
Abstract
The microRNAs (miRNAs) of their hosts play an important role in regulating both the innate and adaptive immune responses to Cryptosporidium parvum infection. The mechanisms of autophagy and apoptosis are important components of the defense system against C. parvum infection. In this study, we investigate the role of miRNA-199a-3p in regulating MTOR-mediated autophagy and apoptosis in HCT-8 cells induced by C. parvum. The expression of miR-199a-3p increased at 3, 6 and 12 hours postinfection (hpi) but decreased at 24 and 48 hpi. The upregulation of miR-199a-3p promoted autophagy and apoptosis and limited the parasite burden in HCT-8 cells after C. parvum infection. The downregulation of miR-199a-3p inhibited the autophagy and apoptosis induced by C. parvum and enhanced the parasite burden in HCT-8 cells. A luciferase reporter showed that MTOR was a target gene of miR-199a-3p. Suppressed expression of MTOR by small interfering RNA (siRNA) promoted autophagy and apoptosis and limited C. parvum burden in HCT-8 cells. Co-transfection with miR-199a-3p inhibitor or si-mTOR revealed that miR-199a-3p regulates autophagy and apoptosis in HCT-8 cells through MTOR, to resist C. parvum infection. In conclusion, intestinal epithelial cells defend against C. parvum infection by regulating their autophagy and apoptosis through the miR-199a-3p-MTOR axis.
Similar content being viewed by others
Introduction
Cryptosporidium is considered the second leading cause of diarrhea and death in children and is one of the world’s most important zoonotic protozoa1. To date, 49 valid species of Cryptosporidium are recognized, 20 of which have been identified in humans, with Cryptosporidium parvum and Cryptosporidium hominis most frequently detected2,3. Cryptosporidium parvum primarily infects the gastrointestinal tracts of animals and humans. It undergoes excystation in the gastrointestinal tract to release infective sporozoites, which blunt the intestinal villi and cause crypt hyperplasia, cytoskeletal remodeling, epithelial chemokine secretion, and so on4,5. At present, the mechanism underlying the intracellular survival of C. parvum in cells and the cause of these cytopathic effects are poorly understood, which is the major factor limiting effective treatment6. Further study of the Cryptosporidium invasion mechanism should facilitate the search for new therapeutic drug targets.
MiRNAs are a highly conserved group of small noncoding RNAs that modulate the expression of their target genes, which are associated with various biological and cellular processes7,8. They regulate gene expression at the posttranscriptional level by binding to the 3’ -untranslated regions (3’ -UTRs) of their target mRNAs, either perfectly or imperfectly, thus regulating a number of cellular processes, including cell proliferation, apoptosis, and autophagy7,9. For example, miR-20a and miR-106b control ULK1 expression; Beclin 1 is regulated by miR-30a; miR-145 regulates the expression of BCL2 interacting protein 3 (BNIP3) to regulate autophagy ; and miR-26a-5p silences PTEN gene expression to regulate apoptosis10,11,12,13. MiRNAs are important players in the regulation of the immune response of host epithelial cells after C. parvum infection. Let-7i regulates the expression of Toll-like receptor 4 and contributes to the immune response of cholangiocytes against infection with C. parvum14. The infection of epithelial cells by C. parvum activates NF-κB signaling to increase the transcription of the miR-27b gene, thereby inhibiting KH-type splicing regulatory protein (KSRP) and reducing the burden of C. parvum15. MiR-26a and miR-30a regulate autophagy through the MAPK signaling pathway in response to C. parvum infection in vitro16. Therefore, in-depth study of the mechanism of miRNAs regulation of epithelial immune resistance to C. parvum infection will provide new ideas for the development of new drugs and vaccines.
Autophagy is a highly conserved cellular catabolic pathway that degrades and recycles long-lived proteins and damaged organelles through lysosomal digestion17. Apoptosis is the orderly and autonomous death of cells, which involves the activation, expression, and regulation of a number of genes, and is very important in maintaining the stability of the intracellular environment18. Research has shown that autophagy and apoptosis not only play an important role in maintaining the homeostasis of the intracellular environment, but also plays an important immune function in the pathogenic microbe-host interaction19. For example, Plasmodium infection of erythrocytes induces autophagy, which is not conducive to parasite proliferation, and human cells resist Toxoplasma infection by undergoing autophagy20,21. However, autophagy promotes the in vitro propagation of Neospora caninum in caprine endometrial epithelial cells22. Cryptosporidium parvum infection reduces the levels of phosphorylated MTOR (p-MTOR), promotes the autophagy of intestinal epithelial cells, and induces apoptosis in biliary epithelia via a Fas/Fas-ligand-dependent mechanism, disturbing parasite reproduction23,24. Although many studies have shown that C. parvum infection of epithelial cells changes the expression of many miRNAs and simultaneously activates the host immune response, the mechanisms of the immune responses involved in the regulation of different miRNAs differ25,26. Our research group previously investigated the expression profile of miRNAs after C. parvum infection of HCT-8 cells, and identified some significantly upregulated or downregulated miRNAs. A bioinformatic analysis predicted that miR-199a-3p targets the MTOR signaling molecule and suggested a potential role for the miR-199a-3p–MTOR axis in regulating the autophagy and apoptosis of intestinal epithelial cells induced by C. parvum. In this study, we focused on the regulatory function of miR-199a-3p in response to C. parvum infection.
Results
Intracellular proliferation of C. parvum inhibited by the complete autophagy of host cells
To confirm the induction of autophagy in HCT-8 cells by C. parvum, western blotting was used to detect the changes in autophagy-associated proteins, Beclin 1, LC3, and P62(SQSTM1), at 3, 6, 12, 24, and 48 hours postinfection (hpi). Compared with uninfected cells, Beclin 1 and LC3 increased from 3 to 48 hpi and their expression was highest at 12 hpi, whereas P62 decreased slightly, but not significantly (Fig. 1A). At 12 hpi, vesicles containing C. parvum trophozoites were observed with transmission electron microscopy, accompanied by autophagic lysosomes (Supplementary Fig. 1A). To confirm the formation of autophagosomes, we used LC3 protein labeled singly with fluorescent enhanced green fluorescent protein (EGFP) to localize the autophagosomes, and detected the formation of autophagosomes after C. parvum infection with fluorescence microscopy. In the blank control group, EGFP–LC3 was uniformly distributed in the cytoplasm without aggregation. In the infected cells, EGFP–LC3 began to accumulate in the cytoplasm at 3 hpi, and EGFP–LC3 aggregation was most obvious at 12 hpi, indicating the greatest degree of autophagy (Supplementary Fig. 1B). And we detected C. parvum and a large number of autophagosomes coexisting in the cytoplasm at 12 hpi (Fig. 1B).

A Expression levels of autophagy-related proteins LC3, P62, and Beclin 1 in HCT-8 cells at different time points after C. parvum sporozoites infection, detected with western blotting. The protein levels of LC3-II, P62, and Beclin 1 relative to GAPDH levels were determined with densitometry(n = 3 biologically independent samples). B Cells were transfected with plasmid stably expressing EGFP-LC3 and then exposed to equal numbers of C. parvum sporozoites for 12 h. C. parvum were stained with Pan Cp followed by goat anti-mouse IgG Alexa Fluor 594, and the nucleus was stained with DAPI. The spatial relationship between C. parvum and autophagosome was observed by confocal microscopy (red dots: C. parvum; green dots: autophagosome, Scale bars, 2 µm). C HCT-8 cells were pretreated with 500 nM Rapamycin (Rap) or 30 µM Chloroquine (CQ) for 4 h, and then infected with C. parvum sporozoites for 12 h. The expression of LC3 and P62 in the cell pellets was determined with western blotting(n = 3 biologically independent samples). D Cells were transfected with plasmid stably expressing mCherry-GFP-LC3 and then exposed to equal numbers of C. parvum sporozoites for 12 h (n = 3 biologically independent samples). The autophagic flux was evaluated with confocal microscopy. Numbers of yellow and free red dots in each group cell were quantified (free red dots: autophagic lysosomes; yellow dots: autophagosome, Scale bar, 5 µm). E Effects of 500 nM Rap and 30 µM CQ on the propagation of C. parvum in HCT-8 cells. HCT-8 cells were pretreated with 500 nM Rap or 30 µM CQ for 4 h and infected with C. parvum sporozoites, which were quantified with RT–qPCR assays at 12 and 24 hpi(n = 3 biologically independent samples). All data are presented as the means ± SD of three independent experiments. Different groups were compared with a t test. (*P < 0.05, **P < 0.01, ***P < 0.001).
To demonstrate the increase of autophagosome was induced by C. parvum infection rather than the impaired lysosome function, Chloroquine (CQ, 30 μM) and Rapamycin (500 nM) were used to treat HCT-8 cells for 4 h prior to C. parvum infection to inhibit and promote the occurrence of autophagy, respectively. At 12 hpi, the autophagy-related proteins LC3 and P62 were detected to evaluate the level of autophagy caused by the C. parvum infection. It was observed that in the CQ treatment group, the expression level of LC3 decreased and the expression level of p62 increased, and the opposite phenomenon was observed for the Rapamycin treatment group, which were consistent with the autophagy inhibitor and activator features of Chloroquine and Rapamycin, respectively. It was worth noting that when C. parvum sporozoites were added to the CQ-treated HCT-8 cells, the expression levels of LC3 and P62 proteins were significantly increased, and when the sporozoites were added to the Rapamycin-treated HCT-8 cells, the expression of LC3 was further increased and P62 was further reduced (Fig. 1C). The change of expression levels of the autophagy-related proteins following C. parvum infection indicated that the formation of autophagosomes was caused by C. parvum infection. (Fig. 1C). Autophagy and the localization of lysosomes were analyzed with the autophagy double-labeling technique to assess the autophagy flux in C. parvum-infected cells. Compared with the control group, free red dots (autophagic lysosomes) and yellow dots (autophagosome) increased significantly in the infected group, indicating that C. parvum promotes autophagic flux (Fig. 1D). Reverse transcription–quantitative PCR (RT–qPCR) was used to detect the parasite burden in the HCT-8 cells treated with Rapamycin and Chloroquine at different time points. It was shown that the parasite burden was reduced by the autophagy inducer Rapamycin and was increased by the autophagy inhibitor Chloroquine in HCT-8 cells (Fig. 1E). Confirming the effect of autophagy mechanism on intracellular proliferation of C. parvum. These experiments showed that the host cells underwent complete autophagy to reduce the burden of C. parvum infection.
Cryptosporidium parvum infection induces miR-199a-3p expression in HCT-8 cells
Our research group previously used the RNA-seq technology to sequence the whole transcriptome of HCT-8 cells in the early stages of C. parvum infection (3 hpi and 12 hpi), and found that the expression of miR-199a-3p was significantly increased at 12 hpi. RT-qPCR also showed that the expression of miR-199a-3p was significantly increased at 3, 6, and 12 hpi, but significantly reduced at 24 and 48 hpi compared with that in uninfected cells (Fig. 2A).

parvum load after infection. A Expression levels of miR-199a-3p in HCT-8 cells at different times after C. parvum sporozoites infection, determined with RT-qPCR (n = 3 biologically independent samples). B Expression efficiency of miR-199a-3p in HCT-8 cells transfected with miR-199a-3p mimic or miR-199a-3p inhibitor for 24 h, determined with RT-qPCR. C Effect of miR-199a-3p on autophagy of cells infected with C. parvum. Cells were transfected with miR-199a-3p mimic or miR-199a-3p inhibitor for 24 h and then exposed to equal numbers of C. parvum sporozoites for 12 h. Expression levels of autophagy-related proteins LC3, P62, and Beclin 1 were determined with western blotting. Protein levels of LC3-II, P62, and Beclin 1 relative to GAPDH levels were determined with densitometry(n = 3 biologically independent samples). D Effect of miR-199a-3p on autophagic flux in C. parvum-infected cells. Cells were cotransfected with miR-199a-3p mimic or miR-199a-3p inhibitor and a plasmid encoding mCherry–EGFP–LC3 for 24 h, and then exposed to equal numbers of C. parvum sporozoites for 12 h (n = 3 biologically independent samples). The autophagic flux was evaluated with confocal microscopy. Numbers of yellow and free red dots in each group cell were quantified (free red dots: autophagic lysosomes; yellow dots: autophagosome, Scale bar, 5 µm). E Effect of miR-199a-3p on apoptosis of cells infected with C. parvum. Cells were transfected with miR-199a-3p mimic or miR-199a-3p inhibitor for 24 h and then exposed to equal numbers of C. parvum sporozoites for 12 h. Cell apoptosis was detected with flow cytometry. The apoptosis rate is the sum of O1-LR and O1-UR (n = 3 biologically independent samples). F Cells were transfected with miR-199a-3p mimic or miR-199a-3p inhibitor for 24 h and then exposed to equal numbers of C. parvum sporozoites for 2 h to determine the initial attachment and cellular invasion of C. parvum with RT-qPCR. The medium was replaced with fresh medium at 3 hpi infection with C. parvum and the infected cells cultured for a further 12 or 24 h to evaluate the parasite burden after invasion(n = 3 biologically independent samples). All data presented are the means ± SD of three independent experiments. Different groups were compared with a t test (*P < 0.05, **P < 0.01, ***P < 0.001).
MiR-199a-3p reduces C. parvum infection by regulating autophagy and apoptosis in HCT-8 cells
Previous studies have shown that miRNAs are involved in the regulation of host cell autophagy and apoptosis. To confirm the role of miR-199a-3p in regulating the mechanisms of autophagy and apoptosis in HCT-8 cells, we transfected cells with an miR-199a-3p mimic or miR-199a-3p inhibitor to upregulate or downregulate its expression, respectively (Fig. 2B). The levels of autophagy and apoptosis were then determined after C. parvum infection at 12 hpi. Western blotting showed that, compared with the miR-199a-3p mimic normal control (NC) group, the expression of Beclin 1 and LC3 were increased and P62 was reduced in the miR-199a-3p mimic group, indicating that the overexpression of miR-199a-3p promoted autophagosome formation. When compared with the miR-199a-3p inhibitor normal control (NC) group, the expression of Beclin 1 and LC3 were reduced and P62 level was unchanged in the miR-199a-3p inhibitor group, indicating that the inhibited expression of miR-199a-3p reduced autophagosome production (Fig. 2C). Both EGFP-LC3 and C. parvum polyclonal antibody (Pn CP) were used to locate autophagosomes and the parasites, and the production of autophagosomes was detected after interfering with the expression of miR-199a-3p. The results showed that when the expression of miR-199a-3p was promoted, the production of autophagosomes was significantly increased, and when the expression of miR-199a-3p was inhibited, the production of autophagosomes was significantly reduced (Supplementary Fig. 2). Autophagy and lysosome localization showed that free red dots (autophagic lysosomes) and yellow dots (autophagosome) increased significantly in the miR-199a-3p mimic group compared with the miR-199a-3p mimic NC group, whereas the miR-199a-3p inhibitor group showed the opposite effect, suggesting that miR-199a-3p promotes the autophagic flux induced by C. parvum (Fig. 2D). Flow cytometry revealed that the rate of apoptosis was significantly increased in HCT-8 cells transfected with the miR-199a-3p mimic compared with that in the miR-199a-3p mimic NC cells, and that apoptosis was significantly reduced in the miR-199a-3p-inhibitor-treated cells (Fig. 2E), confirming that miR-199a-3p promoted the apoptosis induced by C. parvum infection.
We also used RT-qPCR to investigate the functional role of miR-199a-3p in regulating the C. parvum burden. MiR-199a-3p had no effect on the parasite infection load at 2 hpi, whereas the miR-199a-3p mimic group showed a significant reduction in the parasite load at 12 and 24 hpi, and the miR-199a-3p inhibitor group showed a significant increase in the parasite load (Fig. 2F). These experimental results indicated that miR-199a-3p limited the C. parvum burden in HCT-8 cells.
MiR-199a-3p negatively regulates the expression of MTOR
TargetScan, miRWalk, and miRBase were used to predict the potential target gene(s) of miR-199a-3p. Binding sites for miR-199a-3p were predicted on MTOR (Fig. 3A). The luciferase activity assay was carried out to initially determine the regulation of MTOR by miR-199a-3p. For cells transfected with the pmiGLO vector containing the target sites of wild-type (WT) MTOR, the transfection of miR-199a-3p mimic significantly reduced the luciferase activity. However, luciferase activities for cells transfected with the mutated MTOR (MTOR-MUT) and the vector alone didn’t show significant change after being transfected with miR-199a-3p (Fig. 3B). Furthermore, the overexpression of miR-199a-3p reduced MTOR mRNA and protein levels, whereas opposite results were observed in cells transfected with the miR-199a-3p inhibitor (Fig. 3C, D). These results demonstrated that miR-199a-3p targeted MTOR and inhibited its expression, suggesting that miR-199a-3p regulated autophagy and apoptosis in HCT-8 cells by targeting MTOR in response to C. parvum infection.

A Binding sites of miR-199a-3p on MTOR predicted with TargetScan. B Binding of miR-199a-3p to MTOR was detected with a dual-luciferase assay in HCT-8 cells cotransfected with miR-199a-3p or miR-199a-3p NC and pmirGLO–MTOR-WT or pmirGLO–MTOR-MUT for 48 h (n = 5 biologically independent samples). C, D Cells were transfected with miR-199a-3p mimic or miR-199a-3p inhibitor for 24 h and then exposed to equal numbers of C. parvum sporozoites for 12 h. MTOR mRNA and protein expression levels were detected with RT–qPCR and western blotting, respectively. Densitometric levels of MTOR protein signals and mRNA levels were quantified and expressed relative to GAPDH (n = 3 biologically independent samples). All data are presented as the means ± SD of three independent experiments. Different groups were compared with a t test (*P < 0.05, **P < 0.01, ***P < 0.001).
MTOR signal is involved in the regulation of C. parvum-induced autophagy and apoptosis in HCT-8 cells
RT-qPCR showed that the expression of MTOR was significantly reduced at 3, 6 and 12 hpi, but did not change at 24 hpi and increased significantly at 48 hpi compared with that in uninfected cells (Fig. 4A). However, western blotting detected no significant changes in the level of total MTOR protein (Supplementary Fig. 3A). The determination of p-MTOR expression levels showed that C. parvum significantly reduced the phosphorylation of the MTOR protein (Fig. 4B). We designed and synthesized three pairs of small interfering RNAs (siRNAs, si-MTORs) to inhibit MTOR expression, and selected the most effective si-MTOR and optimized its concentration (Supplementary Fig. 3B). We then investigated the role of the MTOR signal in regulating autophagy, apoptosis, and the parasite burden in HCT-8 cells after C. parvum infection. The knockdown of MTOR with siRNA promoted autophagy and increased the autophagic flux of HCT-8 cells after C. parvum infection (Fig. 4C and E, Supplementary Fig. 4). The increase in the apoptosis rate was confirmed with flow cytometry (Fig. 4F). Moreover, the parasite load was significantly lower in cells transfected with si-MTOR than in cells transfected with si-NC at 12 and 24 hpi (Fig. 4D). These results demonstrated that C. parvum induced autophagy and apoptosis in HCT-8 cells through MTOR signaling.

A MTOR mRNA expression was detected with RT–qPCR(n = 3 biologically independent samples). B Levels of phosphorylated MTOR (p-MTOR) and total MTOR were analyzed with western blotting 12 h after HCT-8 cells were infected with C. parvum sporozoites. Densitometry was used to compare the relative levels of p-MTOR protein and total MTOR protein, and the data were normalized to GAPDH(n = 3 biologically independent samples). C Cells were transfected with si-mTOR or si-NC for 24 h and then exposed to equal numbers of C. parvum sporozoites for 12 h. Expression levels of autophagy-related proteins LC3, P62, and Beclin 1 were determined with western blotting. Protein levels of LC3-II, P62, and Beclin 1 were determined relative to GAPDH with densitometry(n = 3 biologically independent samples). D Cells were transfected with si-MTOR or si-NC for 24 h and then exposed to equal numbers of C. parvum sporozoites for 3 h before the medium was replaced with fresh medium. The infected cells were cultured for 12 or 24 h to evaluate the parasite burden with RT–qPCR after cell invasion(n = 3 biologically independent samples). E Cells were cotransfected with si-MTOR or si-NC and plasmid encoding mCherry–EGFP–LC3 for 24 h, and then exposed to equal numbers of C. parvum sporozoites for 12 h(n = 3 biologically independent samples). The autophagic flux was evaluated with confocal microscopy. Numbers of yellow and free red dots in each group cell were quantified (free red dots: autophagic lysosomes; yellow dots: autophagosome, Scale bar, 5 µm). F Cells were transfected with si-MTOR or si-NC for 24 h and then exposed to equal numbers of C. parvum sporozoites for 12 h. Cell apoptosis was then detected with flow cytometry. The apoptosis rate is the sum of O1-LR and O1-UR(n = 3 biologically independent samples). All data are presented as the means ± SD of three independent experiments. Different groups were compared with a t test (*P < 0.05, **P < 0.01, ***P < 0.001).
MiR-199a-3p regulates cell autophagy and apoptosis and reduces the Cryptosporidium parvum burden in HCT-8 cells by targeting MTOR
The above results demonstrated that the transfection of miR-199a-3p inhibitors promoted the expression of MTOR, which subsequently inhibited the autophagy and the autophagic flux of HCT-8 cells to reduce the apoptosis rate and increase the burden of C. parvum. Meanwhile, si-MTOR inhibited the expression of MTOR and promoted autophagy and increased the autophagic flux of HCT-8 cells, resulting in the increase of apoptosis after C. parvum infection. To verify whether si-MTOR would reverse the regulatory effect caused by miR-199a-3p inhibitor, we simultaneously transfected HCT-8 cells with an miR-199a-3p inhibitor and si-MTOR. Compared with cells cotransfected with the miR-199a-3p inhibitor NC and si-NC, expressions of LC3 and Beclin 1 were significantly reduced and the expression of P62 was significantly increased in cells transfected with an miR-199a-3p inhibitor and si-NC, indicating that the autophagic intensity of the cells was reduced by the miR-199a-3p inhibitor. However, when HCT-8 cells were co-transfected with the miR-199a-3p inhibitor and si-MTOR, the inhibition of LC3 and Beclin1 expression induced by the miR-199a-3p inhibitor was eased and the increase of P62 expression was also significantly limited compared with the miR-199a-3p inhibitor and si-NC co-transfection group, illustrating that si-MTOR reversed the inhibition of autophagy caused by the miR-199a-3p inhibitor (Fig. 5A). Similarly, flow cytometry confirmed that the co-transfection of the miR-199a-3p inhibitor and si-NC reduced the apoptosis rate, whereas when the miR-199a-3p inhibitor and si-MTOR were co-transfected, the inhibition of the apoptosis was alleviated (Fig. 5B). Furthermore, the expression of miR-199a-3p was detected under the condition of miR-199a-3p inhibitor and si-MTOR co-transfection, and the inhibited miR-199a-3p expression induced by the miR-199a-3p inhibitor showed significant recovery in the presence of si-MTOR (Fig. 5C). Compared with HCT-8 cells transfected with negative controls of miR-199a-3p inhibitor and MTOR, significant increases of C. parvum burden were observed for cells transfected with the miR-199a-3p inhibitor and for cells co-transfected with the miR-199a-3p inhibitor and si-MTOR. However, the increase of parasite burden in cells co-transfected with the miR-199a-3p inhibitor and si-MTOR was smaller than that in cells transfected with the miR-199a-3p inhibitor, suggesting that the si-MTOR inhibitor reversed the increase of parasite burden caused by the miR-199a-3p inhibitor (Fig. 5D). These results indicated that miR-199a-3p targets MTOR to regulate autophagy and apoptosis in HCT-8 cells in order to reduce the C. parvum burden.

parvum burden in HCT-8 cells by targeting MTOR. A Cells were cotransfected with miR-199a-3p inhibitor or miR-199a-3p inhibitor NC and si-mTOR or si-NC for 24 h, and then exposed to equal numbers of C. parvum sporozoites for 12 h. Expression levels of autophagy-related proteins LC3, P62, and Beclin 1 were determined with western blotting. Protein levels of LC3II, P62, and Beclin 1 were determined relative to GAPDH protein levels with densitometry(n = 3 biologically independent samples). B Cells were cotransfected with miR-199a-3p inhibitor or miR-199a-3p inhibitor NC and si-MTOR or si-NC for 24 h, and then exposed to equal numbers of C. parvum sporozoites for 12 h. Cell apoptosis was then detected with flow cytometry. The apoptosis rate is the sum of O1-LR and O1-UR(n = 3 biologically independent samples). C Expression efficiency of miR-199a-3p in HCT-8 cells cotransfected with miR-199a-3p inhibitor and si-MTOR for 24 h, determined with RT–qPCR. D Cells cotransfected with miR-199a-3p inhibitor or miR-199a-3p inhibitor NC and si-MTOR or si-NC for 24 h were then exposed to equal numbers of C. parvum sporozoites for 3 h. The medium was then replaced with fresh medium. The infected cells were cultured for 12 or 24 h to evaluate the parasite burden after cell invasion, with RT-qPCR(n = 3 biologically independent samples). All data are presented as the means ± SD of three independent experiments. Different groups were compared with a t test (*P < 0.05, **P < 0.01, ***P < 0.001).
Discussion
Autophagy is widely reported to play an important role in the host response to infection by most intracellular pathogens, including bacteria, viruses, and parasites. There is evidence that infection by protozoan parasites induces the autophagy of the host cells, thereby necessitating immune escape related to the autophagic flux22,27. Trypanosoma cruzi activates the early steps of host autophagy, inducing the formation of autophagosomes. However, no autophagic lysosomes are observed in the infected cells, indicating that host autophagy is incomplete after Trypanosoma cruzi infection. This suggests that the parasite has mechanisms that allow it to evade autophagic capture28. The invasion of host cells by Toxoplasma triggers epidermal growth factor receptor (EGFR) signaling, inhibiting lysosomal degradation and thus allowing parasite survival. However, when an EGFR tyrosine kinase inhibitor (TKI) is added to previously infected cells, autophagosomal LC3 and lysosomal LAMP-1 co-capture Toxoplasma, through the autophagy-associated proteins ULK1 and Beclin 1, and lysosomal enzymes then kill the parasite29. Host cell activation of the EGFR–PI3K–AKT pathway induces autophagy, thereby reducing the C. parvum load in cells30. In the present study, complete cell autophagy was activated during C. parvum infection, which reduced the burden of C. parvum in HCT-8 cells. Cryptosporidium parvum not only induced the formation of a large number of autophagosomes, but also promoted the fusion of these autophagosomes with lysosomes. Finally, we used an autophagy inducer (Rapamycin) and a lysosome inhibitor (Chloroquine) to treat HCT-8 cells before their infection with C. parvum. Chloroquine significantly promoted the proliferation of the intracellular parasite, whereas in contrast, the number of C. parvum in HCT-8 cells treated with rapamycin was significantly reduced. These findings suggest that complete autophagy hinders the intracellular reproduction of C. parvum. Similar previously reported evidence showed that host cells used autophagy to reduce C. parvum infection16,30.
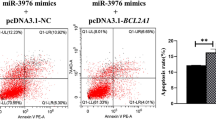
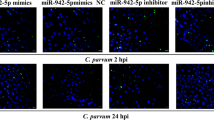
MiRNAs play an important regulatory role in the response of epithelial cells to C. parvum infection. Cryptosporidium parvum-infected cholangiocytes induce miR-125b-1, miR-21, miR-30b, and miR-23b-27b-24-1 expression, and the inhibition of these miRNAs increases the C. parvum burden in cholangiocytes, although the underlying mechanism remains unclear31. Recent studies have demonstrated the biological functions of some miRNAs in response to C. parvum infection32. Cryptosporidium parvum regulates HCT-8 cell autophagy to facilitate host survival by inhibiting miR-26a and promoting miR-30a expression16. MiR-3976 expression is induced by C. parvum infection, and miR-3976 regulates cell apoptosis by targeting BCL2A1, thus reducing the C. parvum burden in HCT-8 cells24. In the present study, miR-199a-3p expression was induced by C. parvum infection, the upregulation of miR-199a-3p promoted autophagy and apoptosis and limited the parasite burden in HCT-8 cells after C. parvum infection, thereby indicated that miR-199a-3p plays an important regulatory role in the response to C. parvum infection. The activation of the TLR/NF-κB signaling pathway in host cells can trigger the transcriptional regulation of miRNA genes, and the regulatory functions of miRNAs in response to C. parvum infection have important relationships with the downstream molecules in this pathway33. The epithelial defense response to C. parvum is increased by the upregulation of TLR4 expression, which is regulated by let-7i14. MiR-942-5p, which targets the IFI27 gene, regulates HCT-8 cell apoptosis through a TRAIL-dependent pathway in the early stage of C. parvum infection34. Many studies have shown that miR-199a-3p inhibited the MTOR signal, thus acting as a potential regulatory factor35,36,37,38,39. In the present study, the regulatory relationship between miR-199a-3p and MTOR, their regulation on host cell autophagy and apoptosis and parasite burden after C. parvum infection were investigated. It was found that miR-199a-3p targeted and inhibited the expression of MTOR, resulting in the promotion of autophagy and apoptosis of HCT-8 cells to reduce the C. parvum burden.
Cryptosporidium parvum is an intracellular protozoan parasite, in this study, we only quantified the effect of autophagy on the infection of host cells by C. parvum. Therefore, the mechanism by which autophagy directly impairs the development of C. parvum requires further research. The autophagy and apoptosis mechanisms play important roles in the immune response to pathogen infection, but the crosstalk between them is extremely complex40,41. Our study has only demonstrated that miR-199a-3p had two functions, regulating the autophagy and apoptosis of epithelial cells, in response to C. parvum infection. However, miR-199a-3p also plays a regulatory role in the crosstalk between autophagy and apoptosis, which warrants further investigation and future studies should determine the mechanisms underlying the role of miR-199a-3p in the host’s anti-C. parvum response in vivo. In conclusion, our results suggested that miR-199a-3p expression was induced by C. parvum infection, which targeted and inhibited the expression of MTOR and subsequently reduce the C. parvum burden by promoting cell autophagy and apoptosis. This study may provide new intervention targets for the prevention and treatment of C. parvum infection.
Methods
Cell culture model and cryptosporidium parvum infection
Human ileocecal adenocarcinoma (HCT-8) cells (American Type Culture Collection, Manassas, VA, USA) were cultured in Dulbecco’s modified Eagle’s Medium supplemented with 10% fetal bovine serum, 4 mmol/l L-glutamine, 100 U/mL penicillin, and 100 U/mL streptomycin at 37°C under 5% CO2. This cell line is mainly used as a model of C. parvum infection in vitro.
Cryptosporidium parvum strain IIdA19G1 was kept in our laboratory, passaged through newborn dairy calves every 3–4 months, and stored in 2.5% K2Cr2O7 solution at 4°C after purification. The neonatal calves were purchased from Ruiya Animal Husbandry Co., Ltd (Zhengzhou, China). Cryptosporidium parvum oocysts were first incubated and sterilized in 0.25% sodium gluconate solution, centrifuged at 13,500 × g for 5 min, and washed three times and suspended in phosphate-buffered saline (PBS). The oocysts were then incubated in a mixture of 0.25% trypsin and 0.75% taurocholate for 2 h at 37°C, with shaking every 30 min. As described above, the oocysts were washed three times with PBS and suspended in PBS. The sporozoites were then added to a monolayer of cells in either a 6- or 12-well plate in a sporozoite:cell ratio of 4:1.
RT-qPCR
HCT-8 cells were transfected or treated with the miR-199a-3p mimic, miR-199a-3p inhibitor, negative control (NC), and si-MTOR or si-NC for 24 h, and then stimulated with sporozoites (sporozoite:cell = 4:1) for 12 h. HCT-8 cells were stimulated with sporozoites in a sporozoite:cell ratio of 4:1. After stimulation with sporozoites for 3 h, the HCT-8 cells were thoroughly rinsed with fresh medium, cultured, and collected at 0, 3, 6, 12, 24, or 48 h. The samples were washed three times with PBS and the total RNA was extracted with TRIzol Reagent (Invitrogen, Waltham, MA, USA), according to the manufacturer’s instructions, which were then treated with recombinant DNase I (Takara, Kyoto, Japan). Housekeeping genes glyceraldehyde 3-phosphate dehydrogenase (GAPDH) and U6 were used as the references for cDNA and miRNA quantification, respectively. cDNA and miRNA expression were determined with SYBR® Green Realtime PCR Master Mix (Toyobo). Differences (fold changes) in gene expression were calculated with the 2−ΔΔCt method to determine the relative expression of each RNA. The primer sequences used in the present study are listed in Supplementary Table S1.
Cell transfection
The autophagy detection plasmid encoding doubly labeled LC3 (Mcherry–EGFP– LC3), the miR-199a-3p mimic, the miR-199a-3p inhibitor, and the negative control (NC) were synthesized by GenePharma (Shanghai, China). Three pairs of siRNAs directed to the MTOR gene (si-h-MTOR_010, genOFFTM st-h-MTOR_001, and genOFFTM st-h-MTOR_002) and the control siRNA (si-NC) were obtained from RiboBio (Guangzhou, China). The autophagy detection plasmid encoding singly fluorescently labeled LC3 (EGFP–LC3) is maintained in our laboratory. HCT-8 cells were seeded in 6- or 12-well cell culture plates. When the number of cells per well in each plate reached 80% of the total number, Lipofectamine 2000 Transfection Reagent (Invitrogen, Carlsbad, CA, USA) was used to transfect them with the appropriate constructs, according to the manufacturer’s protocol. Serum-free culture medium was added and the cells incubated for 6 h. The medium was then changed to DMEM containing 10% fetal bovine serum, 4 mmol/L glutamine, 100 U/mL penicillin, and 100 U/mL streptomycin. The cells were incubated at 37°C under 5% CO2 for 24 h. Mcherry-EGFP-LC3 and EGFP-LC3 (1.6 μg each in 12-well plates), miR-199a-3p mimic or NC (2.5 μL each 12-well plates or 5 μL each 6-well plates), miR-199a-3p inhibitor or NC inhibitor (4 μL each in 12-well plates or 8 μL each in 6-well plates), and si-MTOR (5 μL each in 12-well plates or 10 μL each in 6-well plates) were then added. For cotransfection, each gene was transfected with the same quantity of vector. The sequences of the siRNAs, miRNA mimic, and miRNA inhibitor are presented in Supplementary Table S2.
Western blotting
HCT-8 cells were stimulated with sporozoites in a sporozoite:cell ratio of 4:1. After stimulation for 3 h, the cells were thoroughly rinsed with fresh medium, cultured, and collected after 0, 3, 6,12, 24, or 48 h. The HCT-8 cells were transfected with the miR-199a-3p mimic, miR-199a-3p inhibitor, negative control (NC), si-MTOR, or si-NC for 24 h, and the cells were stimulated with sporozoites (sporozoite:cell = 4:1) for 12 h. The total protein was extracted from the cells with RIPA Lysis Buffer (Solarbio Life Sciences, Beijing, China) containing 1 mM phenylmethylsulfonyl fluoride (PMSF) and phosphatase inhibitors (Beijing Solarbio Science & NCM Biotech, Suzhou, China). The protein concentrations were measured with a Pierce™ BCA Protein Assay Kit (Thermo Fisher Scientific, Waltham, MA, USA). A 20 μg sample of protein was separated by 6%, 10%, or 15% SDS-PAGE and then transferred to polyvinyl difluoride (PVDF) membranes (0.45 μm or 0.22 μm) (Millipore, Billerica, MA, USA). The membranes were blocked with 5% skimmed milk (in PBS containing Tween 20 [PBST]) for 2 h and then incubated overnight with the primary antibody at 4°C. The antibodies used included rabbit antibodies directed against LC3B(cat. no. 3868 S), P62(cat. no. 5114 S), Beclin 1(cat. no. 3495 S) (diluted 1:1,000; Cell Signaling Technology, Danvers, MA, USA), MTOR(cat. no. 2983 S) and p-MTOR(cat. no. 5536 S) (diluted 1:1,000; Cell Signaling Technology), and a mouse antibody directed against human GAPDH (diluted 1:2000; Immunoway, Tennyson, TX, USA). The membranes were then incubated with a horseradish-peroxidase-conjugated secondary goat anti-rabbit or goat anti-mouse antibody (diluted 1:5,000; Immunoway) at room temperature for 1–2 h. The protein bands were visualized with an enhanced chemiluminescence (ECL) system, and quantified with the ImageJ software (NIH, USA).
Autophagy detection
HCT-8 cells were transfected with the EGFP–LC3/mCherry–EGFP–LC3-encoding plasmid and the miR-199a-3p mimic, miR-199a-3p inhibitor, negative control (NC), si-MTOR, or si-NC for 24 h, after which the cells were stimulated with sporozoites (sporozoite:cell = 4:1) for 12 h. Add 500-1000 μL (4%) of paraformaldehyde onto the slide and 0.3% Triton-100 was transvented for 10 min, washed with PBS 3 times, then closed with 1% BSA for 1-2 h, incubated with C. parvum polyclonal antibody (1:500), and incubated at 4°C overnight, washed with PBS to remove primary antibody and incubated with sheep anti-mouse (594) secondary antibody, and incubated at room temperature for 2 h. The cell nuclei were stained with 4’6-diamidino-2-phenylindole (DAPI) (Solarbio) for 20 min. The distribution of EGFP–LC3/mCherry–EGFP–LC3 in the cells was observed each group under a confocal microscope. The average numbers of EGFP-mCherry-LC3 (yellow-green) and mCherry-LC3B (free red) dots in the cells each group field were plotted.
Flow-cytometry detection of cell apoptosis
Cells were washed with PBS at 12 hpi, digested with EDTA-free pancreatic enzymes for 5 min, and centrifuged at 800 × g for 5 min at room temperature. The supernatant was removed and the cells were washed twice with PBS. Using a PE Annexin V Apoptosis Detection Kit I (BD, Denver, CO, USA), the cells were resuspended in 500 µL of precooling 1 × binding buffer to a cell density of 105–106 cells/mL, and 5 µL of annexin V-phycoerythrin (PE) and 5 µL of 7-aminoactinomycin D were added to them. The suspension was incubated for 15 min in the dark at room temperature. The cellular apoptosis rate was determined as the percentage of apoptotic cells in late or early phase relative to the total number of cells. using flow cytometry (Beckman Coulter, Brea, CA, USA). The apoptosisrate is the sum of O1-LR and O1-UR. All experiments were conducted independently three times.
Dual luciferase assay
To perform the luciferase reporter assay in HCT-8 cells, the 3’-UTR fragment of MTOR, which contains target sites for miR-199a-3p, was amplified and cloned into the pmirGLO control vector (Promega, Madison, WI, USA). The sequences of the MTOR-WT 3’-UTR and MTOR-MUT 3’-UTR uopare presented in Supplementary Table S3 Supplementary Fig. 6. The empty dual luciferase reporter plasmids were used as the negative control. For the luciferase assay, HCT-8 cells were cotransfected with the reporter plasmid and the miR-199a-3p mimic or miR-NC mimic using Lipofectamine 2000 Transfection Reagent (Invitrogen). Forty-eight hours after transfection, the cells were lysed, and the luciferase activity was assayed with the Renilla Luciferase Assay System (Promega), according to the manufacturer’s protocol, and the Dual-GLO® Luciferase Assay System (Promega). The relative luciferase activity was calculated as the ratio firefly luciferase signal/Renilla luciferase signal and normalized to the activity of the cotransfected Renilla luciferase. All experiments were performed independently three times.
Drug treatment and propagation of C. parvum
Rapamycin is an effective and specific MTOR inhibitor that effectively activates autophagy42, whereas chloroquine is an inhibitor of autophagy, and alters lysosomal acidity to inhibit lysosomal degradation43. Different concentrations of rapamycin (100, 200, 500, 1000, or 2,000 nM) and chloroquine (10, 20, 30, 50, or 100 μM) were blended with cell culture medium and added to cells in culture. After 24 h, Cell Counting Kit-8 (CCK8) (TargetMol, Shanghai, China) was used to detect cell viability and thus inhibitor toxicity. The appropriate concentrations were selected for subsequent tests (Supplementary Fig. 2A).
Generating the pan Cp polyclonal antiserum
Antigen for the pan Cp polyclonal antiserum was generated by excysting 2.5 × 108 bleached oocysts in 0.75% sodium taurocholate and 0.25 trypsin at 37 °C, for 2 h., excysted oocysts were then freeze-thawed 6 times (3 min on dry ice mixed with ethanol, then 3 min at 37 °C). After centrifugation, the supernatant was removed and mixed with 400 microliters of PBS, and then add equal volume of Freund’s Complete Adjuvant (FCA)/Freund’s Incomplete Adjuvant (FIA). Four BALB/c 6-8 weeks female mice were injected subcutaneously with 200 μL antigen with FCA and boosted three times at 21-day intervals with 200 μL antigen in FIA. All immunofluorescent staining experiments used the pan Cp antiserum at a 1:500 dilution. The present study was approved by the Independent Ethics Committee of Henan Agricultural University (approval no. IEC-HENAU-20180323-06). We have complied with all relevant ethical regulations for animal use.
Statistics and reproducibility
All statistical analyses were performed with GraphPad Prism 8.0.2 (GraphPad Software, San Diego, CA, USA). Differences between two groups were analyzed with an unpaired t-test with Bonferroni’s correction, and differences among multiple groups were analyzed with nonparametric one-way ANOVA. All data are expressed as means ± standard deviations (SD). All experiments were performed with at least three biological replicates. For all analyses, P < 0.05 was considered significant. *P < 0.05, **P < 0.01 and ***P < 0.001.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The source data behind the graphs in the paper can be found in Supplementary Data 1.
References
O’Hara, S. P. & Chen, X. M. The cell biology of Cryptosporidium infection. Microbes Infect. 13, 721–730 (2011).
Feng, Y., Ryan, U. M. & Xiao, L. Genetic diversity and population structure of cryptosporidium. Trends Parasitol. 34, 997–1011 (2018).
Ryan, U. M., Feng, Y., Fayer, R. & Xiao, L. Taxonomy and molecular epidemiology of Cryptosporidium and Giardia a 50 year perspective (1971-2021). Int. J. Parasitol. 51, 1099–1119 (2021).
McCole, D. F., Eckmann, L., Laurent, F. & Kagnoff, M. F. Intestinal epithelial cell apoptosis following Cryptosporidium parvum infection. Infect. Immun. 68, 1710–1713 (2000).
Deng, M., Rutherford, M. S. & Abrahamsen, M. S. Host intestinal epithelial response to Cryptosporidium parvum. Adv. drug Deliv. Rev. 56, 869–884 (2004).
Innes, E. A., Chalmers, R. M., Wells, B. & Pawlowic, M. C. A one health approach to tackle cryptosporidiosis. Trends Parasitol. 36, 290–303 (2020).
Jonas, S. & Izaurralde, E. Towards a molecular understanding of microRNA-mediated gene silencing. Nat. Rev. Genet. 16, 421–433 (2015).
Saliminejad, K., Khorram Khorshid, H. R., Soleymani Fard, S. & Ghaffari, S. H. An overview of microRNAs: biology, functions, therapeutics, and analysis methods. J. Cell Physiol. 234, 5451–5465 (2019).
O’Connell, R. M., Rao, D. S., Chaudhuri, A. A. & Baltimore, D. Physiological and pathological roles for microRNAs in the immune system. Nat. Rev. Immunol. 10, 111–122 (2010).
Jing, R., Zhong, Q. Q., Long, T. Y., Pan, W. & Qian, Z. X. Downregulated miRNA-26a-5p induces the apoptosis of endothelial cells in coronary heart disease by inhibiting PI3K/AKT pathway. Eur. Rev. Med. Pharmacol. Sci. 23, 4940–4947 (2019).
Chen, J. et al. MicroRNA-30a ameliorates hepatic fibrosis by inhibiting Beclin1-mediated autophagy. J. Cell. Mol. Med. 21, 3679–3692 (2017).
Liu, H. et al. Mitophagy protects SH-SY5Y neuroblastoma cells against the TNFα-induced inflammatory injury: Involvement of microRNA-145 and Bnip3. Biomed. Pharmacother. 109, 957–968 (2019).
Wu, H. et al. MiR-20a and miR-106b negatively regulate autophagy induced by leucine deprivation via suppression of ULK1 expression in C2C12 myoblasts. Cell. Signal. 24, 2179–2186 (2012).
Chen, X. M., Splinter, P. L., O’Hara, S. P. & LaRusso, N. F. A cellular micro-RNA, let-7i, regulates Toll-like receptor 4 expression and contributes to cholangiocyte immune responses against Cryptosporidium parvum infection. J. Biol. Chem. 282, 28929–28938 (2007).
Zhou, R., Gong, A. Y., Eischeid, A. N. & Chen, X. M. miR-27b targets KSRP to coordinate TLR4-mediated epithelial defense against Cryptosporidium parvum infection. PLoS Pathog. 8, e1002702 (2012).
Jiang, H. et al. Cryptosporidium parvum regulates HCT-8 cell autophagy to facilitate survival via inhibiting miR-26a and promoting miR-30a expression. Parasites Vectors 15, 470 (2022).
Glick, D., Barth, S. & Macleod, K. F. Autophagy: cellular and molecular mechanisms. J. Pathol. 221, 3–12 (2010).
Elmore, S. Apoptosis: a review of programmed cell death. Toxicol. Pathol. 35, 495–516 (2007).
Crighton, D., Wilkinson, S. & Ryan, K. M. DRAM links autophagy to p53 and programmed cell death. Autophagy 3, 72–74 (2007).
Krishnamurthy, S., Konstantinou, E. K., Young, L. H., Gold, D. A. & Saeij, J. P. The human immune response to Toxoplasma: Autophagy versus cell death. PLoS Pathog. 13, e1006176 (2017).
Joy, S. et al. Basal and starvation-induced autophagy mediates parasite survival during intraerythrocytic stages of Plasmodium falciparum. Cell death Discov. 4, 43 (2018).
Zhao, S. S. et al. Neospora caninum infection activated autophagy of caprine endometrial epithelial cells via mTOR signaling. Vet. Parasitol. 304, 109685 (2022).
Priyamvada, S. et al. Cryptosporidium parvum infection induces autophagy in intestinal epithelial cells. Cell. Microbiol. 23, e13298 (2021).
Li, J. et al. MiR-3976 regulates HCT-8 cell apoptosis and parasite burden by targeting BCL2A1 in response to Cryptosporidium parvum infection. Parasites Vectors 16, 221 (2023).
Wang, C. et al. MicroRNA expression profile of HCT-8 cells in the early phase of Cryptosporidium parvum infection. BMC Genomics 20, 37 (2019).
Sun, L. et al. Whole transcriptome analysis of HCT-8 cells infected by Cryptosporidium parvum. Parasites Vectors 15, 441 (2022).
Subauste, C. S. Autophagy in immunity against Toxoplasma gondii. Curr. Top. Microbiol. Immunol. 335, 251–265 (2009).
Onizuka, Y. et al. Inhibition of autolysosome formation in host autophagy by Trypanosoma cruzi infection. Acta Tropica 170, 57–62 (2017).
Lopez Corcino, Y. et al. Toxoplasma gondii induces prolonged host epidermal growth factor receptor signalling to prevent parasite elimination by autophagy: Perspectives for in vivo control of the parasite. Cell. Microbiol. 21, e13084 (2019). Epub 2019 Jul 17.
Yang, H. et al. Cryptosporidium parvum maintains intracellular survival by activating the host cellular EGFR-PI3K/Akt signaling pathway. Mol. Immunol. 154, 69–79 (2023).
Zhou, R. et al. NF-kappaB p65-dependent transactivation of miRNA genes following Cryptosporidium parvum infection stimulates epithelial cell immune responses. PLoS Pathog. 5, e1000681 (2009).
Zhou, R., Feng, Y. & Chen, X. M. Non-coding RNAs in epithelial immunity to Cryptosporidium infection. Parasitology 141, 1233–1243 (2014).
Feng, R. et al. Cryptosporidium parvum downregulates miR-181d in HCT-8 cells via the p50-dependent TLRs/NF-κB pathway. Vet. Parasitol. 305, 109710 (2022).
Xie, F. et al. MiR-942-5p targeting the IFI27 gene regulates HCT-8 cell apoptosis via a TRAIL-dependent pathway during the early phase of Cryptosporidium parvum infection. Parasites vectors 15, 291 (2022).
Jia, L. et al. MiRNA-199a-3p regulates C2C12 myoblast differentiation through IGF-1/AKT/mTOR signal pathway. Int. J. Mol. Sci. 15, 296–308 (2013).
Li, T. et al. Formononetin ameliorates the drug resistance of Taxol resistant triple negative breast cancer by inhibiting autophagy. Am. J. Transl. Res. 13, 497–514 (2021).
Fu, J. et al. miR-199a-3p is involved in estrogen-mediated autophagy through the IGF-1/mTOR pathway in osteocyte-like MLO-Y4 cells. J. Cell Physiol. 233, 2292–2303 (2018).
Deleyto-Seldas, N. & Efeyan, A. The mTOR-Autophagy Axis and the Control of Metabolism. Front. Cell Dev. Biol. 9, 655731 (2021).
Wu, W. et al. Improvement of myocardial cell injury by miR-199a-3p/mTOR axis through regulating cell apoptosis and autophagy. J. Immunol. Res. 2022, 1642301 (2022).
Kasprowska-Liśkiewicz, D. The cell on the edge of life and death: Crosstalk between autophagy and apoptosis. Postepy Hig. Med. Dosw. (Online) 71, 825–841 (2017).
Mehrbod, P. et al. The roles of apoptosis, autophagy and unfolded protein response in arbovirus, influenza virus, and HIV infections. Virulence 10, 376–413 (2019).
Gong, Y., Wu, J., Yang, R., Zhang, L. & Ma, Z. Rapamycin-induced autophagy plays a pro-survival role by enhancing up-regulation of intracellular ferritin expression in acute lymphoblastic leukemia. Exp. Oncol. 42, 11–15 (2020). 14067.
Bryant, K. L. et al. Combination of ERK and autophagy inhibition as a treatment approach for pancreatic cancer. Nat. Med. 25, 628–640 (2019). Combination of ERK and autophagy inhibition as a treatment approach for pancreatic cancer.
Acknowledgements
This research was supported by grants of the National Key Research and Development Program of China (2022YFD1800200, 2023YFD1801200), and the Science Fund for Distinguished Young Scholars of Henan Province (222300420010). We thank International Science Editing (http://www.internationalscienceediting.com) for editing this manuscript.
Author information
Authors and Affiliations
Contributions
RJ.W. and LoX.Z. conceived and designed the experiments; SB.W. and TR.S. performed the experiments; JF.L., LY.W., XY.L., JJ.X., LL.S., YF.Z., and LiJ.Z. analyzed the data and prepared figures; SB.W. and RJ.W. wrote the paper. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
The present study was approved by the Independent Ethics Committee of Henan Agricultural University (approval no. IEC-HENAU-20180323-06). We have complied with all relevant ethical regulations for animal use.
Peer review
Peer review information
Communications Biology thanks William Witola and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editors: Nishith Gupta and Johannes Stortz.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wu, S., Shao, T., Xie, J. et al. MiR-199a-3p regulates HCT-8 cell autophagy and apoptosis in response to Cryptosporidium parvum infection by targeting MTOR. Commun Biol 7, 924 (2024). https://doi.org/10.1038/s42003-024-06632-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-024-06632-5
- Springer Nature Limited