
Abstract
Language is supported by a distributed network of brain regions with a particular contribution from the left hemisphere. A multi-level understanding of this network requires studying its genetic architecture. We used resting-state imaging data from 29,681 participants (UK Biobank) to measure connectivity between 18 left-hemisphere regions involved in multimodal sentence-level processing, as well as their right-hemisphere homotopes, and interhemispheric connections. Multivariate genome-wide association analysis of this total network, based on genetic variants with population frequencies >1%, identified 14 genomic loci, of which three were also associated with asymmetry of intrahemispheric connectivity. Polygenic dispositions to lower language-related abilities, dyslexia and left-handedness were associated with generally reduced leftward asymmetry of functional connectivity. Exome-wide association analysis based on rare, protein-altering variants (frequencies <1%) suggested 7 additional genes. These findings shed new light on genetic contributions to language network organization and related behavioural traits.
Similar content being viewed by others


Introduction
The degree of sophistication in verbal communicative capacities is a uniquely defining trait of human beings compared to other primates. A distinctive feature of the neurobiology of language is hemispheric dominance, which is probably rooted in structural and functional asymmetries of the prenatal and infant brain1,2,3,4,5,6,7. There is some evidence for more pronounced structural and functional lateralization in relation to language as development progresses8,9, although recent precision functional imaging has indicated adult-like lateralization of the language network already by the age of 4 years10. In any case, leftward hemispheric dominance is ultimately found in around 85 percent of adults11. Most remaining adults have no clear dominant hemisphere for language, while roughly one percent show rightward hemispheric language dominance11. The left-hemisphere language network comprises various distributed regions including hubs in the inferior frontal gyrus and superior temporal sulcus12,13. However, to a lesser extent, the right hemisphere homotopic regions are also active during language tasks, especially during language comprehension rather than production13,14.
Language-related cognitive performance is highly heritable15,16,17,18,19,20, and genetic factors also play a substantial role in susceptibility to language-related neurodevelopmental disorders such as childhood apraxia of speech21, developmental language disorder (previously referred to as specific language impairment) and dyslexia22,23,24. In addition, hemispheric dominance for language builds on structural and functional asymmetries that are already present in neonates4. This suggests an early developmental basis for such asymmetries that is driven by a genetic developmental program25,26,27.
Genome-wide association studies (GWAS) in tens or hundreds of thousands of individuals have begun to identify individual genomic loci associated with language- and/or reading-related performance19, dyslexia24, brain structural asymmetry27 and/or left- or mixed-handedness28. Handedness is a behavioual manifestation of brain asymmetry with subtle and complex relations to hemispheric language dominance and language-related cognition and disorders11,24,29. The implicated genes in these GWAS tend to be most strongly expressed in the embryonic and fetal brain rather than postnatally. All together, these findings suggest that genetic contributions to inter-individual variation in language-related performance, and functional and structural brain asymmetries, exert their effects mostly early in life.
The genetic variants identified so far explain only a small proportion of the heritable variance in language-related performance or its structural underpinnings in the brain. A complementary approach to finding genes involved in language is to measure functional connectivity within the network of regions that support language in the brain, in many thousands of individuals, in order to perform well-powered GWAS. There are no existing datasets of this size that have collected functional imaging data during language task performance, but resting state functional connectivity is predictive of task-related functional activation30,31,32 and also reveals meaningful organization of the human cortex33,34. The resting state functional connectivity approach involves identifying similarities between different brain regions in terms of their time course variation in the deoxyhemoglobin to hemoglobin ratio during the resting state, i.e., while participants are awake but not performing any particular task during functional magnetic resonance imaging (fMRI). The task-free nature of resting state fMRI makes it insensitive to choices in task design that can affect lateralization estimates14, and is potentially more useful for studying the language network as a whole rather than circuits activated by one specific task. In addition, task-based fMRI has tended to find generally less heritable measures compared to resting state fMRI35, making the latter perhaps more suitable for genetic investigation.
Previous work by Mekki et al.36 found 20 loci in a genome-wide association study of functional language network connectivity based on resting state fMRI. The 25 brain regions used in their analyses to capture the brain’s language network were defined based on a meta-analysis of language-task activation across multiple previous task fMRI studies37. Of these 25 brain regions, 20 are in the left hemisphere and only 5 in the right hemisphere. The 25 regions were then analyzed jointly with no further attention to hemispheric differences. However, given the early developmental basis of functional asymmetries4, we reasoned that it may be informative for genetic association analysis to consider connectivity and hemispheric differences between all bilateral pairs of involved regions. For the present study we therefore chose a functional atlas with left and right hemisphere homotopies38, developed in the BIL&GIN cohort, which consists of ~300 young adults roughly balanced for handedness. In previous work in this cohort, a core language network was defined in right handers (N=144) based on three language tasks (reading, listening, and language production) and a resting state paradigm12. A consensus multimodal language network called SENSAAS was defined, consisting of 18 regions in the left hemisphere that were active during all three language tasks.
For the purpose of the present gene mapping study, the right hemisphere homotopic regions were also included, yielding 36 regions in total (18 per hemisphere). We derived functional connectivity measures between these 36 regions (Supplementary Fig. 1 for study design) in 29,681 participants from the UK Biobank who had genetic and brain imaging data available, yielding 630 intra- and interhemispheric connectivity measures and 153 hemispheric differences between left and right intrahemispheric connectivity. We then investigated multivariate associations of these functional connectivity phenotypes with common genetic variants, as well as polygenic scores for language-related abilities19, dyslexia24 and left-handedness28.
In addition, we hypothesized that rare, protein-altering variants could also contribute to functional language connectivity, with relatively large effects in the few people who carry them. Such variants could give more direct clues to biological mechanisms underlying the formation of the brain’s language network. Previous large-scale genetic studies of both brain29,36 and cognitive or behavioral language-related traits19,20,24 only analyzed common genetic variants (allele frequency in the population ≥1%). Tentative evidence for rare variant associations with right-hemisphere language dominance, involving actin cytoskeleton genes, was found in an exploratory study of 66 unrelated participants39. The first exome-wide association studies of the UK Biobank40,41 included structural brain imaging metrics, but not functional metrics. Therefore, the possible contributions of rare protein-coding variants to functional language connectivity had yet to be investigated in a biobank-sized data set, prior to the present study.
Results
After quality control (see Methods, section “Sample-level quality control”) we included 29,681 participants from the UK Biobank between ages 45 and 82 years, for whom single nucleotide polymorphism (SNP) genotyping array data, exome sequences, and resting state fMRI data were available, and that were in a previously defined ‘white British’ ancestry cluster42 (by far the largest single cluster in the data set). For these participants we derived 630 Pearson correlations between the time courses of the 36 regions in the language network (hereafter language network edges) and 153 hemispheric differences between left and right intrahemispheric homotopies (L-R, hereafter hemispheric differences) (Supplementary Fig. 1 and Methods, section “Imaging data preprocessing and phenotype derivation”). Positive hemispheric differences correspond to stronger connectivity on the left and negative hemispheric differences correspond to stronger connectivity on the right. We excluded language network edges or hemispheric differences with no significant heritability (nominal p ≤ 0.05) for subsequent analyses (see Supplementary Fig. 2 and Methods, section “Heritability analysis”), which left 629 edges and 103 hemispheric differences (Supplementary Data 1), among which the median SNP-based heritability was 0.070 (min: 0.018, max: 0.165) for language network connectivity and 0.026 (min: 0, max: 0.070) for hemispheric differences.
Common genetic variant associations with language network connectivity and asymmetry
The 629 heritable language network edges were entered into a multivariate genome-wide association scan (mvGWAS) with 8,735,699 biallelic SNPs (genome build hg19) that passed variant quality control (see Methods, sections “Genetic variant-level QC” and “Common variant association testing”), using the MOSTest software43 (see Methods, section “Common variant association testing”), after controlling for potential confounders including age and sex (Methods). Using the standard GWAS multiple comparison threshold (5 × 10−8), 14 independent genomic loci showed significant multivariate associations with language network edges (Fig. 1A, Supplementary Data 2, Supplementary Fig. 3). Subsequent gene mapping based on positional, eQTL and chromatin interaction information of SNPs (using FUMA44) found 111 associated genes (of which 40 were protein-coding, Supplementary Data 3). In addition, tissue expression analysis with MAGMA45 showed preferential expression of language network associated genetic effects in prenatal development in the Brainspan gene expression data46, which was significant at 21 weeks post conception but also generally elevated prenatally (Fig. 1C, Supplementary Data 4). Enrichment analysis against 11,404 gene sets (gene ontology and other curated sets)47,48 found no significant associations after correction for multiple comparisons, and cross-tissue enrichment analysis with respect to postmortem whole-body expression levels from GTEx49 also found no significantly higher expression in any particular tissue of the body (Supplementary Fig. 4 and Supplementary Data 5).

Associations with language network connectivity and asymmetry, for genetic variants with population frequencies ≥ 1 percent. Multivariate GWAS Manhattan plots for language network edges (A) and hemispheric differences (B). The genome is represented along the X axis of each Manhattan plot, with chromosomes in ascending numerical order and their p-to-q arms arranged from left to right. The Y axis of each Manhattan plot shows the pointwise significance of multivariate association, and each dot represents a single variant in the genome. The horizontal dashed line represents the threshold p ≤ 5 × 10−8 for genome-wide multiple-testing correction. Genes associated with language network edges (C) and hemispheric differences (D) tend to be most strongly expressed in prenatal brain tissue compared to postnatal brain tissue, according to MAGMA analysis of the Brainspan gene expression database. PCW: post conception week. YRS: years. The horizontal dashed line represents the threshold for multiple testing correction across all developmental stages separately. Underlying univariate beta weights for the three most significant lead SNPs for language network edges (E, from top to bottom: N = 29,681; N = 29,503; and N = 29,681 respectively), and the three most significant lead SNPs for hemispheric differences (F, from top to bottom: N = 29,444; N = 29,503 and N = 29,505 respectively). Red indicates a positive association of a given edge or hemispheric difference with increasing numbers of the minor allele of the genetic variant, and blue indicates a negative association. Plots for all lead SNPs can be found in Supplementary Fig. 8.
To probe the genetic effects on language network connectivity of our lead multivariate findings, we plotted the underlying univariate beta effect estimates across connectivity measures for each of the 14 lead SNPs, and assessed using t-tests whether the effects generally involved increased or decreased connectivity, or differed for left versus right intrahemispheric connections, or intra- versus interhemispheric connections (see Methods, section “Descriptive analysis of overall directions of effects”, Fig. 1E, Supplementary Fig. 8, Supplementary Data 6–8). We will illustrate our findings with the three most significant loci. These showed heterogeneous effects on language network connectivity (Fig. 1E). Lead SNP rs35124509 of the most significantly associated genomic locus on chromosome 3 was an exonic SNP in the EPHA3 gene, where minor allele carriers (C, minor allele frequency (MAF) = 0.39) had on average generally reduced connectivity (t = −6.673, p = 5.52 × 10−11), i.e., lower time series correlations between regions, compared to non-carriers (Fig. 1E, Supplementary Fig. 5, Supplementary Fig. 8, Supplementary Data 6-8). However, connectivity could also be higher on average for a minority of network edges in these variant carriers (Fig. 1E, Supplementary Fig. 8, Supplementary Data 6). No global differences were observed for left versus right intrahemispheric connections, or intra- versus interhemispheric connections for this SNP (Supplementary Data 7). For the second most significantly associated genomic locus, minor allele carriers (T, MAF = 0.21) of lead SNP rs2279829 (on chromosome 3) displayed increased connectivity (t = 14.606, p = 8.27 × 10−42) on average compared to non-carriers (Fig. 1E, Supplementary Fig. 6, Supplementary Fig. 8, Supplementary Data 6–8). This SNP was located upstream from the ZIC4 gene (Supplementary Fig. 5). No global differences were observed for left versus right intrahemispheric connections, or intra- versus interhemispheric connections for this SNP (Supplementary Data 8). Lead SNP rs2274224 of the third most significantly associated genomic locus (on chromosome 10) is an exonic SNP in PLCCE1:PLCE1-AS1, (Supplementary Fig. 7–8). Carriers (C, MAF = 0.44) had a stronger global increase in intrahemispheric connectivity than in interhemispheric connectivity (t = 4.5878, p = 5.41 × 10−6) compared to non-carriers (Fig. 1E, Supplementary Fig. 7, 8, Supplementary Data 8). Brain spatial pattern plots for all 14 lead SNPs can be found in Supplementary Fig. 8, and univariate betas, p-values and t-statistics in Supplementary Data 6–8.
Separately, 103 heritable hemispheric differences were also entered into a single mvGWAS, using the same procedure as for the language network edges. Three independent genomic loci were significantly associated with hemispheric differences (Fig. 1B, Supplementary Data 9–11, Supplementary Fig. 11), all of which were located on chromosome 3, and had also shown significant associations in the mvGWAS of language network edges. Lead SNP rs7625916, a different SNP in the same broader locus that encompasses EPHA3, showed a heterogeneous pattern in hemispheric differences for carriers of the minor allele (A, MAF = 0.40) (1F). This SNP was located in an intergenic region of RP11-91A15.1 (Supplementary Fig. 12). The lead SNP of the second locus rs2279829, located upstream of ZIC4 was the same as for the language network edge results. Carriers of minor effect allele (C, MAF = 0.39) displayed heterogeneous changes in hemispheric differences (Fig. 1F, Supplementary Fig. 13). The lead SNP for the third locus, rs13321297, located in an intronic region near TBC1D5, was associated with a broadly rightward shift in hemispheric differences (t = −8.767, p = 4.314 × 10−14) for carriers of the minor allele (A, MAF = 0.31, Supplementary Fig. 14). A full overview can be found in Supplementary Data 9–15. Using gene-based association mapping in FUMA we identified nine genes associated with hemispheric differences, of which four were protein-coding, namely EPHA3, TBC1D5, ZIC1 and ZIC4. Tissue expression of genes associated with hemispheric differences, using MAGMA as implemented in FUMA, was enriched prenatally in the Brainspan developmental data46, reaching significance at post-conception week 21 (Fig. 1D, Supplementary Data 11). Analysis of postmortem cross-tissue expression levels from GTEx49, and gene set analysis against 11,404 ontology and other curated sets47,48, showed no significant associations after correction for multiple comparisons (Supplementary Fig. 15 and Supplementary Data 12).
Sensitivity analyses that additionally included covariate effects of mean whole-brain functional connectivity (for the language network mvGWAS) or mean whole-brain hemispheric differences (for the hemispheric difference mvGWAS) yielded almost identical results (Supplementary Fig. 9–10, 16, 17). In principle, treating a heritable measure such as mean whole-brain functional connectivity as a covariate can bias GWAS analysis50, when such a measure is a collider rather than confound in genetic association testing. This is why we did not include such covariates in our main analysis.
Polygenic scores for language-related abilities, dyslexia and handedness
We used PRS-CS51 to calculate genome-wide polygenic scores for language-related abilities19, dyslexia24 and left-handedness28 for each of the 29,681 UK Biobank participants, using summary statistics from previous large-scale GWAS of these traits in combination with UK Biobank genotype data (see Methods, section “Associations with genetic predispositions” for details). Note that the previous GWAS of language-related abilities19 was a multivariate GWAS that considered several language-related traits that had been quantitatively assessed with different neuropsychological tests: word reading, nonword reading, spelling, and phoneme awareness. After controlling for covariates, polygenic disposition towards higher language-related abilities in the UK Biobank individuals was weakly negatively correlated with polygenic disposition towards dyslexia (r = −0.138, p = 3.504 × 10−126). Polygenic disposition towards left-handedness was not correlated with polygenic disposition as regards language-related abilities (r = −0.008, p = 0.147) or dyslexia (r = −0.005, p = 0.310).
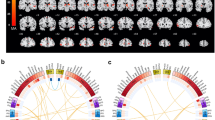
We then used canonical correlation analysis (CCA) in combination with permutation testing to estimate overall associations of polygenic scores with language network edges and hemispheric differences (see Methods, section “Associations with genetic predispositions”, Supplementary Fig. 18 for the null distributions, and Supplementary Data 16–19 for loadings and descriptive analysis of overall effect directions). Polygenic disposition to higher language-related abilities showed a significant multivariate association with language network edges (canonical correlation r = 0.160, p = 3 × 10−4) and with hemispheric differences (canonical correlation r = 0.076, p = 9.9 × 10−5). The canonical correlation loadings showed that polygenic disposition to higher language-related abilities was most notably associated with stronger left-hemisphere connectivity (t = 7.700, p = 1.924 × 10−13), with less impact on right-hemisphere connectivity, which also meant a generally leftward shift in hemispheric differences (Fig. 2A).

Multivariate associations with genome-wide polygenic dispositions to higher language-related abilities, dyslexia and left-handedness, for (A) the language network and (B) its hemispheric differences. Shown are the loading patterns on the first mode of six different CCA decompositions. Red indicates a positive association between polygenic score and brain phenotype, whereas blue indicates a negative association. N = 29,681 participants.
Polygenic disposition to dyslexia also showed significant canonical correlations with language network edges (r = 0.177, p = 9.9 × 10−5) and hemispheric differences (r = 0.078, p = 2 × 10−4), where especially interhemispheric connectivity was higher in those with higher polygenic disposition for this developmental reading disorder (t = −7.701, p = 5.278 × 10−14, Fig. 2A). In terms of hemispheric differences, higher polygenic disposition to dyslexia was associated with a broadly rightward shift in asymmetry of connectivity (Fig. 2B).
Polygenic disposition to left-handedness also showed significant canonical correlations: r = 0.154 (p = 2.16 × 10−2) for language network edges and r = 0.067 (p = 2.44 × 10−2) for hemispheric differences. Higher polygenic disposition to left-handedness was associated most notably with increased interhemispheric (t = −8.583, p = 7.258 × 10−17) and right intrahemispheric connectivity (t = −3.471, p = 5.940 × 10−4), which in terms of hemispheric differences corresponds to a broadly rightward shift in asymmetry of connectivity (Fig. 2B).
Rare, protein-coding variants and functional connectivity
The previous analyses were all based on genetic variants with population frequencies > 1 percent. We next performed a gene-based, exome-wide association scan based on protein-coding variants with frequencies <1%, using REGENIE52. We used the SKAT-O gene-based test53 for each of over 18,000 protein-coding genes with respect to 629 language network edges and 103 hemispheric differences as phenotypes, and separately using either broad (inclusive) or strict filtering for the predicted functional impacts of exonic variants (see Methods, section “Exome-wide scan” for details). Per gene we identified the lowest association p value across phenotypes (Tippet’s method), and then applied an empirical exome-wide significance threshold of 2.5 × 10−7 to account for multiple testing across genes and phenotypes (previously established using randomized phenotypes and exome data from UK Biobank, and applied in the context of thousands of phenotypes54). Five genes, NIBAN1 (p = 2.356 × 10−7), MANEAL (p = 1.338 × 10−7), SLC25A48 (p = 4.263 × 10−8), DUSP29 (p = 2.494 × 10−7) and TRIP11 (p = 2.183 × 10−7), were associated with language network edges under a broad filter (Fig. 3A, Supplementary Fig. 19, Supplementary Data 20-21) and 2 genes, WDCP (p = 2.064 × 10−7) and DDX25 (p = 2.011 × 10−8), were associated with hemispheric differences with a strict filter (Fig. 3B, Supplementary Fig. 20 and Supplementary Data 22–23).

Associations of rare protein-coding variants with language network edges or hemispheric differences. SKAT-O -LOG10 p-values for genes significantly associated with the language network edges (A) and hemispheric differences (B). C, D. Distribution of -LOG10 p-values for the significantly associated genes across all brain phenotypes. E. RNA expression values are shown over time for all four genes that were available from the Brainspan dataset (see Supplementary Data 29). Each dot represents expression levels at one timepoint in one location in the brain from one sample. Trend averages (line) and variance (shading) are shown. N = 29,681 participants.
For each of these 7 genes, the associations were based on multiple rare genetic variants present across multiple participants (Supplementary Data 24). The gene with the most distributed association pattern across functional connectivity measures of the language network was MANEAL, located on chromosome 1. Rare variants in this gene were most significantly associated with interhemispheric functional connectivity between the left middle temporal gyrus (G_Temporal_Mid-4-L) and the right supplementary motor area (G_Supp_Motor_Area-3-R), with p = 1.34 × 10−7. SKAT-O testing is flexible for testing association when individual genetic variants might have varying directions and sizes of effects on phenotypes, but its output does not provide direct insight into these directions and effect sizes in the aggregate. We therefore followed up with a burden analysis (see Methods, section “Exome-wide scan”) and found that an increased number of rare protein-coding variants in MANEAL was associated with generally decreased language network connectivity (t = −31.542, p = 1.356 × 10−131, Supplementary Fig. 21, Supplementary Data 25, 26).
Another gene with a distributed association pattern was DDX25, where rare variants were associated with multiple hemispheric differences. The hemispheric difference with the most significant association to this gene was for connectivity between the inferior frontal sulcus (S_Inf_Frontal-2) and the supplementary motor area (G_Supp_Motor_Area-2), with p = 2.01 × 10−8. Follow-up burden analysis showed that an increased number of DDX25 variants that were predicted to be deleterious was associated with a generally rightward shift in intrahemispheric connectivity asymmetry (t = −11.809, p = 8.458 × 10−21, Supplementary Fig. 22, Supplementary Data 27-28), which was most strongly for the connectivity between between the inferior frontal sulcus (S_Inf_Frontal-2) and the supplementary motor area (G_Supp_Motor_Area-2) (z = −4.1405).
The five remaining genes, NIBAN1, SLC25A48, DUSP29, TRIP11 and WDCP did not display widespread associations with respect to language network connectivity measures or hemispheric differences (Fig. 3C, D), but rather were driven by one or a few individual edges or hemispheric differences.
Discussion
Studying the genetics of language-related brain traits, such as language network functional connectivity in the resting state, can yield clues to developmental and neurobiological mechanisms that support the brain’s functional architecture for language. In this study we report common genetic variant, polygenic and exonic rare variant associations with language network functional connectivity, and/or hemispheric differences of connectivity. We found 14 genomic loci associated with language network edges and 3 of these loci were also associated with hemispheric differences. EPHA3 was the most significantly associated gene based on common genetic variants. A polygenic disposition for higher language-related abilities was associated with a leftward shift in functional connectivity asymmetry, while polygenic dispositions to dyslexia and left-handedness were associated with rightward shifts in functional connectivity asymmetry. Lastly, exome-wide scanning suggested five genes associated with language network edges and 2 genes with hemispheric differences on the basis of rare, protein-coding variants. MANEAL and DDX25 showed distributed association profiles across multiple regional brain connectivity measures.
The most significant association we found was on the 3p11.1 locus, near the EPHA3 gene, which codes for ephrin type-A receptor 3. EPHA3 is involved in developmental processes such as neurogenesis, neural crest cell migration, axon guidance and fasciculation55,56,57 and is preferentially expressed 8–24 weeks post-conception. This genomic locus has previously shown association with individual differences in both resting state functional connectivity35,36,58 and white matter connectivity36,59 in the frontotemporal semantic network. Here we add to the literature that this locus is also associated with hemispheric differences of language network functional connectivity, although with leftward shifts for some homotopic pairs of connections and rightward shifts for others, without an overall average trend towards one hemisphere. EPHA3 may therefore be involved in the development of left-right asymmetries in the brain that support hemispheric specialization for language.
A second locus associated with language network connectivity and asymmetry was located in 3p24.3, near the TBC1D5 gene, which codes for subunit TBC1 domain family member 5. This gene may act as a GTPase-activating protein for Rab family protein(s), and is expressed in all tissues, including the brain60. TBC1D5 is involved in cell processes related to macroautophagy and receptor metabolism. Recent studies have found associations of this gene with functional language network connectivity36, white matter61, dyslexia24, and health-related associations with Parkinson’s Disease62 and schizophrenia63. Again, here we add an association with hemispheric differences that implies a role in development of the left-right axis in the brain that supports language lateralization.
In total, of the 14 genomic loci we found, 12 were previously reported in other GWAS of brain traits35,36,58,59. Two loci that have no previous literature associated with them in the GWAS Catalog64 were a locus on the pseudo-autosomal part of the X and Y chromosome, with rs2360257 as lead SNP, and a locus on 3q22.2, with rs143322006 as lead SNP. The latter is intergenic to EPHB1, and therefore this novel finding underscores a potential role of ephrin receptors in functional connectivity of the brain’s language network. The well-known functions of ephrins in axon guidance for nerve fiber tract formation are likely to be relevant in this context.
The other 12 loci were found in two prior GWAS studies of functional connectivity36,58, both of which differed from each other and from the present study in terms of connectomic methodologies. This suggests that connectome methodological choices only partially influence the discovery of genetic loci, i.e., some genetic influences on brain functional connectivity can be relatively robustly detected across different methodological choices. Six out of 14 loci were also found in a study of the white matter connectome59, which confirms that functional and structural connectivity have partially overlapping genetic architectures.
The overlap of significant loci from the present study with those found in GWAS studies of dyslexia, language-related abilities and handedness was more limited. The 3p24.3 locus from the present study was found in a large GWAS for dyslexia24, and the 17q21.31 locus was also associated with left-handedness65. This limited overlap probably relates, at least in part, to limited statistical power in these different GWAS studies of cognitive and behavioral traits to identify particular loci at genome-wide significant levels (i.e., not surpassing stringent multiple testing correction for genome-wide association testing, even if they might have shown associations to a lesser extent). Similarly, studies have also reported a limited number of overlapping genome-wide significant loci between psychiatric disorders and structural brain traits59,66,67. It is also possible that some genetic variants with influences on functional connectivity of the brain’s language network are not relevant to individual differences in language-related cognition or behavior. This may reflect that there are functionally relevant aspects of brain network architecture for language-related cognition which are not captured by resting fMRI and/or a parcel-based approach to its analysis. Nonetheless, our analysis of polygenic scores (discussed further in the section below) clearly indicates that genetic influences on language-related abilities, dyslexia and left-handedness are also associated with functional connectivity and asymmetry within the language network.
Furthermore, the genes we identified through genetic analysis of language network connectivity in the present study are likely to be involved in fetal development of the brain’s language network and its lateralization, as evidenced by our analysis of gene expression data in the brain across the lifespan. This is consistent with reports of the prenatal appearance of molecular and structural brain asymmetries (reviewed by ref. 5), and also with studies that have detected leftward functional lateralization of auditory or language networks in infants and young children (see refs. 5,6,10 and the Introduction). It is therefore likely that much of the heritable variance in language network functional connectivity in the adult brain is established early in life.
Genome-wide polygenic scores for language-related abilities, dyslexia, or left-handedness were significantly but subtly associated at the population-level with language network functional connectivity and asymmetry. These subject-level polygenic scores quantify the cumulative effects of common genetic variants from across the genome on a given trait. The leftward shift of asymmetry in people with polygenic dispositions to higher language-related abilities is consistent with functional asymmetry reflecting an optimal organization for language processing. Although language performance and functional language lateralization do not seem to be strongly correlated in healthy adults68,69, an absence of clear hemispheric language dominance has been reported to associate with slightly reduced cognitive functioning across multiple domains70.
The rightward shift in asymmetry of language network connectivity with higher polygenic disposition to dyslexia is in line with some previous studies in smaller samples that suggested decreased left hemisphere language dominance in dyslexia, although this previous evidence was often inconsistent and inconclusive71,72,73,74. This association also converges in its direction with the association of TBC1D5 with hemispheric differences described above. Our study therefore illustrates how large-scale brain imaging genetic analysis of genetic disposition to a human cognitive disorder can inform on the neurobiological correlates of the disorder, even when carried out using general population data.
The rightward shift in asymmetry of language network functional connectivity with higher polygenic scores for left-handedness that we observed is consistent with increased right hemisphere language dominance in left-handers11,29,75. Causality cannot be determined in a cross-sectional dataset of the kind used in our study. For example, genetic disposition may affect prenatal brain development in ways that alter functional asymmetries, and this seems likely given that many of the relevant genes are upregulated in the prenatal brain, and that functional asymmetries already exist in neonates4. However, some functional asymmetries may also follow, or be reinforced through, behaviors that are influenced by genetic disposition28. Consistent with this latter possibility, a meta-analysis of neuroimaging studies of dyslexia suggested that reduced left-hemisphere dominance is only present in adults and not in children72. The UK Biobank consists of middle-aged and older adults, but future studies of polygenic risk for dyslexia should test the association with brain connectiviy in younger samples, to help address the developmental/aging questions.
It is important to recognize that gene-brain associations in general population data are usually subtle28,76 and also that canonical correlations tend to increase with the number of variables, due to higher degrees of freedom77. However, as we only used the first canonical mode and only tested a single polygenic score on one side of the correlation in each analysis (versus multiple brain traits on the other side), then the freedom of the canonical correlation was relatively restricted. The permutation test that we used showed that all multivariate associations with polygenic scores were greater than expected by chance. Furthermore, the first canonical mode has previously been shown to be the most replicable78 as it captures the most variance. Cross-validation in canonical correlation analysis is often employed for supervised model evaluations, but our use here was unsupervised and descriptive, for which there is no clear procedure for model evaluation77. Our interest was to describe the most accurate overall association between polygenic disposition to a given trait and brain functional connectivity measures in the available sample.
We report associations of five genes, NIBAN1, MANEAL, SLC25A48, DUSP29 and TRIP11, with language network connectivity and two genes, WDCP and DDX25, with hemispheric differences on the basis of rare, protein-coding variants from exome sequence data. No previous rare variant associations have been reported with any of these seven genes40,41, but MANEAL has been previously implicated in a GWAS of mathematical ability based on common genetic variants79, which testifies broadly to its relevance for cognitive function. The protein encoded by MANEAL is found in the Golgi apparatus80 and may regulate alpha-mannosidase activity. Previous work has shown relatively high expression of this gene in the brain compared to various other tissues60. DDX25 is a DEAD box protein with the Asp-Glu-Ala-Asp motif, involved in RNA processing. Tissue expression for DDX25 is also relatively high in the brain or testis compared to other tissues60. The roles of these seven genes in brain development and function remain to be studied, for example using model systems such as cerebral organoids or knockout mice.
The exome-wide association analysis that we used here involved mass univariate testing with respect to brain connectivity measures, rather than multivariate modeling. For common genetic variants, several multivariate association frameworks have been developed, one of which we used here for our common variant GWAS (MOSTest)43. Such methods generally provide increased statistical power to detect effects compared to mass univariate testing, when genetic variants are associated with phenotypic covariance. However, such multivariate methods are currently lacking for application to the study of rare, protein-coding variants in Biobank-scale samples, where the effects of individual variants must be aggregated at the gene level and computational feasibility is an important consideration. The development of new multivariate methods for exome-wide analysis is required. As the findings in our exome-wide association scan only surpassed the multiple testing correction threshold by a small amount, we regard these findings as tentative until they might be replicated in the future in other datasets.
Resting state functional connectivity does not provide a direct measurement of language lateralization. In this study we quantified resting state functional connectivity between regions that were previously found to be involved in language on the basis of fMRI during sentence-level reading, listening and production tasks12, and also where left-right homotopic regions were defined for the investigation of hemispheric differences. The use of full correlations as connectivity measures, as is common in the field, means that an increase in connectivity between a pair of regions can also be indirect through other regions81. Another caveat is that individual anatomical differences may seep into functional connectivity measures when a hard parcellation is used81,82. However, as the literature has shown more broadly, structural brain properties can make meaningful contributions to functional connectivity and it might not be possible to fully disentangle the two83,84,85,86.
Issues with respect to our chosen methods for genetic association testing have been discussed above. A general point is that we used one large discovery sample of 29,681 participants to maximize power in our GWAS, polygenic association analysis, and exome-wide scan. This did not allow for a discovery-replication design. However, using the largest available sample leads to the most accurate estimate of any possible association, including of its effect size. In light of this, the utility of discovery-replication designs has declined in relevance with the rise of biobank-scale data87.
A limitation of the UK Biobank is that participation is on a voluntary basis, which has led to an overrepresentation of healthy participants rather than being fully representative of the general population76,88.
In conclusion, we report 14 genomic loci associated with language network connectivity or its hemispheric differences based on common genetic variants. Polygenic dispositions to lower language-related abilities, dyslexia and left-handedness were associated with generally reduced leftward asymmetry of functional connectivity in the language network. Exome-wide association analysis based on rare, protein-altering variants (frequencies ≤1 %) suggested 7 additional genes. These findings shed new light on the genetic contributions to language network connectivity and its hemispheric differences based on both common and rare genetic variants, and reveal genetic links to language- and reading-related abilities and hemispheric dominance for hand preference.
Methods
Participants
Imaging and genomic data were obtained from the UK Biobank42 as part of research application 16066 from primary applicant Clyde Francks. The UK Biobank received ethical approval from the National Research Ethics Service Committee North West-Haydock (reference 11/NW/0382), and all of their procedures were performed in accordance with the World Medical Association guidelines. Informed consent was obtained for all participants89. Analyses were conducted on 29,681 participants that remained after quality control of genotype, exome and imaging data (see below).
Imaging data
Brain imaging data were collected as described previously90,91. In this analysis resting state fMRI data were used (UK Biobank data-field 20227, February 2020 release90,91). Identical scanners and software platforms were used for data collection (Siemens 3T Skyra; software platform VD13). For collection of rs-fMRI data, participants were instructed to lie still and relaxed with their eyes fixed on a crosshair for a duration of 6 min. In that timeframe 490 datapoints were collected using a multiband 8 gradient echo EPI sequence with a flip angle of 52°, resulting in a TR of 0.735 s with a resolution of 2.4 × 2.4 × 2.4 mm3 and field-of-view of 88 × 88 × 64 voxels. Our study made use of pre-processed image data generated by an image-processing pipeline developed and run on behalf of UK Biobank (see details below).
Genetic data
Genome-wide genotype data (UK Biobank data category 263) was obtained by the UK Biobank using two different genotyping arrays (for full details see ref. 42). Imputed array-based genotype data contained over 90 million SNPs and short insertion-deletions with their coordinates reported in human reference genome assembly GRCh37 (hg19). In downstream analyses we used both the unimputed and imputed array-based genotype data in different steps (below).
Exome sequencing data were obtained and processed as described in more detail elsewhere40,54,92 (UK Biobank data category 170, genome build GRCh38). Briefly, the IDT xGen Exome Research Panel v.1.0 was used to capture exomes. Samples were sequenced using the Illumina NovaSeq 6000 platform with S2 (first 50,000 samples) or S4 (remaining samples) flow cells and were processed by the UK Biobank team according to the OQFE Protocol (https://hub.docker.com/r/dnanexus/oqfe). Analyses using individual-level exome data (UK Biobank data field 23157) were conducted on the Research Analysis Platform (https://UKBiobankiobank.dnanexus.com).
Sample-level quality control
Sample-level quality control at the phenotypic and genetic level was conducted on 40,595 participants who had imaging, genotype and exome data available. In phenotype sample-level quality control, participants were first excluded with imaging data labeled as unusable by UK Biobank quality control. Second, participants were removed based on outliers (here defined as 6× interquartile range (IQR)) in at least one of the following metrics: discrepancy between rs-fMRI brain image and T1 structural brain image (UK Biobank field 25739), inverted temporal signal-to-noise ratio in preprocessed and artifact-cleaned preprocessed rs-fMRI (data fields 25743 and 25744), scanner X, Y, and Z brain position (fields 25756, 25757 and 25758) or in functional connectivity asymmetries (see section “Imaging data preprocessing and phenotype derivation”). Third, participants with missing data in the connectivity matrices were excluded. In total 3472 participants were excluded in the phenotype QC.
Subsequently, in genetic sample-level quality control, only participants in the pre-defined white British ancestry cluster were included (data-field 22006)42, as this was the largest single cluster in terms of ancestral homogeneity—an important consideration for some of the genetic analyses that we carried out (below). Furthermore, participants were excluded when self-reported sex (data-field 31) did not match genetically inferred sex based on genotype data (data-field 22001) or exome data, when sex chromosome aneuploidy was suspected (data-field 22019), or when exclusion thresholds were exceeded in heterozygosity (≥0.1903) and/or genotype missingness rate (≥0.05) (data-field 22027). Finally, one random member of each pair of related participants (up to third degree, kinship coefficient ≥0.0442, pre-calculated by UK Biobank) was removed from the analysis. This led to the further exclusion of 7442 participants. In total 29,681 participants were included in all further analyses.
Imaging data preprocessing and phenotype derivation
Preprocessing was conducted by the UK Biobank and consisted of motion correction using MCFlirt93, intensity normalization, high-pass filtering to remove temporal drift (sigma = 50.0 s), unwarping using fieldmaps and gradient distortion correction. Structured scanner and movement artifacts were removed using ICA-FIX.94,95,96 Preprocessed data were registered to a common reference template in order to make analyses comparable (the 6th generation nonlinear MNI152 space, http://www.bic.mni.mcgill.ca/ServicesAtlases/ICBM152NLin6).
On the local compute cluster at the MPI for Psycholinguistics, network connectivity was derived based on the AICHA atlas38. Key properties of the AICHA atlas are its homotopies. For each of the 192 parcels left and right hemisphere functional homotopies were defined. Of these 192 pairs, 7 regions were previously excluded from the atlas due to poor signal on the outside of the brain38, leaving 185 parcel pairs. Time courses were extracted from the AICHA atlas using invwarp and applywarp from FSL (v. 5.0.1097) and mri_segstats from Freesurfer (v.6.0.098). Correlations between time courses were derived with numpy (v.1.13.1) using Python 2.7 and were transformed to z-scores using a Fisher transform in order to achieve normality. In addition, only the upper diagonal values were used. These values can be considered a measure of connection strength between two regions. Functional hemispheric differences (L-R) were derived for each connection, and outliers (6 × IQR) were excluded. Previous work identified 18 regions as part of the core language network in multiple language processing domains (reading, listening and speaking12). These 18 regions and their homotopies were used in this analysis.
Two different types of imaging-derived phenotypes (IDPs) were extracted and used in genetic analyses. First, all 630 Z-transformed correlation values were included, including both intra- and interhemispheric connectivity. Second, for all intrahemispheric connectivity edges, hemispheric differences (L-R) were included, yielding 153 edge hemispheric differences. In total this yielded 783 new IDPs for further analysis.
Genetic variant-level QC
Four different genetic datasets were prepared, as needed for four different analysis processes:
-
1.
Array-based genotype data were filtered, maintaining variants with linkage disequilibrium (LD) ≤0.9, minor allele frequency (MAF) ≥ 0.01, Hardy-Weinberg Equilibrium test p-value ≥ 1 × 10−15 (see52), and genotype missingness ≤0.01 for REGENIE step 1 (below).
-
2.
Imputed genotype data were filtered, maintaining bi-allelic variants with an imputation quality ≥0.7, Hardy-Weinberg Equilibrium test p-value ≥1 × 10−7 and genotype missingness ≥0.05 for association testing in MOSTest (below).
-
3.
For genetic relationship matrices SNPs were only used if they were bi-allelic, had a genotype missingness rate ≤0.02, a Hardy Weinberg Equilibrium p-value ≥1 × 10−6, an imputation INFO score ≥0.9, a MAF ≥0.01, and a MAF difference ≤0.2 between the imaging subset and the whole UK Biobank were used.
-
4.
For exome sequence data, only variants in the 39 Mbp exome sequencing target regions were retained (UK Biobank resource 3803), excluding variants in 100 bp flanking regions for which reads were not checked for coverage and quality standards in the exome processing pipeline. Monoallelic variants (marked with a ‘MONOALLELIC’ filter flag) were also removed. Then, individual-level genotypes were set to no-call if the read depth was ≤7 (for single nucleotide variants) or ≤10 (for indel variant sites) and/or if the genotype quality was ≤20. Variant-level filtering comprised removal of variants sites with an average GQ (which is the Phred-scaled probability that the call is incorrect) across genotypes ≤35, variant missingness rate ≥0.10, minor allele count (MAC) ≤1, and/or low allele balance (only for variants with exclusively heterozygous genotype carriers; ≤0.15 for SNV sites, ≤0.20 for INDEL variant sites). Transition-transversion ratios were calculated prior to and after variant-level filtering as an indicator of data quality. Filtered pVCF files were converted to PLINK binary format, dropping multi-allelic variants, and then merged per chromosome. For the X chromosome, pseudo-autosomal regions (PAR1: start - base pair 2781479, PAR2: base pair 155701383 – end, genome build GRCh38) were split off from the rest of chromosome X. Any heterozygous haploid genotypes in the non-PAR chr X were set to missing.
Statistics and reproducibility
Heritability analysis
Genetic relationship matrices (GRMs) were computed for the study sample using GCTA v. 1.93.0beta99. In addition to the previous sample-level quality control, individuals with a genotyping rate ≤0.98 and one random individual per pair with a kinship coefficient ≥0.025 derived from the GRM were excluded from heritability analysis. The SNP-based heritability of each of the 783 newly derived IDPs was estimated using genome-based restricted maximum likelihood (GREML) in GCTA v. 1.93.0beta99. IDPs with heritabilities that passed a nominal significance threshold of p ≤0.05 were included in subsequent analysis, similarly to previous studies36,59 and in line with recommendations for mvGWAS43.
Common variant association testing
Multivariate common variant association testing (mvGWAS) was performed using the MOSTest toolbox43 for all heritable measures, separately for all 629 heritable language network edges and all 103 heritable hemispheric differences. MOSTest fully accounts for the multivariate nature by estimating the correlation structure on permuted genotype data and then computing the Mahalanobis norm as the sum of squared de-correlated z-values across univariate GWAS summary statistics and then fitting a null distribution using a gamma cumulative density function to extrapolate beyond the permuted data to significant findings. The multivariate z-statistic from MOSTest is always positive and does not provide information on directionality. We used imputed genotype array data and the following covariates: sex, age, age2, age × sex, the first 10 genetic principle components that capture genome-wide ancestral diversity, genotype array (binary variable) and various scanner-related quality measures (scanner X, Y and Z-position, inverted temporal signal to noise ratio and mean displacement as an indication of head motion) (see Supplementary Table 1 for UK Biobank field IDs). For sensitivity analyses we also included additional covariate effects of mean whole-brain functional connectivity (for the language network mvGWAS) or mean whole-brain hemispheric differences (for the hemispheric difference mvGWAS). Genome-wide significant variants were annotated using the online FUMA platform (version 1.5.2)44. MAGMA (version 1.08)45 gene analysis in FUMA was used to calculate gene-based p-values and for gene-property analyses, to investigate potential gene sets of interest47,48 and to map the expression of associated genes in a tissue-specific49 and time-specific46 fashion. Gene sets smaller than 10 were excluded from the analysis, due to risk for statistical inflation.
Associations with genetic predispositions
In order to understand how language network edges and hemispheric differences relate to genetic predisposition for language-related abilities (quantitatively assessed in up to 33,959 participants from the GenLang consortium)19, dyslexia (51,800 cases and 1,087,070 controls) from 23andMe, Inc.24 and left-handedness (33,704 cases and 272,673 controls) from UK Biobank participants without imaging data28, we used polygenic scores and canonical correlation analysis (CCA) for each polygenic score separately. Polygenic scores were calculated with PRS-CS51, which uses a Bayesian regression framework to infer posterior effect sizes of autosomal SNPs based on genome-wide association summary statistics. PRS-CS was applied using default parameters and a recommended global shrinkage parameter phi = 0.01, combined with LD information from the 1000 Genomes Project phase 3 European-descent reference panel. PRS-CS performed in a similar way to other polygenic scoring methods, with noticeably better out-of-sample prediction than an clumping and thresholding approach100,101. Before entering polygenic scores into a CCA analysis, they were residualised for these covariates: sex, age, age2, age × sex, the first 10 genetic principle components that capture genome-wide ancestral diversity, genotype array (binary variable) and various scanner-related quality measures (scanner X, Y and Z-position, inverted temporal signal to noise ratio and mean displacement as an indication of head motion) (see Supplementary Table 1 for UK Biobank field IDs). Polygenic scores were then normalized using quantile_transform from scikit-learn v.1.0.1 and entered into a CCA analysis, also using scikit-learn. As correlation values in CCA tend to increase with the number of variables, we permuted the polygenic scores 10,000 times to build a null distribution of correlation values between IDPs and permuted polygenic scores and tested whether the correlation values of the first mode were outside the 95th percentile of the null distribution.
Exome-wide scan
For rare variant association testing REGENIE v.3.2.1 was used52. In brief, REGENIE is a two-step machine learning method that fits a whole genome regression model and uses a block-based approach for computational efficiency. In REGENIE step 1, array-based genotype data were used to estimate the polygenic signal in blocks across the genome with a two-level ridge regression cross-validation approach. The estimated predictors were combined into a single predictor, which was then decomposed into 23 per-chromosome predictors using a leave one chromosome out (LOCO) approach, with a block size of 1000, 4 threads and low-memory flag. Phenotypes were transformed to a normal distribution in both REGENIE step 1 and 2. Covariates for both steps included sex, age, age2, age × sex, the first 10 genetic principle components that capture genome-wide ancestral diversity, genotype array (binary variable) and various scanner-related quality measures (scanner X, Y and Z-position, inverted temporal signal to noise ratio and mean displacement as an indication of head motion) (see Supplementary Table 1 for UK Biobank field IDs). Common and rare variant association tests were run conditional upon the LOCO predictor in REGENIE step 2. Functional annotation of variants was conducted using snpEff v5.1d (build 2022-04-19)102. Physical position in the genome was used to assign variants to genes and were annotated with Ensembl release 105. Combined Annotation Dependent Depletion (CADD) Phred scores for variants were taken from the database for nonsynonymous functional prediction (dbNSFP) (version 4.3a)103 using snpSift 5.1d(build 2022-04-19). Variants were then classified for downstream analysis based on their functional annotations to either be included in a “Strict” or “Broad” filter or be excluded from further analysis. The “Strict”-filter only included variants that were annotated with a “High” impact on a canonical gene transcript (variant types include highly disruptive mutations like frameshifts) outside of the 5% tail end of the corresponding protein (high-impact variants in the 5% tail ends usually escape nonsense-mediated decay) or a “Moderate” effect on a canonical gene transcript combined with CADD Phred score ≥20 (these include likely deleterious protein-altering missense variants). The second “Broad” set of variants also included “High” annotated variants affecting alternative gene transcripts outside of 5% tail ends, “Moderate” annotated variants that affected canonical or alternative gene transcripts with CADD Phred scores of at least 1, and “Modifier” variants that affected canonical or alternative gene transcripts with CADD Phred scores of at least 1 (see Supplementary Table 2). A higher CADD score entails higher predicted deleteriousness of a SNP104. In REGENIE step 2, we performed a gene-based SKAT-O test53 with strict and broad variant filters based on functional annotation with all heritable IDPs. A SKAT-O test is most appropriate in our study design as we had no a priori hypothesis about the direction of the genetic effect. Multivariate exome testing was conducted separately for language network edges and hemispheric differences by using Tippet’s method which involves taking the lowest p-value across the phenotypes of interest. This was previously used as validation method for development of MOSTest43 and was shown to be less sensitive than multivariate genetic association testing in common variants. We adjusted for the exome-wide gene-based multiple comparison burden using an empirical p-value threshold for Type 1 error control from previous work (2.5 × 10−741). This was computed as 0.05 × the average p value from 300 random phenotypes with varying heritabilities and UK Biobank exome data and approximates 0.05 expected false positives per phenotype. We then followed up significant results using (i) burden testing for assessing the effect of genetic mutation burden on brain connectivity and (ii) confirmatory variant-level association testing on the significant genes to describe which variants drove the gene-based associations.
Descriptive analysis of overall directions of effects
In order to test for overall patterns in the directions of genetic effects across multiple connections (for SNPs, polygenic scores, or gene-based rare variant burden scores), we performed the following t-tests (as implemented in the python module scipy v. 1.9.3) on the effect measures, i.e., z-scores (for SNPs or burden scores) or mode loadings (for polygenic scores):
-
1.
For whether effects involved a general increase or decrease across 629 network connectivity edges, we tested whether there was a significant difference from zero using a one-sample two-tailed t-test. A positive t-value indicates an average increase in connectivity, a negative t-value indicates an average decrease in connectivity.
-
2.
For whether effects differed on 153 left versus 153 right (i.e., homotopic) intrahemispheric edges, we used a two-sample two-tailed t-test. A positive t-value indicates generally stronger left intrahemispheric connectivity, a negative t-value indicates generally stronger right intrahemispheric connectivity.
-
3.
For whether effects differed on 306 intrahemispheric edges versus 323 interhemispheric edges, we used a two-sample two-tailed t-test. A positive t-value indicates stronger intrahemispheric connectivity, a negative t-value indicates stronger interhemispheric connectivity.
-
4.
For whether effects involved general increases or decreases in 103 hemispheric differences (L-R), we tested for a significant difference from zero using a one-sample two-tailed t test. A positive t value indicates stronger left intrahemispheric connectivity, a negative t-value indicates stronger right intrahemispheric connectivity.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The primary data used in this study are from the UK Biobank. These data can be provided by UK Biobank pending scientific review and a completed material transfer agreement. Requests for the data should be submitted to the UK Biobank: https://www.ukbiobank.ac.uk. Specific UK Biobank data field codes are given in the Methods section. Other publicly available data sources and applications are cited in the Methods section. We have made our mvGWAS summary statistics available online within the GWAS catalog: https://ebi.ac.uk/gwas/. Numerical source data for figures 1A and B can be found in the summary statistics as deposited in GWAS Catalog https://ebi.ac.uk/gwas/. Numerical source data for figures 1C-F can be found in Supplementary Data 4, 6, 11 and 13. Numerical source data for Fig. 2 can be found in Supplementary Data 16 and 18. Numerical source data for Fig. 3 can be found in Supplementary Data 21, 23 and 29.
Code availability
This study used openly available software and codes, specifically GCTA (https://cnsgenomics.com/software/gcta/#GREML105), MOSTest (https://github.com/precimed/mostest), FUMA, MAGMA (https://ctg.cncr.nl/software/magma, as implemented in FUMA), PRS-CS (https://github.com/getian107/PRScs), REGENIE (https://rgcgithub.github.io/regenie/install/106). Custom code for this study is available from https://github.com/jsamelink/langnet_paper107. All other data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials.
References
Dehaene-Lambertz, G., Dehaene, S. & Hertz-Pannier, L. Functional neuroimaging of speech perception in infants. Science 298, 2013–2015 (2002).
Telkemeyer, S. et al. Sensitivity of newborn auditory cortex to the temporal structure of sounds. J. Neurosci. 29, 14726–14733 (2009).
Telkemeyer, S. et al. Acoustic processing of temporally modulated sounds in infants: evidence from a combined near-infrared spectroscopy and EEG study. Front. Psychol. 2, 62 (2011).
Williams, L. Z. J. et al. Structural and functional asymmetry of the neonatal cerebral cortex. Nat. Hum. Behav. 7, 942–955 (2023).
Schmitz, J., Güntürkün, O. & Ocklenburg, S. Building an asymmetrical brain: the molecular perspective. Front. Psychol. 10, 982 (2019).
Dehaene-Lambertz, G. The human infant brain: a neural architecture able to learn language. Psychonom. Bull. Rev. 24, 48–55 (2017).
Perani, D. et al. Neural language networks at birth. Proc. Natl Acad. Sci. 108, 16056–16061 (2011).
Olulade, O. A. et al. The neural basis of language development: changes in lateralization over age. Proc. Natl Acad. Sci. 117, 23477–23483 (2020).
Qi, T., Schaadt, G. & Friederici, A. D. Cortical thickness lateralization and its relation to language abilities in children. Dev. Cogn. Neurosci. 39, 100704 (2019).
Ozernov-Palchik, O. et al. Precision fMRI reveals that the language network exhibits adult-like left-hemispheric lateralization by 4 years of age (2024).
Mazoyer, B. et al. Gaussian mixture modeling of hemispheric lateralization for language in a large sample of healthy individuals balanced for handedness. PLOS ONE 9, e101165 (2014).
Labache, L. et al. A SENtence Supramodal Areas AtlaS (SENSAAS) based on multiple task-induced activation mapping and graph analysis of intrinsic connectivity in 144 healthy right-handers. Brain Struct. Funct. 224, 859–882 (2019).
Malik-Moraleda, S. et al. An investigation across 45 languages and 12 language families reveals a universal language network. Nat. Neurosci. 25, 1014–1019 (2022).
Bradshaw, A. R., Thompson, P. A., Wilson, A. C., Bishop, D. V. M. & Woodhead, Z. V. J. Measuring language lateralisation with different language tasks: a systematic review. PeerJ 5, e3929 (2017).
Dale, P. et al. Genetic influence on language delay in two-year-old children. Nat. Neurosci. 1, 324–328 (1998).
Le Guen, Y., Amalric, M., Pinel, P., Pallier, C. & Frouin, V. Shared genetic aetiology between cognitive performance and brain activations in language and math tasks. Sci. Rep. 8, 17624 (2018).
Newbury, D. F., Bishop, D. V. M. & Monaco, A. P. Genetic influences on language impairment and phonological short-term memory. Trends Cogn. Sci. 9, 528–534 (2005).
Andreola, C. et al. The heritability of reading and reading-related neurocognitive components: a multi-level meta-analysis. Neurosci. Biobehav. Rev. 121, 175–200 (2021).
Eising, E. et al. Genome-wide analyses of individual differences in quantitatively assessed reading- and language-related skills in up to 34,000 people. Proc. Natl Acad. Sci. 119, e2202764119 (2022).
Verhoef, E., Shapland, C. Y., Fisher, S. E., Dale, P. S. & St Pourcain, B. The developmental origins of genetic factors influencing language and literacy: Associations with early-childhood vocabulary. J. Child Psychol. Psychiatry 62, 728–738 (2021).
Eising, E. et al. A set of regulatory genes co-expressed in embryonic human brain is implicated in disrupted speech development. Mol. Psychiatry 24, 1065–1078 (2019).
Deriziotis, P. & Fisher, S. E. Speech and language: translating the genome. Trends Genet. 33, 642–656 (2017).
Bates, T. C. et al. Genetic and environmental bases of reading and spelling: a unified genetic dual route model. Read. Writ. 20, 147–171 (2007).
Doust, C. et al. Discovery of 42 genome-wide significant loci associated with dyslexia. Nat. Genet. 54, 1621–1629 (2022).
de Kovel, C. G. F., Carrión-Castillo, A. & Francks, C. A large-scale population study of early life factors influencing left-handedness. Sci. Rep. 9, 584 (2019).
Francks, C. Exploring human brain lateralization with molecular genetics and genomics. Ann. N. Y. Acad. Sci. 1359, 1–13 (2015).
Sha, Z. et al. The genetic architecture of structural left–right asymmetry of the human brain. Nat. Hum. Behav. 5, 1226–1239 (2021).
Sha, Z. et al. Handedness and its genetic influences are associated with structural asymmetries of the cerebral cortex in 31,864 individuals. Proc. Natl Acad. Sci. 118, e2113095118 (2021).
Wiberg, A. et al. Handedness, language areas and neuropsychiatric diseases: insights from brain imaging and genetics. Brain 142, 2938–2947 (2019).
Tavor, I. et al. Task-free MRI predicts individual differences in brain activity during task performance. Science 352, 216–220 (2016).
Joliot, M., Tzourio-Mazoyer, N. & Mazoyer, B. Intra-hemispheric intrinsic connectivity asymmetry and its relationships with handedness and language Lateralization. Neuropsychologia 93, 437–447 (2016).
Labache, L., Ge, T., Yeo, B. T. T. & Holmes, A. J. Language network lateralization is reflected throughout the macroscale functional organization of cortex. Nat. Commun. 14, 3405 (2023).
Smith, S. M. et al. Correspondence of the brain’s functional architecture during activation and rest. Proc. Natl Acad. Sci. 106, 13040–13045 (2009).
Margulies, D. S. et al. Situating the default-mode network along a principal gradient of macroscale cortical organization. Proc. Natl Acad. Sci. 113, 12574–12579 (2016).
Elliott, L. T. et al. Genome-wide association studies of brain imaging phenotypes in UK Biobank. Nature 562, 210–216 (2018).
Mekki, Y. et al. The genetic architecture of language functional connectivity. NeuroImage 249, 118795 (2022).
Yarkoni, T., Poldrack, R. A., Nichols, T. E., Van Essen, D. C. & Wager, T. D. Large-scale automated synthesis of human functional neuroimaging data. Nat. Methods 8, 665–670 (2011).
Joliot, M. et al. AICHA: An atlas of intrinsic connectivity of homotopic areas. J. Neurosci. Methods 254, 46–59 (2015).
Carrion-Castillo, A. et al. Genome sequencing for rightward hemispheric language dominance. Genes, Brain Behav. 18, e12572 (2019).
Backman, J. D. et al. Exome sequencing and analysis of 454,787 UK Biobank participants. Nature 599, 628–634 (2021).
Karczewski, K. J. et al. Systematic single-variant and gene-based association testing of thousands of phenotypes in 394,841 UK Biobank exomes. Cell Genom. 2, 100168 (2022).
Bycroft, C. et al. The UK Biobank resource with deep phenotyping and genomic data. Nature 562, 203–209 (2018).
van der Meer, D. et al. Understanding the genetic determinants of the brain with MOSTest. Nat. Commun. 11, 3512 (2020).
Watanabe, K., Taskesen, E., van Bochoven, A. & Posthuma, D. Functional mapping and annotation of genetic associations with FUMA. Nat. Commun. 8, 1826 (2017).
de Leeuw, C. A., Mooij, J. M., Heskes, T. & Posthuma, D. MAGMA: generalized gene-set analysis of GWAS data. PLOS Comput. Biol. 11, e1004219 (2015).
Sunkin, S. M. et al. Allen Brain Atlas: an integrated spatio-temporal portal for exploring the central nervous system. Nucleic Acids Res. 41, D996–D1008 (2013).
Subramanian, A. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl Acad. Sci. 102, 15545–15550 (2005).
Liberzon, A. et al. Molecular signatures database (MSigDB) 3.0. Bioinformatics 27, 1739–1740 (2011).
THE GTEX CONSORTIUM. The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science 369, 1318–1330 (2020).
Aschard, H., Vilhjálmsson, B. J., Joshi, A. D., Price, A. L. & Kraft, P. Adjusting for heritable covariates can bias effect estimates in Genome-Wide Association studies. Am. J. Hum. Genet. 96, 329–339 (2015).
Ge, T., Chen, C.-Y., Ni, Y., Feng, Y.-C. A. & Smoller, J. W. Polygenic prediction via Bayesian regression and continuous shrinkage priors. Nat. Commun. 10, 1776 (2019).
Mbatchou, J. et al. Computationally efficient whole-genome regression for quantitative and binary traits. Nat. Genet. 53, 1097–1103 (2021).
Lee, S. et al. Optimal unified approach for rare-variant association testing with application to small-sample case-control whole-exome sequencing studies. Am. J. Hum. Genet. 91, 224–237 (2012).
Szustakowski, J. D. et al. Advancing human genetics research and drug discovery through exome sequencing of the UK Biobank. Nat. Genet. 53, 942–948 (2021).
Pasquale, E. B. Eph-ephrin bidirectional signaling in physiology and disease. Cell 133, 38–52 (2008).
Gibson, D. A. & Ma, L. Developmental regulation of axon branching in the vertebrate nervous system. Development 138, 183–195 (2011).
Gerstmann, K. & Zimmer, G. The role of the Eph/ephrin family during cortical development and cerebral malformations. Med. Res. Arch. 6 (2018).
Zhao, B. et al. Common variants contribute to intrinsic human brain functional networks. Nat. Genet. 54, 508–517 (2022).
Sha, Z., Schijven, D., Fisher, S. E. & Francks, C. Genetic architecture of the white matter connectome of the human brain. Sci. Adv. 9, eadd2870 (2023).
Fagerberg, L. et al. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol. Cell. Proteom. 13, 397–406 (2014).
Fan, C. C. et al. Multivariate genome-wide association study on tissue-sensitive diffusion metrics highlights pathways that shape the human brain. Nat. Commun. 13, 2423 (2022).
Nalls, M. A. et al. Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: a meta-analysis of genome-wide association studies. Lancet Neurol. 18, 1091–1102 (2019).
Trubetskoy, V. et al. Mapping genomic loci implicates genes and synaptic biology in schizophrenia. Nature 604, 502–508 (2022).
Sollis, E. et al. The NHGRI-EBI GWAS Catalog: knowledgebase and deposition resource. Nucleic Acids Res. 51, D977–D985 (2023).
Cuellar-Partida, G. et al. Genome-wide association study identifies 48 common genetic variants associated with handedness. Nat. Hum. Behav. 5, 59–70 (2021).
Sha, Z. et al. The overlapping genetic architecture of psychiatric disorders and cortical brain structure (2023).
Roelfs, D. et al. Genetic overlap between multivariate measures of human functional brain connectivity and psychiatric disorders. Nat. Ment. Health 2, 189–199 (2024).
Knecht, S. et al. Behavioural relevance of atypical language lateralization in healthy subjects. Brain 124, 1657–1665 (2001).
Bruckert, L. Is language laterality related to language abilities? http://purl.org/dc/dcmitype/Text (University of Oxford, 2016).
Mellet, E. et al. Weak language lateralization affects both verbal and spatial skills: an fMRI study in 297 subjects. Neuropsychologia 65, 56–62 (2014).
Leonard, C. M. & Eckert, M. A. Asymmetry and dyslexia. Dev.l Neuropsychol. 33, 663–681 (2008).
Richlan, F., Kronbichler, M. & Wimmer, H. Meta-analyzing brain dysfunctions in dyslexic children and adults. NeuroImage 56, 1735–1742 (2011).
van der Mark, S. et al. The left occipitotemporal system in reading: disruption of focal fMRI connectivity to left inferior frontal and inferior parietal language areas in children with dyslexia. NeuroImage 54, 2426–2436 (2011).
Kershner, J. R. Neuroscience and education: cerebral lateralization of networks and oscillations in dyslexia. Laterality 25, 109–125 (2020).
Zago, L. et al. Predicting hemispheric dominance for language production in healthy individuals using support vector machine. Hum. Brain Mapp. 38, 5871–5889 (2017).
Sha, Z., Schijven, D. & Francks, C. Patterns of brain asymmetry associated with polygenic risks for autism and schizophrenia implicate language and executive functions but not brain masculinization. Mol. Psychiatry 26, 7652–7660 (2021).
Wang, H.-T. et al. Finding the needle in a high-dimensional haystack: canonical correlation analysis for neuroscientists. NeuroImage 216, 116745 (2020).
Smith, S. M. et al. A positive-negative mode of population covariation links brain connectivity, demographics and behavior. Nat. Neurosci. 18, 1565–1567 (2015).
Lee, J. J. et al. Gene discovery and polygenic prediction from a genome-wide association study of educational attainment in 1.1 million individuals. Nat. Genet. 50, 1112–1121 (2018).
Gaudet, P., Livstone, M. S., Lewis, S. E. & Thomas, P. D. Phylogenetic-based propagation of functional annotations within the Gene Ontology consortium. Brief. Bioinforma. 12, 449–462 (2011).
Bijsterbosch, J. et al. Challenges and future directions for representations of functional brain organization. Nat. Neurosci. 23, 1484–1495 (2020).
Bijsterbosch, J. D., Valk, S. L., Wang, D. & Glasser, M. F. Recent developments in representations of the connectome. NeuroImage 243, 118533 (2021).
Bignardi, G. et al. Genetic effects on structural and functional properties of sensorimotor-association axis of cortical organization are selectively distinct. https://doi.org/10.1101/2023.07.13.548817 (2024).
Llera, A., Wolfers, T., Mulders, P. & Beckmann, C. F. Inter-individual differences in human brain structure and morphology link to variation in demographics and behavior. eLife 8, e44443 (2019).
Pang, J. C. et al. Geometric constraints on human brain function. Nature 618, 566–574 (2023).
Suárez, L. E., Markello, R. D., Betzel, R. F. & Misic, B. Linking Structure and Function in Macroscale Brain Networks. Trends Cogn. Sci. 24, 302–315 (2020).
Huffman, J. E. Examining the current standards for genetic discovery and replication in the era of mega-biobanks. Nat. Commun. 9, 5054 (2018).
Fry, A. et al. Comparison of sociodemographic and health-related characteristics of UK biobank participants with those of the general population. Am. J. Epidemiol. 186, 1026–1034 (2017).
Sudlow, C. et al. UK Biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLOS Med. 12, e1001779 (2015).
Alfaro-Almagro, F. et al. Image processing and quality control for the first 10,000 brain imaging datasets from UK Biobank. NeuroImage 166, 400–424 (2018).
Miller, K. L. et al. Multimodal population brain imaging in the UK Biobank prospective epidemiological study. Nat. Neurosci. 19, 1523–1536 (2016).
Van Hout, C. V. et al. Exome sequencing and characterization of 49,960 individuals in the UK Biobank. Nature 586, 749–756 (2020).
Jenkinson, M., Bannister, P., Brady, M. & Smith, S. Improved optimization for the robust and accurate linear registration and motion correction of brain images. NeuroImage 17, 825–841 (2002).
Beckmann, C. & Smith, S. Probabilistic independent component analysis for functional magnetic resonance imaging. IEEE Trans. Med. Imaging 23, 137–152 (2004).
Griffanti, L. et al. ICA-based artefact removal and accelerated fMRI acquisition for improved resting state network imaging. NeuroImage 95, 232–247 (2014).
Salimi-Khorshidi, G. et al. Automatic denoising of functional MRI data: combining independent component analysis and hierarchical fusion of classifiers. NeuroImage 90, 449–468 (2014).
Jenkinson, M., Beckmann, C. F., Behrens, T. E. J., Woolrich, M. W. & Smith, S. M. FSL. NeuroImage 62, 782–790 (2012).
Fischl, B. FreeSurfer. NeuroImage 62, 774–781 (2012).
Yang, J., Lee, S. H., Goddard, M. E. & Visscher, P. M. GCTA: a tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 88, 76–82 (2011).
Ni, G. et al. A comparison of ten polygenic score methods for psychiatric disorders applied across multiple cohorts. Biol. Psychiatry 90, 611–620 (2021).
Zheutlin, A. B. et al. Penetrance and pleiotropy of polygenic risk scores for schizophrenia in 106,160 patients across four health care systems. Am. J. Psychiatry 176, 846–855 (2019).
Cingolani, P. et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 6, 80–92 (2012).
Liu, X., Li, C., Mou, C., Dong, Y. & Tu, Y. dbNSFP v4: a comprehensive database of transcript-specific functional predictions and annotations for human nonsynonymous and splice-site SNVs. Genome Med. 12, 103 (2020).
Rentzsch, P., Witten, D., Cooper, G. M., Shendure, J. & Kircher, M. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 47, D886–D894 (2019).
Yang, J. et al. Jianyangqt/gcta: GCTA. Zenodo (2021).
Mbatchou, J. et al. Rgcgithub/regenie: Regenie v3.2.1. Zenodo (2022).
Amelink, J. Jamelink/langnet_paper: Final submission. Zenodo (2024).
Acknowledgements
This research was funded by the Max Planck Society (Germany), together with grants from the Netherlands Organisation for Scientific Research (NWO) (grant number 054-15-101) and French National Research Agency (ANR, grant No. 15-HBPR-0001-03) as part of the FLAG-ERA consortium project “MULTI-LATERAL”, a Partner Project to the European Union’s Flagship Human Brain Project, and the Language in Interaction consortium (NWO Gravitation grant number 024-001-006). The study was conducted using the UK Biobank resource under application no. 16066 with C.F. as the principal applicant. Our study made use of quality-controlled brain images generated by an image-processing pipeline developed and run on behalf of the UK Biobank. The funders had no role in study design, data collection and analysis, and the decision to publish or preparation of the manuscript. The authors thank Else Eising, Giacomo Bignardi and Tristan Looden for their thoughts on the methodology. The authors thank Fabrice Crivello and Antonietta Pepe for their involvement in the inception of this project. The authors would like to thank the research participants and employees of 23andMe, Inc. for making this work possible.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
Conceptualization—J.S.A, M.C.P., X.Z.K., M.J., S.E.F., C.F; Methodology—J.S.A, X.Z.K., Z.S., D.S., A.C.C., B.M., S.S-N., M.J.; Software—J.S.A., X.Z.K., D.S, M.J.; Formal analysis—J.S.A., M.C.P., X.Z.K., D.S., Z.S.; Data curation—J.S.A., X.Z.K., D.S.; Writing - original draft—J.S.A.; Writing—review & editing—M.C.P., X.Z.K., A.C.C., S-S.N, Z.S., D.S., B.M., M.J., S.E.F., C.F.; Visualization—J.S.A.; Project administration—C.F.; Resources - S.E.F, C.F.; Funding acquisition—C.F., S.E.F., M.J.; Supervision—S.E.F., C.F.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks Gesa Schaadt and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editors: Sahar Ahmad and Joao Valente.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Amelink, J.S., Postema, M.C., Kong, XZ. et al. Imaging genetics of language network functional connectivity reveals links with language-related abilities, dyslexia and handedness. Commun Biol 7, 1209 (2024). https://doi.org/10.1038/s42003-024-06890-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-024-06890-3
- Springer Nature Limited