays to determine covalent conjugation of our aptamer library. SDS-PAGE is a commonly used technique that denatures proteins and resolves them by size. Therefore, any shift in apparent molecular weight by SDS-PAGE is due to covalent modification and not reversible binding. Assessment of the NHS-modified portion of the library (ssDNA region after step 3, Fig. 2) for covalent binding in excess monoclonal mouse IgG Ab revealed a high degree of conjugation compared to a naïve random library, as determined by SDS-PAGE gel shift assay with fluorescence detection of the DNA (Fig. 3A). To determine whether we had simply selected for improved CuAAC functionalization, we reacted the modified libraries with PEG5K-amine (methoxy-PEG5000-amine) which results in a stepwise decrease in migration reflective of the number of reactions between the NHS ester and large primary amine. This resulted in a significant shift in a naïve library indicating extensive PEG5000 functionalization of the library, whereas the selected library displayed only conjugation numbers of 1 or 2. Interestingly, the library covalently bound to mouse IgG2a and mouse IgG2b, with the latter less so than the former. Also, importantly, the selected library showed significant covalent binding to the Fc fragment of mouse IgG1 compared with the naïve random library (Fig. 3A). When an excess of NHS-DNA was reacted with IgG1, a distinct two-phase gel shift was apparent in a non-reducing gel, in agreement with the occupation of two sites within the Fc dimer (Fig. 3B). Monitoring the DNA in this reaction shows 2 major shifted bands, again consistent with specific labeling of one or two sites in the dimeric Fc fragment.

A Gel shift assay comparing IgG covalent binding by a naïve random library (top) and a selected library after round 6 (bottom). DNA was CuAAC-modified with sulfo-HSAB, reacted with 150 nM mouse IgG (or Fc) or 8 mM PEG5K-amine, then imaged by fluorescence. The banding pattern after reaction with PEG5K-amine indicates successful modification of the library with NHS esters. B Non-reducing SDS-PAGE gel shift assay monitoring the same gel for protein (left) and DNA (right). ssDNA library from round 6 of selection was CuAAC-modified with sulfo-HSAB and reacted with 100 ng mouse IgG1 in a library:heavy chain ratio of ~5:1. The lane containing only NHS-modified library contains 1/10th the amount reacted with the IgG1 in the last lane.
Two aptamer families dominated the library after selection
Deep sequencing was performed to determine individual clone sequences comprising the library after round 6. Two sequence families (~553,000 and ~354,000 reads) made up ~93% of all the reads (Table 1). The two families (referred to as family 1 and family 2) contained highly conserved motifs encompassing two NHS ester modification sites (T bases in the sequenced variable region), strongly suggesting a role for these short sequences in covalent binding. Family 1 was largely composed of 2 sequences differing at the 5’ end of the random region, and we will refer to the most populous sequence in this family as Clone 1. Family 2 was largely composed of a single sequence with variability at 2 distinct positions where the more common adenosine was replaced with guanosine ~1/3 of the time (Table 1) and we will refer to the most populous sequence in this family as Clone 2.
Covalent aptamer binding is dependent on Cu2+ ions
An initially puzzling feature of the library was high variability of conjugation efficiency with different modified DNA preparations, and complete inhibition of conjugation by the presence of micromolar concentrations of bovine serum albumin (BSA) in the reaction. Further testing led to a realization that the reaction is dependent on micromolar concentrations of Cu2+ ions (Figs. 3 and 4). This dependence can be explained by a variable amount of residual copper remaining in solution after copper-catalyzed click chemistry modification of the DNA library (using ~1 mM CuSO4), even after the reaction was buffer exchanged twice with a gel filtration spin column (standard procedure). Apparently, the residual Cu+ re-oxidized to Cu2+ and aided in covalent binding of the library. Inhibition by BSA can be explained by a well-documented high affinity copper binding site on serum albumin that is implicated in copper homeostasis in humans and other mammals38. Including a copper concentration higher than that of the BSA recovered covalent binding of Clone 1 (Fig. 4). Based on Fig. 4 and the faint shifted band generated by reaction with BSA, the specificity coefficient for Clone 1 reaction with 150 nM IgG1 over 3 µM BSA in the presence of saturating CuSO4 can be estimated to be 200. Copper-dependence of covalent binding was also confirmed for Clone 2 with a similar experiment (Supplementary Fig. S1). The optimal copper concentration (with limiting aptamer concentration) is ~5 µM (Supplementary Fig. S2). Copper-dependence of the conjugation reaction is reminiscent of work by Rosen et al.18, where a previously reported metal binding site was used to localize covalent linkage of DNA to the Fc fragment of mouse IgG1. It is possible that covalent aptamers obtained in this study are binding to the same copper-Ab complex. Copper interactions with DNA are well-documented39,40,41, and the presence of cationic copper in the complex is likely to reduce electrostatic repulsion between highly anionic DNA and near-neutral Fc fragment. This copper binding site is proposed to exist in IgG in species ranging from rabbit to human18, providing a potential simplified path toward further isolation of Fc-specific aptamers for IgGs from other species used for monoclonal antibody production.

Covalent conjugation is abolished in the presence of BSA and is re-established when the copper(II) concentration exceeds the BSA concentration. Each table column indicates the reagents that were combined in a reaction and loaded in the lane below. The same gel was imaged for DNA by fluorescence (left) before Coomassie staining for protein (right). The pre-stained protein ladder is brightly visible in the left gel image because it fluoresces under high laser intensity at the same wavelength as the IRDye700 fluorophore used for DNA detection. The 72 kDa protein ladder band was pink before Coomassie staining and poorly detected by the fluorescence imager.
Truncation experiments reveal structure-dependence
Clones 1 and 2 were verified to covalently bind to mouse IgG1, with Clone 1 containing a higher maximum binding fraction (40%) compared with Clone 2 (30%), consistent with their relative abundance in the deep sequencing data (Supplementary Figs. S3 and S4). Maximum binding fraction is a parameter often used to describe aptamers because aptamer folding is rarely 100% for any given sequence32,42,43. However, in this study, maximum binding fraction is also affected by the purity of the sulfo-HSAB reagent, the efficiency of NHS ester modification and background hydrolysis of the NHS ester at the reaction pH (7.2) in which the typical half-life is on the order of hours44. When reacted with 8 mM PEG5K-amine to estimate NHS ester modification efficiency, 45% and 39% Clone 1 reacted once and twice, respectively, whereas 54% and 15% of Clone 2 reacted once and twice, respectively (Supplementary Fig. S5). This suggests that neither clone was saturated with NHS esters at each possible modification site, although we cannot rule out occupation by poorly PEG5K-amine-reactive NHS esters. It also supports our library design limiting the number of modification sites in the random region to 2-3 as extensive heterogeneity in NHS modification may limit the reproducibility for any given clone in the library and lower its enrichment factor for each round of selection. The dissociation constant (KD), defined as the target concentration at which half-maximal binding is observed, is a parameter typically determined for aptamers. Because the interaction is irreversible, we cannot determine KD; however, we can estimate that the Ab concentration at which covalent binding is half-maximal for Clone 1 is ≤10 nM and Clone 2 is ≤20 nM. Stepwise truncations and other modifications were performed to determine the importance of structure in covalent bond formation. This involved the systematic removal of sequence from either side of the 76mer molecule and determination of covalent binding using SDS-PAGE and fluorescence detection. For folding predictions, we included sequence adjacent to the primer site that would have been part of the SELMA library, specifically sequence forming part of the stem region in Fig. 1. Truncation experiments suggest the requirement of larger closed structures45 in both Clone 1 and Clone 2 for efficient binding, as evidenced by a significant loss in covalent binding by truncations eliminating these features (binding is summarized in Fig. 5; please refer to the quantitative binding curves in Supplementary Figs. S3 and S4). In the major variant for Clone 1, the closed structure is likely formed by two base pairs but may be aided by adjacent sequence with partial complementarity. The minor Clone 1 variant is predicted to form a similarly closed structure with 6 base pairs with an intervening mismatch (Supplementary Fig. S6). Minimized structures for both Clone 1 and Clone 2 were confirmed to retain a significant amount of covalent binding (Table 1 and Supplementary Figs. S3 and S4).

Deduced minimized structures are shaded within the full-length construct. E denotes EdU. T in the minimized structures represents EdU bases, where T was used as a base surrogate for mFold structure prediction. A qualitative score from + (weak covalent conjugation) to +++++ (strong covalent conjugation) was assigned to each construct based on binding assays detailed in Supplementary Figs. S3 and S4.
We also asked whether both NHS ester modifications were necessary for covalent binding by Clone 1 and Clone 2. We therefore replaced EdU incorporations with thymidine in two separate constructs and measured binding (Fig. 5 and Supplementary Fig. S7). Both Clone 1 and Clone 2 displayed reduced covalent binding when the first EdU was replaced by thymidine. When the second EdU was replaced by thymidine, both clones displayed an inverse binding correlation with diminished covalent binding at higher IgG concentrations. This suggests that these EdU-to-T variants are sequestered in a non-productive complex in the presence of high concentrations of IgG.
Clones 1 and 2 target the Fc of mouse IgG1
We wanted to determine whether Clones 1 and 2 target the Fc of mouse IgG1. To this end, we reacted each clone with recombinant mouse IgG1 Fc fragment or Fab fragment isolated from mouse serum total IgG. Clone 1 showed a strong shift on non-reducing SDS-PAGE upon reaction with the Fc fragment at the lowest concentration tested (20 nM) (Fig. 6). When reacted with mouse IgG Fab, a shifted band was barely visible at 200 nM concentration (Fig. 6). Similarly, Clone 2 showed a strong shift upon reaction with recombinant Fc and showed minimal gel shift upon reaction with mouse Fc (Supplementary Fig. S8). Collectively, this data suggests that both clones specifically bind to mouse IgG1 Fc.

Clone 1 was incubated with either recombinant mouse IgG1 Fc or Fab isolated from total mouse serum IgG with 5 µM Cu2+ at the indicated concentrations, run on non-reducing SDS-PAGE and imaged by fluorescence. A strong shifted band is observed upon reaction with mouse IgG1 Fc whereas a faint shifted band is observed upon reaction with Mouse IgG Fab.
Clones 1 and 2 covalently bind to IgGs from other species
A broader panel of IgGs were tested and Clone 1 and Clone 2 showed degrees of cross-reactivity (Supplementary Fig. S9). Both clones also bound to mouse IgG2a. Clone 1 showed covalent binding to mouse IgG2b where Clone 2 conjugation was almost undetectable. Notably, Clone 1 and Clone 2 efficiently conjugated with rat IgG2b while they differed in rat IgG1 and IgG2a conjugation, with which Clone 2 retained some covalent binding while Clone 1 did not. Neither clone showed detectable cross reactivity with rabbit IgG nor human IgG2.
Application to antibody-oligonucleotide conjugation
The utility of CAs in AOC formation is realized when exogenous ODNs are attached to them, enabling site specific attachment of DNA to antibodies with CAs acting as adapters (Fig. 1). EdU-containing oligonucleotides were ordered from a commercial oligonucleotide synthesis company that contained both the CA sequences and ODN tag sequence. As mentioned previously, DNA folding prediction suggested that a minor variant of Clone 1 (13% of the library) formed a more stable closed loop structure containing the core aptamer sequence within it (Supplementary Fig. S6). A more stable structure could provide an ideal aptamer for generating AOCs because it decreases the chance that the CA functional core is affected by neighboring sequence. Therefore, a tagged, stem-loop variant of the sequence was ordered and modified with sulfo-HSAB by CuAAC and reacted with mouse IgG1. Gel electrophoresis and protein staining showed that the sequence was efficient at conjugating with the IgG1 site-specifically, with ~95% of the heavy chains modified, with no observable shift of the light chain (Fig. 7). Clone 2 is predicted to form a stable hairpin flanked by conserved sequence enclosed in a larger stem-loop structure (Fig. 5). We therefore designed an aptamer core with a single base substitution to strengthen the enclosing stem structure and attached exogenous ODN sequence to the 5’ end of this oligonucleotide.

5 µg of RPA-T4 mouse IgG1 antibody were conjugated with a tagged covalent aptamer (Proximity ligation CA-ODN 1), PEG8K-precipitated and approximately 2 µg of the re-solubilized product were run in reducing conditions. The unconjugated reduced RPA-T4 antibody was run for comparison. HC Heavy Chain, HC* conjugated Heavy Chain, LC Light Chain.
Proximity ligation assay
Functionality of aptamer-guided site-specific AOCs is critical to their broad use. To examine this, the constructs detailed above were conjugated with two commercially available monoclonal anti-HIV p24 mouse antibodies (IgG1 and IgG2a) and used in a solid-phase proximity ligation assay (Supplementary Fig. S10). Excess covalent aptamer was removed from Clone 1 and Clone 2 AOCs by selective precipitation of the AOCs in 20% PEG8000. In the proximity assay, two AOCs recognize non-overlapping epitopes on p24 protein, bringing their attached ODNs in close proximity with one another. Solid-phase recovery of AOC-p24-AOC complexes was accomplished by annealing of a biotinylated, photocleavable oligonucleotide to one of the AOCs and binding with streptavidin-coated magnetic beads. After washing the beads, elution of complexes was performed by irradiation with 350 nm (UV-A) light. The addition of a splint oligonucleotide and DNA ligase resulted in the formation of a long DNA that is amplifiable by quantitative PCR and directly related to the amount of p24 in solution. This resulted in sensitive detection of p24 with a detection limit, defined as 3 standard deviations above the control, between 10 and 100 pg/ml (Fig. 8).

CA-ODN-conjugated anti-p24 monoclonal antibodies targeting non-overlapping epitopes were mixed with different concentrations of p24, captured, washed and ligated before quantitative PCR determination. Cycle threshold (Ct) is defined as the number of PCR cycles required to amplify to a detectable level of fluorescence. ΔCt is the difference in cycle threshold (Ct) relative to the control (no p24).
DNA-PAINT
DNA-PAINT is a super-resolution microscopy technique that requires the use of AOCs, where transient hybridization of fluorophore-labeled imager strands to the DNA tag enables the localization of epitopes at nanometer resolution6. We asked whether covalent aptamers could be useful in DNA-PAINT. We conjugated a covalent aptamer containing a 25-base adapter region (Supplementary Fig. S11) that was then hybridized to a secondary adapter sequence carrying a docking strand (Supplementary Table S1). DNA-PAINT super-resolution imaging using an anti-beta tubulin-CA complex for detection resulted in specific staining of microtubule filaments (Fig. 9). However, we note that the labeling density of filaments has not resulted in the same quality as achieved previously46. Thus, further optimization of antibody-CA conjugates for cellular staining is required.

Left: DNA-PAINT overview image of microtubules stained with an antibody that is conjugated to an aptamer modified with a DNA-PAINT docking strand. Right: Zoom-in of the region highlighted in yellow. Scale bars: 5 µm (left), 1 µm (right).
Discussion
Here we report the selection of chemically modified aptamers that covalently bind to the Fc region of native mouse monoclonal antibodies. These aptamers can serve as adapters for the generation of antibody-DNA conjugates that can be used in PCR and microscopy-based protein detection assays. The rationale for NHS ester-modified aptamer libraries is that transient yet specific interactions between the modified aptamer and the target protein can position the NHS ester for efficient reaction with a primary amine, namely that on a lysine side chain or the N-terminus of the protein. Hydroxysulfosuccinimidyl-4-azidobenzoate (sulfo-HSAB) has a short, rigid benzene linker (between azide and NHS ester) and is compatible with copper-catalyzed azide-alkyne cycloaddition. The rigidity of the linker is proposed to minimize the number of conformations that can be sampled by the NHS ester, thereby minimizing non-specific reactions that can result from random collision with primary amines from non-cognate parts of the target protein or other proteins. As in a previous study of RNA CAs37, aptamers were not explicitly selected for covalent binding. Rather, when a library is modified with NHS ester warheads, it is likely that covalent binding is the simplest solution to the applied selective pressure (binding in general).
Macromolecule reactant concentration is a kinetic driver of bioconjugation reactions. Conjugation specificity is obtained only if the antibody and covalent aptamer are at concentrations that favor the specific reaction and disfavor conjugation resulting from random collision of an antibody surface lysine/N-terminus with the NHS ester of the aptamer. For the production of antibody-DNA conjugates, we carried out our reactions at an aptamer-heavy chain ratio of 5:1 and at a concentration ratio of ~40 µM/8 µM. While higher concentrations are possible, they may lead to more non-specific reaction enabled by the increase in random collisions between aptamer and biomolecule. Likewise, more dilute reactants may result in lower levels of nonspecific conjugation. Clones 1 and 2 were ~half maximally covalently bound near 20 nM heavy chain concentration and therefore, the range of concentrations allowing specific conjugation is flexible.
A surprising finding was copper dependence of our covalent aptamers. Previous work by the Gothelf laboratory18 sheds light on our observations and agrees with the existence of a copper binding site on the IgG that enables interaction with our DNA aptamers. The location of the copper binding site has not been unambiguously identified, however Gothelf et al. predict the presence of the site at a histidine-rich patch on the Fc fragment. Thus far, we have been unsuccessful at identifying the exact location of aptamer conjugation on the mouse IgG1 using mass spectrometry, a significant hindrance being the highly charged aptamer and ambiguity of aptamer stumps after nuclease digestion making LC-MS/MS identification difficult. Covalent conjugation of both Clone 1 and Clone 2 with IgGs from other animals (rat, hamster, human) suggests that optimized covalent aptamers may be isolated for these IgGs. Rat IgG2b appears to conjugate well with both Clone 1 and Clone 2. There are no obvious distinguishing characteristics of this subtype, however there is likely a set of common features between this and mouse IgG1 and IgG2a that enable efficient covalent conjugation. Collectively, these results suggest that covalent aptamers optimized for other species/subtypes may be obtained and used for the generation of AOCs, and such studies are underway.
DNA-PAINT images obtained in this study were grainy. One potential reason for less-than-optimal DNA-PAINT is the lack of signal amplification that occurs using an unmodified primary antibody and DNA-conjugated secondary antibody. We also observe grainy images using primary antibodies conjugated with DNA using other conjugation methods. Another potential explanation for sub-optimal DNA-PAINT is our use of a particularly long ODN sequence, upon which we hybridized another DNA strand that contained the landing site for the imaging strands. The rod-like hybridized duplex (20 bp) may cause steric hindrance for binding, even though it is located at the Fc. Lastly, the 5x R1 adapter strand may interact with the covalent aptamer, potentially occluding R1 landing sites. Further testing of different constructs (particularly ones that minimize bulkiness and secondary structure) may produce more effective conjugates for DNA-PAINT.
We can categorize covalent aptamers into two groups, Type-1 or Type-2, where Type-1 CAs are obtained by post-SELEX addition of covalently reactive warheads to aptamers21,25,47,48,49, and Type-2 CAs are obtained by arming SELEX libraries with covalently reactive warheads and selecting CAs directly (both types reviewed by Yang et al.50). In addition to this report, Type-2 CAs have been obtained in a few examples37,51,52,53,54. They are inherently more difficult to obtain because the SELEX strategy must accommodate chemically modified libraries as well as chemical crosslinking to the target. However, they have the benefit of being selected directly for covalent binding, which can result in improved efficiency and specificity. In support of this notion, Qin et al. recently attached SuFEx warheads to constant adenosine phosphorothioate nucleotides in an otherwise random aptamer library containing canonical cytosine, guanosine and thymidine nucleotides, and selected for CAs that bind to SARS-CoV-2 spike protein or human complement C551. Warhead-protein conjugates were cleaved from the DNA by alkaline hydrolysis, allowing for efficient PCR amplification. The authors observed a disconnect between unmodified aptamer affinity and covalent conjugation efficiency, noting the importance of selecting CAs directly from chemically modified libraries for optimal selectivity and conjugation efficiency51.
The conjugation method presented here can be added to a short list of technologies enabling efficient site-specific attachment of oligonucleotides to native antibodies13,14,15,16,18. There are features of this CA-based method that could make it broadly useful. First, the conjugation reaction is rapid, where close to 100% functionalization can be accomplished in as little as 15 min. Second, the entire aptamer-ODN construct can be synthesized at an oligonucleotide production facility, where solid-phase synthesis of the EdU-containing oligonucleotide followed by CuAAC with HSAB can be routinely performed. The end user can then simply mix the aptamer-ODN construct with their antibody of interest. An alternative strategy could be to include the CuAAC reaction in the preparation of a novel phosphoramidite building block. Following deprotection, sulfo-NHS functionalization can be carried out using well-established 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) chemistry. This approach may benefit from simpler conditions (no need for low oxygen CuAAC) and enable the processing of larger quantities of NHS-activated aptamer since nonspecific copper-DNA interactions, which can interfere with CuAAC, are avoided. Lastly, the lack of protein-based adapter molecules or glycan processing allows for a more native-like antibody for in vivo application, for example for the testing of antibody-drug conjugates using short, minimally immunogenic CAs as adapters. A full assessment of effector function and serum nuclease resistance for aptamer-modified antibodies is warranted for these uses.
We anticipate that sulfo-HSAB-modified covalent aptamers will have utility in preparative site-specific conjugation (as in this report), proteomics or specific cell surface protein labeling when total soluble protein concentration and buffer can be controlled. In contrast, covalent aptamers isolated from sulfo-HSAB-modified SELEX libraries will likely have limited utility in vivo. For example, albumin in human serum is present at approximately 0.6 mM- a concentration at which bovine serum albumin will readily react with Clone 1. That said, activated esters with lower electrophilicity or increased steric shielding around the ester may allow for greater specificity in conjugation, and we are exploring this possibility.
Materials and methods
Materials
A complete list of oligonucleotides and antibodies used in this study can be found in Supplementary Tables S1 and S2, respectively. EdUTP and EdU-containing oligonucleotides were purchased from Baseclick GMBH. Oligonucleotides with only canonical bases were purchased from Integrated DNA Technologies. Taq polymerase, ligase, Bst 2.0 warmstart polymerase, canonical dNTPs, hydrophilic streptavidin beads and protein ladder were purchased from New England Biolabs (NEB). Sephadex G-50 was purchased from Cytiva. Sulfo-HSAB was purchased from Gbiosciences. THPTA was purchased from Lumiprobe. Methoxy-PEG5000-amine (PEG5K-amine) was purchased from Creative PEGWorks.
Methods
Library generation and selection
The starting library strand (50 pmol) and regeneration hairpin (75 pmol) (Supplementary Table 1) were combined in a 1X ligation buffer (New England Biolabs (NEB)), heated to 95 °C and slowly cooled to 55 °C and put on ice before the addition of 1000 units of T4 DNA ligase in 1× buffer (New England Biolabs (NEB)) and 10 mM dithiothreitol and incubated at room temperature overnight. The ligation mix (20 µl) was added to a 50 µl reaction containing 1X thermopol buffer (NEB), 200 µM each of dATP, dCTP, dGTP and EdUTP, and 8 units of Bst 2.0 warmstart DNA polymerase (NEB) followed by incubation at 60 °C for 2 min to extend the strand with EdUTP-containing DNA. The reaction was immediately desalted using a gel filtration spin column loaded with Sephadex-G50. To the desalted material, 1X thermopol buffer (final concentration), 200 µM each of dATP, dCTP, dGTP and dTTP, 75 pmol strand displacement primer, and 200 µM dNTPs and 8 units of Bst 2.0 warmstart DNA polymerase (NEB) were added and reaction was incubated at 65 °C for 2 min. The reaction was immediately buffer-exchanged into click buffer (25 mM MES pH 6.0, 5 mM MgSO4) using a gel filtration spin column loaded with Sephadex G-50. This DNA was combined with 1.5 mM THPTA ligand (Lumiprobe) and 1 mM CuSO4 (final concentrations) in a capless 0.5 ml tube. In a separate capless tube, 25 mM sulfo-HSAB in 1X click buffer was prepared. Note: sulfo-HSAB powder must be kept desiccated at 4 °C. In another capless tube, a fresh solution of 25 mM sodium ascorbate was prepared. The three tubes were placed in a two-necked pear-shaped flask (25 ml) fitted with a rubber septum, 16-gauge needles and tubing to allow the flow of argon into one neck and the flow of circulated argon out of the other neck. Foil was placed over the entire system to minimize light degradation of the sulfo-HSAB. Argon flushing was initiated with high flow rate (~2 L/min) for 3 min followed by low flow rate (100 ml/min) for 30 min. At low flow rate, the exhaust septa/needle were removed, 8 µl sulfo-HSAB was transferred to the tube containing the library followed by the transfer of 1 µl sodium ascorbate to the same tube to initiate the reaction, followed by pipette mixing with care taken to minimize the introduction of air from the pipettes into the flask. The reaction was allowed to proceed under low argon flow for 30 min. The reaction was then buffer-exchanged twice into 1X selection buffer (20 mM HEPES pH 7.2, 150 mM NaCl, 2 mM MgSO4 using Sephadex G-50 spin columns.
The library was brought to a total volume of 50 µl with selection buffer to which 100 nM anti-streptavidin mouse IgG1 (Novus Biologicals) or IgG1 clone MOPC-21 (Tonbo Bioscienes) was added for 1 h. Then, the mixture was incubated with 0.02 mg streptavidin magnetic beads (NEB)(for anti-streptavidin target) or Protein A/G magnetic beads (PierceTM)(for MOPC-21 target) for 30 min. The beads were washed twice with selection buffer and resuspended in 20 µl of elution buffer (20 mM Tris pH 8, 150 mM NaCl) in a clear PCR tube and irradiated with 350 nm light (Sylvania F25T8/350BL) at a distance of 1 cm for 10 min with intermittent resuspension of the magnetic beads. Then, a magnet was applied and the supernatant was added to a PCR mix containing 70 pmol biotinylated forward primer, 70 pmol reverse primer, 200 uM each dNTP and 1X thermopol buffer in a total volume of 230 µl. 30 µl was removed to which 0.6 units Taq hot start polymerase (NEB) was added and distributed to 3 tubes which were immediately subjected to thermal cycling. The pilot PCR reactions were retrieved at varying cycle intervals and run on 2% agarose and visualized with ethidium bromide to determine the optimal cycle number for recovery of the library. Then, 4 units of Taq hot start were added to the remaining 200 µl reaction and the PCR was allowed to proceed to the optimal cycle number. The PCR was finished with a 10 min incubation at 72 °C to promote non-templated addition of an adenosine overhang which is critical for efficient ligation in the library regeneration step. The PCR product was then incubated with 6U of exonuclease I and incubated at 37 °C to digest unreacted primer.
Library regeneration
The 200 µl PCR reaction was incubated with 0.2 mg streptavidin magnetic beads, 12.5 mM EDTA and 500 mM NaCl for 30 min and the non-biotinylated strand isolated by incubation of the beads with 40 µl of 100 mM NaOH after washing in Tris/NaCl wash buffer and neutralized by the addition of 4 µl of 1 M HCl and 1 µl of 1 M Tris pH 8.0.
Further rounds of library generation were performed as follows: 14 µl of the neutralized ssDNA was incubated with 0.08 mg streptavidin magnetic beads to remove residual biotinylated DNA. To the DNA, 2 µl of 10X ligase buffer and 20 pmol regeneration hairpin were added. The DNA was annealed by heating to 95°C and then cooled to 55 °C. 1 mM ATP and 10 mM DTT were added and 500U of T4 ligase were added and the ligation (20 µl) was allowed to proceed overnight at room temperature. Further rounds of library generation required 30 pmol of strand displacement primer.
Next -generation sequencing
After 6 rounds of selection, the library was amplified with Illumina adapter forward and reverse primers (Supplementary Table S1), purified with a PCR clean-up kit (Qiagen) and submitted to Admera Health for sequencing with an Illumina MiSeq Nano kit. Sequences were analyzed with Aptasuite55 and summarized in Supplementary Data 1.
Covalent binding assays
EdU-containing sequences were obtained by thermocycled primer extensions in which a 400 nM primer was incubated with 20 nM template in a reaction containing 200 µM dATP, dCTP, dGTP and EdUTP and 0.02 U/µl Hot Start Taq polymerase in 25 µl reactions. The reactions were allowed to cycle between 94 °C, a primer-specific annealing temperature and 72 °C for 12 cycles. 2–3 µl of the crude reaction was then modified with sulfo-HSAB as described above. Various concentrations of IgG, antibody fragment or PEG-amine were incubated with the modified DNA for 1 h at room temperature (20–22 °C) in the presence of 5 µM CuSO4, added immediately at the time of DNA addition, unless stated otherwise. For copper titration, truncation, mutagenesis and cross-species assays, the reactions were terminated by the addition of non-reducing SDS loading buffer (1% SDS, 20 mM Tris pH 8.0, 20 mM EDTA, 5% glycerol final concentration) along with 50 nM fluorophore-labeled imaging strand complementary to a portion of the DNA sequence followed by heating at 98°C and slow cooling (0.1°/sec) to room temperature in a thermocycler. For library-based binding studies, fluorophore-labeled reverse primer was used. For truncation and mutagenesis studies, fluorophore-labeled core sequence complement was used. The reactions were then separated on dual 7%/16% (upper half/lower half) polyacrylamide/0.5X TBE gels containing 0.1% SDS and imaged directly with a Li-Cor Odyssey imager. Fluorescence measurements were made using Li-Cor Odyssey software from which binding fractions were calculated. Uncropped gel images and their corresponding fluorescence measurements can be found in Supplementary Data 2 and Supplementary Data 3, respectively.
Preparation of IgG-DNA conjugates
EdU-containing DNA was functionalized by CuAAC similarly to the library preparation, however different concentrations of reactants (40 µM DNA, 5 mM CuSO4, 5 mM THPTA) were used and sodium ascorbate (2.5 mM final concentration) was added to initiate the reaction of ≤50 µl. After the CuAAC step, 2 µl of 100 mM THPTA was added and three buffer exchanges were performed using Sephadex G-50 spin columns, the first two into click buffer and the third into selection buffer. 2 µl of 100 mM THPTA were added to the sample collection tubes of the first two spin column steps to wash copper ions from the bulk DNA before removal by gel filtration. Following the final spin column step, the DNA was added to Tris- and sodium azide-free antibody in a 5:1 (DNA:heavy chain) ratio in 10 µM CuSO4 and the reaction allowed to proceed for 60 minutes. The conjugation reaction was quenched by the addition of 100 mM Tris pH 8.0 for 1 h. Conjugates were analyzed by reducing SDS-PAGE and staining with colloidal Coomassie blue and imaged with a Li-Cor Odyssey imager with detection at 700 nm.
PEG precipitation was used to remove excess oligonucleotide from the conjugate preparations. To accomplish this, the antibody-DNA conjugation reaction was diluted to a final concentration of 20% PEG8000 (from a ~60 µl reaction volume) with 30% PEG8000. The precipitate was centrifuged at 17,900 G to the bottom of the tube, the liquid removed and the pellet resuspended with 1X selection buffer.
Proximity ligation assay
Solid-phase proximity ligation assay was performed with antibody-DNA conjugates. The first conjugate was formed between the sequence Proxlig CA-ODN 1 (Supplementary Table 1) and anti-p24 clone 39/5.4A (Abcam). The second conjugate was formed between Proxlig CA-ODN 2 and MAB73601 (R&D Systems). Following conjugation, an oligonucleotide (Proximity Ligation Primer 2) was annealed to the second conjugate at ambient temperature to rigidify ODN-2. The assay was performed as follows: Antibody-DNA conjugates were added to PBS + 0.2% Triton x-100 at a final concentration of 2 nM each. A photocleavable biotinylated capture probe was added to a final concentration of 10 nM. The mixture was incubated with varying concentrations of recombinant p24 protein for 1 h at room temperature. Complexes were captured from 25 µl volumes by incubation with 2 µg of hydrophilic streptavidin beads (NEB) and incubated for 30 min with frequent agitation. The beads were washed with PBS + 0.2% Triton X-100 and resuspended with 15 µl 1X ligase buffer, and irradiated for 10 minutes with 350 nm light (as described) with frequent mixing. The supernatant was then mixed with 5 µl containing ligase and a splint oligonucleotide (Supplementary Table S1) in ligase buffer and the ligation allowed to proceed for 1 h before 1 µl was used as template in a qPCR reaction with a Taqman probe (Supplementary Table 1) using the StepOnePlus Real-Time PCR System (Thermofisher) for amplification detection. Samples were run in triplicate.
DNA PAINT
Cells sample preparation for DNA-PAINT imaging
U-2 OS were cultured in McCoy’s 5A medium (Thermo Fisher Scientific) supplemented with 10% FBS. For imaging, cells were seeded 1 day before fixation in glass-bottomed eight-well µ-slides (ibidi, 80827).
Fixatives were preheated to 37 °C before use. U-2 OS cells were first pre-extracted with 0.3% glutaraldehyde and 0.25% Triton X-100 for 90 s, followed by fixation with 3% glutaraldehyde for 10 min. Afterwards, samples were rinsed twice with PBS (Thermo Fisher) and free aldehyde groups were reduced with 0.1% NaBH4 for 5 min. After rinsing four times with PBS, cells were blocked and permeabilized in blocking buffer (1x PBS, 2% BSA, 0.02% Tween-20) supplemented with 0.25% Triton X-100 for 2 h. Then, aptamer-conjugated anti-beta-tubulin antibody (Sigma Aldrich, T8328) was incubated at a concentration of 5 µg/ml in Blocking buffer for 90 min at room temperature. Unbound antibody washed off three times with PBS with an incubation time of 10 min for the last wash. The 5xR1 adapter strand was hybridized to the aptamer at 10 nM for 15 min diluted in imaging buffer, followed by two washes with PBS. Before imaging, 1 nM R1 imager strand diluted in imaging buffer (1xPBS, 500 mM NaCl, pH 7.2) was added to the cells.
DNA-PAINT imaging
Fluorescence imaging was carried out on an inverted microscope (Nikon Instruments, Eclipse Ti2) with the Perfect Focus System, applying an objective-type TIRF configuration equipped with an oil-immersion objective (Nikon Instruments, Apo SR TIRF ×100, NA 1.49, Oil). A 561-nm laser (MPB Communications, 2W, DPSS system) was used for excitation. The laser beam was passed through a cleanup filter (Chroma Technology, ZET561/10) and coupled into the microscope objective using a beam splitter (Chroma Technology, ZT561rdc). Fluorescence was spectrally filtered with an emission filter (Chroma Technology, ET600/50 m) and imaged on an sCMOS camera (Andor, Zyla 4.2 Plus) without further magnification, resulting in an effective pixel size of 130 nm (after 2 × 2 binning). Images were acquired by choosing a region of interest with a size of 512 × 512 pixels. Microtubules were imaged using an exposure time of 100 ms, 15,000 frames, and a laser power density of 200 W/cm2.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Data from this study are available within this article, supplementary information or from the authors upon request. NGS raw sequence data has been deposited to the Sequence Read Archive (accession PRJNA1119072).
References
Dovgan, I. et al. Antibody-Oligonucleotide Conjugates as Therapeutic, Imaging, and Detection Agents. Bioconjug. Chem. 30, 2483–2501 (2019).
Greenwood, C. et al. Proximity assays for sensitive quantification of proteins. Biomol. Detect Quantif. 4, 10–16 (2015).
Spengler, M., Adler, M. & Niemeyer, C. M. Highly sensitive ligand-binding assays in pre-clinical and clinical applications: immuno-PCR and other emerging techniques. Analyst 140, 6175–6194 (2015).
Peterson, V. M. et al. Multiplexed quantification of proteins and transcripts in single cells. Nat. Biotechnol. 35, 936–939 (2017).
Stoeckius, M. et al. Simultaneous epitope and transcriptome measurement in single cells. Nat. Methods 14, 865–868 (2017).
Schnitzbauer, J. et al. Super-resolution microscopy with DNA-PAINT. Nat. Protoc. 12, 1198–1228 (2017).
van Buggenum, J. A. et al. A covalent and cleavable antibody-DNA conjugation strategy for sensitive protein detection via immuno-PCR. Sci. Rep. 6, 22675 (2016).
Wiener, J. et al. Preparation of single- and double-oligonucleotide antibody conjugates and their application for protein analytics. Sci. Rep. 10, 1457 (2020).
Gong, H. et al. Simple Method To Prepare Oligonucleotide-Conjugated Antibodies and Its Application in Multiplex Protein Detection in Single Cells. Bioconjug. Chem. 27, 217–225 (2016).
Li, G. & Moellering, R. E. A Concise, Modular Antibody-Oligonucleotide Conjugation Strategy Based on Disuccinimidyl Ester Activation Chemistry. Chembiochem 20, 1599–1605 (2019).
Konc, J. et al. A Platform for Site-Specific DNA-Antibody Bioconjugation by Using Benzoylacrylic-Labelled Oligonucleotides. Angew. Chem. Int Ed. Engl. 60, 25905–25913 (2021).
Cuellar, T. L. et al. Systematic evaluation of antibody-mediated siRNA delivery using an industrial platform of THIOMAB-siRNA conjugates. Nucleic Acids Res. 43, 1189–1203 (2015).
Fruh, S. M. et al. Site-Specifically-Labeled Antibodies for Super-Resolution Microscopy Reveal In Situ Linkage Errors. ACS Nano 15, 12161–12170 (2021).
Jones, J. A. et al. Oligonucleotide conjugated antibody strategies for cyclic immunostaining. Sci. Rep. 11, 23844 (2021).
Cremers, G. A. O. et al. Efficient Small-Scale Conjugation of DNA to Primary Antibodies for Multiplexed Cellular Targeting. Bioconjug. Chem. 30, 2384–2392 (2019).
Stiller, C. et al. Fast and Efficient Fc-Specific Photoaffinity Labeling To Produce Antibody-DNA Conjugates. Bioconjug. Chem. 30, 2790–2798 (2019).
Hale, J. E. & Beidler, D. E. Purification of humanized murine and murine monoclonal antibodies using immobilized metal-affinity chromatography. Anal. Biochem. 222, 29–33 (1994).
Rosen, C. B. et al. Template-directed covalent conjugation of DNA to native antibodies, transferrin and other metal-binding proteins. Nat. Chem. 6, 804–809 (2014).
Blind, M. & Blank, M. Aptamer Selection Technology and Recent Advances. Mol. Ther. Nucleic Acids 4, e223 (2015).
Stoltenburg, R., Reinemann, C. & Strehlitz, B. SELEX-a (r)evolutionary method to generate high-affinity nucleic acid ligands. Biomol. Eng. 24, 381–403 (2007).
Skovsgaard, M. B. et al. Aptamer-Directed Conjugation of DNA to Therapeutic Antibodies. Bioconjug. Chem. 30, 2127–2135 (2019).
Guo, A. D. et al. Spatiotemporal and global profiling of DNA-protein interactions enables discovery of low-affinity transcription factors. Nat. Chem. 15, 803–814 (2023).
Ivancova, I. et al. Squaramate-Modified Nucleotides and DNA for Specific Cross-Linking with Lysine-Containing Peptides and Proteins. Angew. Chem. Int. Ed. Engl. 58, 13345–13348 (2019).
Monakhova, M. V. et al. Reactive Acrylamide-Modified DNA Traps for Accurate Cross-Linking with Cysteine Residues in DNA-Protein Complexes Using Mismatch Repair Protein MutS as a Model. Molecules 27, 2438 (2022).
Tivon, Y., Falcone, G. & Deiters, A. Protein Labeling and Crosslinking by Covalent Aptamers. Angew. Chem. Int. Ed. Engl. 60, 15899–15904 (2021).
Dadova, J. et al. Azidopropylvinylsulfonamide as a New Bifunctional Click Reagent for Bioorthogonal Conjugations: Application for DNA-Protein Cross-Linking. Chem. Eur. J. 21, 16091–16102 (2015).
Ivancova, I., Leone, D. L. & Hocek, M. Reactive modifications of DNA nucleobases for labelling, bioconjugations, and cross-linking. Curr. Opin. Chem. Biol. 52, 136–144 (2019).
Fantoni, N. Z., El-Sagheer, A. H. & Brown, T. A Hitchhiker’s Guide to Click-Chemistry with Nucleic Acids. Chem. Rev. 121, 7122–7154 (2021).
Odeh, F. et al. Aptamers Chemistry: Chemical Modifications and Conjugation Strategies. Molecules 25, 3 (2020).
Chan, K. Y. et al. Chemical Modifications for a Next Generation of Nucleic Acid Aptamers. Chembiochem 23, e202200006 (2022).
MacPherson, I. S. et al. Multivalent glycocluster design through directed evolution. Angew. Chem. Int Ed. Engl. 50, 11238–11242 (2011).
MacPherson, I. S., Temme, J. S. & Krauss, I. J. DNA display of folded RNA libraries enabling RNA-SELEX without reverse transcription. Chem. Commun. 53, 2878–2881 (2017).
Redman, R. L. & Krauss, I. J. Directed Evolution of 2’-Fluoro-Modified, RNA-Supported Carbohydrate Clusters That Bind Tightly to HIV Antibody 2G12. J. Am. Chem. Soc. 143, 8565–8571 (2021).
Temme, J. S. & Krauss, I. J. SELMA: Selection with Modified Aptamers. Curr. Protoc. Chem. Biol. 7, 73–92 (2015).
Temme, J. S. et al. High temperature SELMA: evolution of DNA-supported oligomannose clusters which are tightly recognized by HIV bnAb 2G12. J. Am. Chem. Soc. 136, 1726–1729 (2014).
Temme, J. S. et al. Directed evolution of 2G12-targeted nonamannose glycoclusters by SELMA. Chem. Eur. J. 19, 17291–17295 (2013).
MacPherson, I. S. & Liedl, T. Chemically “barbed” aptamers selected from a base-modified RNA library. Aptamers 2, 74–81 (2018).
Masuoka, J. et al. Intrinsic stoichiometric equilibrium constants for the binding of zinc(II) and copper(II) to the high affinity site of serum albumin. J. Biol. Chem. 268, 21533–21537 (1993).
Sagripanti, J. L., Goering, P. L. & Lamanna, A. Interaction of copper with DNA and antagonism by other metals. Toxicol. Appl Pharm. 110, 477–485 (1991).
Sorokin, V. A. et al. Studies of formation of bivalent copper complexes with native and denatured DNA. J. Inorg. Biochem 30, 87–99 (1987).
Andrushchenko, V., van de Sande, J. H. & Wieser, H. Vibrational circular dichroism and IR absorption of DNA complexes with Cu2+ ions. Biopolymers 72, 374–390 (2003).
Nimjee, S. M. et al. Rapidly regulating platelet activity in vivo with an antidote controlled platelet inhibitor. Mol. Ther. 20, 391–397 (2012).
Slinger, B. L. & Meyer, M. M. RNA regulators responding to ribosomal protein S15 are frequent in sequence space. Nucleic Acids Res 44, 9331–9341 (2016).
Kalkhof, S. & Sinz, A. Chances and pitfalls of chemical cross-linking with amine-reactive N-hydroxysuccinimide esters. Anal. Bioanal. Chem. 392, 305–312 (2008).
Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 31, 3406–3415 (2003).
Schlichthaerle, T. et al. Bacterially Derived Antibody Binders as Small Adapters for DNA-PAINT Microscopy. Chembiochem 20, 1032–1038 (2019).
Tabuchi, Y., Yang, J. & Taki, M. Inhibition of thrombin activity by a covalent-binding aptamer and reversal by the complementary strand antidote. Chem. Commun. 57, 2483–2486 (2021).
Wang, D. et al. Robust Covalent Aptamer Strategy Enables Sensitive Detection and Enhanced Inhibition of SARS-CoV-2 Proteins. ACS Cent. Sci. 9, 72–83 (2023).
Wang, R. et al. Using modified aptamers for site specific protein-aptamer conjugations. Chem. Sci. 7, 2157–2161 (2016).
Yang, J. et al. bioTCIs: Middle-to-Macro Biomolecular Targeted Covalent Inhibitors Possessing Both Semi-Permanent Drug Action and Stringent Target Specificity as Potential Antibody Replacements. Int J. Mol. Sci. 24, 3525 (2023).
Qin, Z. et al. Discovering covalent inhibitors of protein-protein interactions from trillions of sulfur(VI) fluoride exchange-modified oligonucleotides. Nat. Chem. 15, 1705–1714 (2023).
Smith, D. et al. Sensitivity and specificity of photoaptamer probes. Mol. Cell Proteom. 2, 11–18 (2003).
Smith, D. et al. In vitro selection of RNA-based irreversible inhibitors of human neutrophil elastase. Chem. Biol. 2, 741–750 (1995).
Jensen, K. B. et al. Using in vitro selection to direct the covalent attachment of human immunodeficiency virus type 1 Rev protein to high-affinity RNA ligands. Proc. Natl Acad. Sci. USA 92, 12220–12224 (1995).
Hoinka, J., Backofen, R. & Przytycka, T. M. AptaSUITE: A Full-Featured Bioinformatics Framework for the Comprehensive Analysis of Aptamers from HT-SELEX Experiments. Mol. Ther. Nucleic Acids 11, 515–517 (2018).
Acknowledgements
I.S.M. discloses support for the research of this work from the Hawaii Community Foundation [Medical Research Grant], a developmental grant from University of Washington/Fred Hutch Center for AIDS Research [an NIH funded program under award number AI027757], and a training grant awarded to the Hawaii Center for AIDS [NIH/NHLBI award number K12HL143960].
Author information
Authors and Affiliations
Contributions
N.S. performed selection and covalent binding characterization experiments. S.S. performed DNA-PAINT experiments and analysis. R.J. conceived and oversaw DNA-PAINT experiments. I.S.M. conceived selection and characterization experiments, performed sequencing, covalent binding characterization, proximity assays and data analysis. All authors took part in manuscript preparation.
Corresponding author
Ethics declarations
Competing interests
I.S.M. is an author on a patent application claiming the method of covalent aptamer isolation and specific sequences of covalent aptamers. Patent application title: “Compositions and methods for the synthesis and identification of covalent aptamers.” Author: Iain S. MacPherson. Patent application #: WO-2020102287-A1. Application filed by University of Hawaii (USA). All other authors declare no competing interests.
Peer review
Peer review information
Communications Chemistry thanks Masumi Taki and the other, anonymous, reviewer for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Soxpollard, N., Strauss, S., Jungmann, R. et al. Selection of antibody-binding covalent aptamers. Commun Chem 7, 174 (2024). https://doi.org/10.1038/s42004-024-01255-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42004-024-01255-7
- Springer Nature Limited