
Abstract
Single gamete cell sequencing together with long-read sequencing can reliably produce chromosome-level phased genomes. In this study, we employed PacBio HiFi and Hi-C sequencing on a male Landrace pig, coupled with single-sperm sequencing of its 102 sperm cells. A haplotype assembly method was developed based on long-read sequencing and sperm-phased markers. The chromosome-level phased assembly showed higher phasing accuracy than methods that rely only on HiFi reads. The use of single-sperm sequencing data enabled the construction of a genetic map, successfully mapping the sperm motility trait to a specific region on chromosome 1 (105.40–110.70 Mb). Furthermore, with the assistance of Y chromosome-bearing sperm data, 26.16 Mb Y chromosome sequences were assembled. We report a reliable approach for assembling chromosome-level phased genomes and reveal the potential of sperm population in basic biology research and sperm phenotype research.
Similar content being viewed by others


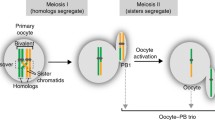
Introduction
The dissimilarity between homologous chromosomes within a diploid genome influences various aspects of genetic analysis, including genome annotation, allele expression, and the exploration of inter-individual homologous chromosome variation1,2,3. Single-cell gamete sequencing has the potential to enable chromosome-scale phased genome assembly and personalized genetic map construction, thereby providing valuable insights into personalized genetics4,5. Due to technological limitations, the fusion of diploid genomes into a pseudo-haploid sequence has been a common practice for an extended period, inadvertently creating challenges for subsequent research endeavors. With the advancement of sequencing techniques, particularly the progress made in long-read sequencing technologies such as Pacific Biosciences (PacBio) and Oxford Nanopore sequencing, the possibility for haplotype assembly using long reads has become apparent. Among the available options for phase assembly software, Falcon-unzip6, and SupernovaTM assembler7 (10x Genomics) make use of the overlap of long reads to determine haplotype phases. However, these methods are restricted to resolving haplotype differences over short distances. Although Falcon-phase can generate longer haplotypes by incorporating Hi-C data, it falls short of achieving chromosome-level phased assembly8,9. Hifiasm can identify parental-specific read bins, enabling the assembly of diploid genomes effectively10. Dipasm utilizes HiFi and Hi-C data to achieve chromosome-level phased assemblies11. However, significant challenges still exist in the pursuit of comprehensive chromosome-level phased genomes, especially in cases where a chromosome contains numerous regions of low heterozygosity. A method that combines stranded short-read RNA-seq with long-read sequencing has demonstrated effectiveness in phasing the human genome12. However, the broader application of this approach has been hindered by the challenge of obtaining strand-specific sequencing data. Fu et al. have developed a methylation-based haplotype phasing method called MethPhaser which they used to phase a human genome13. Shi et al. utilized 12 pollen samples to assemble a pear genome14, while Kirkness et al. used 96 sperm cells to phase the HuRef human genome15, thereby demonstrating the feasibility of gamete-based genome assembly. However, the current limitations of sequencing technologies continue to impede the attainment of high-quality assemblies. Trio binning, which relies on Illumina short reads from both parental genomes to differentiate haplotypes within offspring’s long reads, is utilized to construct complete diploid assemblies1. However, in the case of mammals, the accessibility of parental genetic information is frequently constrained, thereby limiting the applicability of trio binning.
The first reference pig genome of a female Duroc pig was published in 201216, and subsequently, several other pig genomes have been reported9,17,18,19,20,21. However, the absence of chromosome-level phased genomes in pigs persists due to technological limitations. In this study, we sequenced a male Landrace pig using PacBio HiFi and Hi-C technologies and 102 of its sperm with single-cell sequencing technology. A phased genome at the chromosomal level was obtained using phased markers and phased long reads. The assembly exhibited high accuracy in phasing. Additionally, using the sperm data, a genetic map was constructed and then used for studying the sperm motility trait. Moreover, the insights gained from Y chromosome-bearing sperm data proved instrumental in advancing pig Y chromosome assembly. Overall, our research revealed the importance of single sperm sequencing in phased genome assembly and sperm phenotype research.
Results and Discussion
The strategy for obtaining complete haplotypes of sperm
In the mammalian meiosis process, after two rounds of cell division following DNA replication, homologous chromosome pairs exchange some genetic materials, resulting in two haploid cells and then generate four genetically unique gametes (sperm or egg) after the sister chromatids segregate4. Sperm has a parental genotype chimeric composition, as shown in Fig. 1a and Supplementary Fig. 1. There are several methods to obtain haplotypes for sperm data; for example, Carioscia et al. developed the rhapsodi method suitable for low coverage of single-gamete sequence analysis22. Li et al. developed Hapi, which utilizes sperm data to obtain haplotypes by employing the PHMM (pairwise Hidden Markov Model) method23. Lyu et al. developed sgcocaller software, which outperforms the Hapi algorithm in accuracy and performance, providing great efficiency for sperm research24. In this study, we sequenced 102 sperm cells with an average depth of 10.05X and sequenced the blood samples from the donor boar with 95.12X, which allowed us to directly infer sperm haplotypes, as shown in Fig. 1. For example, in Fig. 1b, we can phase the genotype of sperm No. 2–8 by comparing their genotypes with sperm No. 1 (same genotype with same color). Based on the recombination information of sperm 2–8, we can easily identify false recombination sites, as shown by the black horizontal line in Fig. 1. These false recombination sites are characterized by recombination occurring in sperm 2–8 (Fig. 1b). As recombination occurs randomly in the population, these abnormal recombination sites are easily identified, which is actually caused by the true recombination of sperm No. 1, leading to false recombination of all other sperm cells at that location. By correcting these false recombination sites, we can obtain real recombination sites in the population (Fig. 1c, d), and then obtain chromosome-level haplotype markers for subsequent phased genome assembly (Fig. 1e).

Numbers 1-8 represent the chromosomes of No.1-8 sperms, and the parental genotype was indicated by A, B, and orange and blue colors, respectively. The black horizontal lines represent false recombination sites in the population. a The real chromosome recombination map of the eight sperm. b The recombination map after genotyping using sperm No.1 as the reference. The same genotype with No.1 sperm was marked as orange, and the different genotype was marked as blue. False recombination occurs at the horizontal line with all sperm (except No. 1) recombined at this position, which was caused by the real recombination of No. 1 sperm. c The recombination map of No. 2-8 sperm after the false recombination site correction. d The recombination map after the correction of all sperm. e The complete haplotype path is based on the population genotype information, which was indicated in dark orange and dark blue.
Initial genome assembly and acquisition of haplotype markers
For the initial genome assembly, 237.79 Gb (95.12X) Illumina 350 bp insertion library data, 89.94 Gb (35.98X) Hi-C data, and 94.26 Gb (37.70X) Pacbio HiFi data (Supplementary Table 1) were obtained from the blood of the Landrace boar. We employed the Falcon unzip pipeline, yielding an initial assembly with a contig N50 of 29.52 Mb and a length of 2.58 Gb (Supplementary Table 2). Firstly, the Illumina 350 bp insertion library data from the blood sample of the same pig that was aligned to the initial genome assembly and 5.02 million heterozygotes sites across the autosomes were detected. The density distribution of heterozygous sites displayed uneven patterns across chromosomes (Supplementary Fig. 2). Certain regions of chromosomes exhibited lower densities of heterozygous sites when compared with other chromosome regions, reflecting the inbreeding process of Landrace pigs leading to some regions of the sequenced genome becoming homozygous. Subsequently, the initial assembly also served as the reference genome for aligning sequencing data of 102 sperm from the boar. The sequencing data is 2.65Tb, yielding an average sequencing depth of 10.05X (ranging from 4.37X to 25.53X) (Supplementary Table 3). For the heterozygous sites, 99.76% of them were genotyped in the sperm population. The average coverage of heterozygous sites within the sperm was 49.49%, with a range spanning from 18.00% to 67.89% (Supplementary Table 4). Based on the sperm exhibiting the highest sequencing depth (S18–162, it covered 67.89% heterozygous sites), we constructed a bin map (Fig. 2a, analogous to Fig. 1b). This bin map enabled the detection of 19 false recombination sites (Fig. 2a). Upon the rectification of these false recombination sites, we produced the final bin map, comprised of 1,471 bins. These bins facilitated the phasing of heterozygous sites within each bin and subsequently served as haplotype makers for phased genome assembly (Fig. 2b, Supplementary Data 1).

a The preliminary recombination map inferred by using sperm S18-162 as the reference, the chromosomes are divided by the white vertical lines, and the vertical black dotted lines represent the potential false recombination site. b The final recombination map after correcting the false recombination sites and filtering suspicious recombination.
Phased genome assembly
Using the phased markers, we categorized the PacBio HiFi reads into two groups, and the HiFi reads that were not classified were copied into two groups. The sequencing depths for these two groups are 25.42X and 25.26X, respectively (Supplementary Table 5). Employing five assembly software (Hifiasm, Next denovo, wtdbg2, flye, and Hicanu) on the classified HiFi reads, we observed that Hifiasm yielded the highest contig N50 and genome length (Supplementary Table 6). Consequently, we adopted Hifiasm for the final phased genome assembly. In addition, we compared our phased assembly strategy with other established pipelines like Dipasm and Falcon phase. All three strategies resulted in highly continuous and complete genomes (Table 1). While the contig N50 of the single-sperm pipeline was comparatively shorter, it yielded the longest assembly length (2.61 Gb, Table 1). However, upon assessing the output using phased markers, the results showed that Dipasm produced lengthy phased segments without achieving the chromosome level (Fig. 3a). Conversely, the Falcon phase failed to produce long-phased segments (Fig. 3b), echoing previous findings1. By leveraging the single-sperm sequencing method, we successfully obtained a chromosome-level phased genome (Fig. 3c), which exhibited strong collinearity with the Duroc genome (Fig. 3d). To evaluate the completeness of the phased genome, we conducted a Benchmarking Universal Single-Copy Orthologs (BUSCO) analysis against mammalian genes. The Landrace genome exhibited 96.4% completeness and 2% partial completeness among the 4104 vertebrate BUSCOs genes. By comparing the assembly of telomeres and centromeres between the genomes of Landrace and Duroc pigs, we found that the Landrace genome assembles more telomere sequences (Supplementary Fig. 3). This result represented the highest integrity achieved at the chromosomal level among available pig genomes to date (Supplementary Table 7), and the assembly outcomes conclusively demonstrated that the phased chromosomes derived from the single-sperm sequencing method were meticulously assembled, showcasing superior continuity and quality.

a–c The phased marker distribution in Dipasm assembly (a), Falcon phase assembly (b), and single sperm-based assembly (c). d Collinearity analysis of the single sperm assembly and the Duroc genome.
Genome annotation
Comparable to the Duroc genome, approximately 34.09% of the Landrace genome consisted of repeat contents (Supplementary Table 8). Public protein sequences from the genomes of six mammals, which include human, mouse, cattle, dog, goat, and Duroc pig, were employed as queries to search the Landrace genome for homology predictions. Then public data from nine tissues and one pooled RNA library from Landrace pig were used to optimize the gene annotation. Finally, we identified and predicted 21,982 protein-coding genes (PCGs), with functional annotations attributed to 97.82% of them (Supplementary Table 9). Our genomic analysis also revealed the presence of 725 ribosomal RNAs (rRNAs), 4496 transfer RNAs (tRNA), 852 microRNAs (miRNAs), and 1808 small nuclear RNAs (snRNAs) in the Landrace genome (Supplementary Table 10). Moreover, approximately 31.70% of the Landrace genome sequence was annotated as transposable elements (TEs) (Supplementary Table 11).
Assisting assembly of Y chromosomes using Y chromosome-bearing sperm cells
The assembly of the Y chromosome in mammals has historically been challenging due to its highly repetitive sequence characteristics and the presence of homologous regions with the X chromosome25. However, leveraging the Y chromosome markers identified from the Y chromosome-bearing sperm cells (Supplementary Table 12), we were able to select the Y chromosome-specific PacBio HiFi reads. This approach allowed us to exclude the influence of X homologous regions, enabling the construction of a robust and accurate Y chromosome assembly. 66.33 Gb of Y chromosome-specific short reads were obtained for selecting Y-specific long reads. After aligning the 6.37 Gb candidate HiFi reads to the genome and removing the long reads aligned to autosomes, we finally obtained the Y chromosome sequence that spans 26.14 Mb in length and encompasses 57 intact PCG models. We successfully identified the presence of key genes such as ZFY and SRY. In humans, SRY and ZFY are linked to the Y chromosome26. Comparing the Landrace Y chromosome assembly with other published pig genomes at the chromosomal level, our assembly boasts the longest total length of non-N bases, indicative of its superior quality and completeness (Table 2).
Genetic map construction
For the first time, we have successfully constructed both a physical recombination map and a genetic map of pigs using the sperm population. This accomplishment holds significance for comprehending genetic recombination in pigs and facilitating the mapping of sperm-related phenotypes. Upon a comparative analysis of the recombination map and the genetic map, the results showed the recombination rate of the genome regions near the telomere is higher than regions near the centromere regions, aligning with prior research findings27 (Fig. 4). According to previous research, these phenomena reflect the cumulative evolutionary history of recombination28. Our study revealed that each sperm, on average, experienced 18 crossovers. This number contrasts with 12 in mice (inferred 2649 crossovers in 217 sperm)29 and 26 crossovers observed in human sperm27.

The X axes represents the physical location, and the Y axes represents the genetic distance.
Mapping of sperm motility traits
Sperm motility is an essential characteristic that assesses the swimming ability of sperm. To measure sperm motility, a three-tiered grading system was conducted (See method). Utilizing the genetic map and 1471 bin markers (Supplementary Data 1 and 2), we successfully mapped the Quantitative Trait Loci (QTL) associated with sperm motility (Supplementary Table 13, Supplementary Data 3). The results showed that there is a peak with LOD of 3.08 at 107.40 Mb (genetic distance of 72.31 cM) in chromosome 1 (Fig. 5). The significant interval is approximately 5.30 Mb, with a range of 105.40–110.70 Mb (genetic distance of 71.31–73.31 cM) (Supplementary Data 3). However, considering the limited number of sperm samples and the precision of phenotype identification, the QTL results still need further validation. Nevertheless, our analysis validates the feasibility of employing single-sperm sequencing to construct a genetic map for studying sperm-related phenotypes. This approach provides opportunities to investigate the genetic foundations of sperm traits.

The X axes represent the chromosome, and the Y axes represent the LOD values. The dashed line represents the significant threshold of LOD at 2.5. LOD is the log10 likelihood ratio comparing the hypothesis of a QTL at position versus that of no QTL.
Conclusion
In this study, combining Pacbio HiFi sequencing and single-sperm sequencing, a chromosome-level phased assembly of a male Landrace pig was generated. In the assembly process, we developed an approach for inferring parental haplotypes using single-sperm data, as well as assembling the Y chromosome by utilizing Y chromosome-bearing sperm. This enables us to understand the genetic contribution of parental haplotypes and achieve precise reconstruction of the Y chromosome sequence, which is important for understanding male-specific genetic traits and potential diseases. The construction of the pig genetic map and the QTL mapping of sperm motility trait demonstrate the potential of single-gamete sequencing in basic biological scientific research and sperm phenotype research.
Methods
Sample collection and PacBio sequencing
Fresh blood samples from a male Landrace pig (two years old) were collected for genome assembly. High-quality genomic DNA (gDNA) was extracted and assessed for purity and quantity using Nanodrop 1000 (Thermo Fisher) and Qubit (Thermo Fisher) assays. A library with an average insert size of ~15 kb was generated using the SMRTbell Express Template Prep Kit 2.0 (PacBio) and fractionated on the SageELF (Sage Science, Beverly, MA) into narrow library fractions. The library was then sequenced on 4 SMRT Cells 8 M on a Sequel II system (Pacbio) using 30 h movie times. Raw data were processed using the CCS algorithm (version 6.0.0, parameters: --minPasses 3 --all --max-length 50000) to generate highly accurate HiFi reads.
Single sperm sequencing
Mature sperm cells were obtained from freshly ejaculated semen from the same Landrace boar, which was used for genome assembly. The sperms were diluted to ~1/mm^2 using PBS + 1%BSA on a petri dish. After phenotyping, every candidate single sperm was isolated by mouth pipetting and put into a reaction tube. The sperm were washed twice with PBS + 1%BSA before being lysed for 3 h in the lysis buffer, as described in a previous study27. The Multiple Annealing and Looping Based Amplification Cycles (MALBAC) technique was employed for single-cell whole genome amplification, with minor modifications30. For cell lysis, each individual cell was introduced into 5 μL of fresh cell lysis buffer, comprising 15 mM DTT, 10 mM Tris-EDTA, 1 mg/ml Qiagen protease, 0.3% Triton X-100, 200 mM KCL, and 0.25 μM GAT3G primer. The lysed cell underwent centrifugation for 1 min at 7000 rpm, followed by a 3 h incubation at 50 °C and protease inactivation at 70°C for 30 min.
Single-cell whole genome pre-amplification with Multiple Annealing and Looping Based Amplification Cycles (MALBAC). The MALBAC primer featured a common 27-nucleotide sequence: GTG AGT GAT GGT TGA GGT AGT GTG GAG. The process initiated with a pre-amplification, during which 30 μL of amplification mixture I (consisting of 1×Thermopol buffer, 0.1 mM dNTP, 133 mM Mg, 0.33 mM Primer1, and 0.33 mM Primer2) was added to PCR tubes containing the lysed single cell. This mixture was subjected to temperature cycles: 95 °C for 3.5 min, followed by 11 cycles of 4 °C for 50 s, 10 °C for 50 s, 20 °C for 50 s, 30 °C for 50 s, 40 °C for 45 s, 50 °C for 45 s, 65 °C for 4 min, 95 °C for 20 s, 58 °C for 20 s, and a 4 °C pause. The tubes were then rapidly cooled on ice.
In the exponential amplification phase, 30 μL of amplification mixture I (comprising 1×Thermopol buffer, 0.1 mM dNTP, 100 mM Mg, 0.67 μM primer, and 0.067 U/μl DeepVentR (exo-) polymerase) was introduced to PCR tubes with the pre-amplified samples from the previous step. This mixture underwent temperature cycles: 95 °C for 30 s, followed by 17 cycles of (95 °C for 20 s, 58 °C for 30 s, 72 °C for 3 min), then a final step of 72 °C for 5 min, 58 °C for 20 s, and a hold at 4 °C. This process facilitated the exponential amplification of DNA from the single sperm cell.
Following this procedure, the converted DNA was subjected to purification using Zymo-Spin columns (Zymo). Subsequently, the DNA was eluted in 50 μl of elution buffer. To ensure the quality and quantity of the DNA, assessment was performed using a Qubit High-Sensitivity dsDNA kit., the libraries were sequenced on the Illumina Novaseq sequencing platform.
Sperm motility identification
To measure sperm motility, a three-tiered grading system was conducted. Grade 1 sperm: These sperm exhibited progressive motility, representing the highest level of motility. They demonstrated strong swimming abilities, moving swiftly in a straight trajectory. Grade 2 sperm (Non-Linear Motility): This category encompasses sperm that move forward, albeit in a curved or irregular manner. Their motion was not strictly linear. Grade 3 sperm: These sperm were characterized by non-progressive motility. Despite moving their tails, they did not make significant forward progress. Furthermore, within grade 3, there were immotile sperm that showed no movement whatsoever. All sperm were observed under a microscope at a magnification of 100X. Each sperm, after phenotyping, was mouth pipetted into a reaction tube and washed twice with PBS + 1%BSA before being lysed for 3 hours in the lysis buffer for subsequent genome sequencing.
Hi-C library construction
Genomic DNA was extracted from the ear for the purpose of constructing the Hi-C library. The Hi-C fragment libraries were generated with insert sizes ranging from 300 to 700 base pairs (bp), and were subjected to sequence on the Illumina platform. The enzyme DpnII was employed to cleave at the recognition sequence “GATC”. Following sequencing, adapter sequences of the raw reads were trimmed, and paired-end reads with low quality were eliminated to yield clean reads using the fastp program (version 0.19.5)31 with default parameters.
Initial genome assembly
To accomplish a diploid contig assembly of the Landrace genome. The initial genome assembly was generated using the Falcon assembler, followed by FALCON-Unzip6, integrated into the pbassembly tool suite (version 0.0.4). This resulted in a draft assembly consisting of primary contigs representing a partially phased haploid genome and haplotigs that represent phased alternative alleles for a subset of the genome. Two rounds of contig polishing were then performed. For the first round, as part of the FALCON-Unzip pipeline, primary contigs and secondary haplotigs were polished using haplotype-phased reads and the Quiver consensus caller. For the second round of polishing, the primary contigs and haplotigs were concatenated into a single reference and then mapped all raw reads to the combined assembly reference using pbmm2 (version 0.12.0), followed by consensus calling with Arrow (genomic consensus version 2.3.3). After the draft set of contigs was generated, a reference-guided scaffolding strategy was applied with RaGOO software32 based on the Sscrofa11.1 assembly. Finally, pilon (version 1.22)33 was used to correct errors introduced into the assembly from the long-read data.
Sperm genotyping and phased assembly
The Illumina reads from the blood sample of the individual used for genome assembly were aligned to the unphased assembly by using the bwa mem algorithm (version 0.7.15-r1140)34. Then GATK (version 3.7-0-gcfedb67) software was used to detect the heterozygous sites of the whole genome35. To ensure the accuracy of heterozygous site identification, the filtering standard is as follows:
-
1.
Quality value ≥ 30;
-
2.
Sequencing depth of the minor allele ≥ 5;
-
3.
Chi-square test was performed for the proportion of alleles, and the expected segregation ratio was 1:1, P = 0.05.
All sequencing data of single sperm were aligned to the partially phased Landrace genome by using bwa mem algorithm (version: 0.7.15-r1140)34 to get the alignment files. Then samtools mpileup (version 1.7)36 and Bcftools (version 0.1.19-96b5f2294a)37 were used to extract the corresponding alleles based on the heterozygous site information identified by the blood sample.
According to the quality control results, the sperm with the highest sequencing depth was used as a reference for the identification of recombination sites and haplotype inference. As shown in Fig. 1, assuming that sperm 1 is the reference sperm, other sperm can be compared with it to make a preliminary genotypic judgment based on whether the genotype is the same. Since sperm 1 may also have recombination sites, other sperm will present abnormal recombination at the recombination sites of sperm 1 (shown by the black line in Fig. 1) in the population; that is, all other sperm will recombine at this site. Based on this information, we can correct these false recombination sites and obtain complete chromosome haplotype information.
Using the sperm genotyping information, the phased markers and their flanking sequences were aligned to the HiFi reads. According to the haplotype information, HiFi reads were grouped. To obtain high-quality contigs, five approaches were used to test de novo genome assembly quality, including wtdbg2 (version 2.5)38, flye (version 2.8.3-b1695)39, HiCanu (version 2.1.1)40, Hifiasm (version 0.15.4_r343)10 and Nextdenovo (version v2.4.0) (https://github.com/Nextomics/NextDenovo). The parameters used for wtdbg2 were ‘-g 2500 m -x ccs -t 60 --edge-min 2 --rescue-low-cov-edges’. The parameters used for flye were ‘--genome-size 2.5 g --pacbio-corr --iterations 2’. The parameters used for HiCanu were ‘genomeSize=2.5 g useGrid=false maxThreads=60 -pacbio-hifi’. The default parameters were used for hifiasm assembly. Nextdenovo (version v2.4.0) was run with parameters of ‘read_type = hifi input_type = corrected genome_size = 2.5 g’. The assemblies yielded by hifiasm were used to remove heterozygous sequences by Purge haplotigs41 with the parameters -a 70. Subsequently, the Hi-C tech was employed to process contig assembly to obtain chromosome-level genome assembly. Detailed data processing procedures were provided as follows: (1) The paired-end Illumina reads were mapped onto the polished temporary genome assembly by using Hic-Pro (version 2.11.1)42 with default parameters to filter the raw Hi-C reads. Self-ligated, non-ligated, and other invalid reads (such as PCR amplification, random break, and extreme fragments) were discarded. (2) Juicer (version 1.6.2)43 and 3D-DNA (version 180114)44 were applied to cluster the genomic contig sequences into potential chromosomal groups. (3) JuiceBox (version 1.11.8)45 was employed to validate the contig orientation and to remove ambiguous fragments with the help of manual inspection.
Genome completeness assessment
The completeness of the Landrace genome was assessed using the BUSCO program (version 5.0.2)46. The BUSCO analysis included 4104 mammalian genes with the “ -m genome” parameter. The telomere and centromeric repeats were identified by quarTeT47.
Annotation of repeats
The interspersed repeats and low-complexity DNA sequences were identified using two methods, de novo repeat identification and known repeat searching against existing databases. RepeatModeler (version 1.0.8) was used to predict repeat sequences in the Landrace genome, RepeatMasker (version 4.0.7) (http://www.repeatmasker.org/) was then used to search the genome against the de novo transposable element (TE) library. The homology-based approach involved applying commonly used databases of known repetitive sequences, RepeatMasker (version 4.0.7) and the Repbase database48 were used to identify TE repeats in the assembled genome, and TEs were identified at both the DNA and protein levels, RepeatMasker was applied for DNA-level identification and RepeatProteinMasker was used to perform protein-level identification.
Gene prediction and annotation
Protein sequences in the genome of six mammals, including human, mouse, cattle, dog, goat, and Duroc pig, were downloaded from the Ensembl database. Besides, the protein sequences of Luchuan pig were downloaded from the China National GenBank (CNGB; https://db.cngb.org/) under the accession of CNP0001159. Subsequently, these protein sequences were used as queries to search against the Landrace genome using GeMoMa (version 1.8)49. Homology predictions were denoted as “Homology-set”. To optimize the genome annotation, the raw reads of nine tissues and one pool RNA libraries from a Landrace pig (NCBI accession numbers: SRR3160015, SRR3160012, SRR3160008, SRR3160011, SRR3160014, SRR3160009, SRR3160017, SRR3160013, SRR3160010, and SRR3160016) were downloaded for further analyses. All raw reads were assessed using fastp (version 0.19.5)31. Then clean reads were mapped to the assembly using Hisat2 (version 2.0.1)50. The output bam files were merged using Samtools (version 1.10)36. Stringtie (version 1.2.2)51 and TransDecoder (version 3.0.1) (https://github.com/TransDecoder/TransDecoder) were employed to assemble the transcripts and identify candidate coding regions into gene models. Gene models created by RNA-seq were denoted as Stringtie -set. All gene models predicted were combined by EvidenceModeler (EVM)52 into a non-redundant set of gene structures. Finally, the produced gene models were refined with the Program to Assemble Spliced Alignment (PASA) (version 2.4.1)53. The integrated gene set was translated into amino-acid sequences. By using Diamond program (version 0.9.30.131)54 with an E-value cutoff of 1e-05, the amino-acid sequences were aligned to three public protein databases, SwissProt55, Kyoto Encyclopedia of Genes and Genomes (KEGG)56 and NCBI nonredundant database (NR). Moreover, BLAST57 was applied to search against Translation of European Molecular Biology Laboratory (Trembl) databases (E-value 1e-05). At last, we search protein domains through InterProScan (version 5.30)58 program. The Gene Ontology (GO) terms for each gene were extracted with InterProScan v5.30.
Noncoding RNAs (ncRNAs) annotation
Four types of ncRNAs were annotated in the Landrace genome, including miRNA, tRNA, rRNA, and snRNA. The tRNA genes were predicted by tRNAscan-SE (version 1.3.1)59 with eukaryote parameters. The rRNA fragments were predicted by searching against vertebrate rRNA sequences using BLAST (version 2.2.24) with an E-value of 1e-5. The miRNA and snRNA genes were obtained by INFERNAL (version 1.1.1)60.
Sperm-assisted Y chromosome assembly
First, 40 Y chromosome-bearing sperm cells were selected from the semen for high-throughput sequencing. By using the bwa mem algorithm (version 0.7.15-r1140)34, the clean reads of these 40 Y chromosome-bearing sperm cells were aligned to the Landrace genome. According to the alignment results, the Y chromosome-specific reads were obtained with samtools software (version 1.7) after removing the autosome alignment36. Next, the Y chromosome-specific reads were mapped to HiFi reads to obtain Y chromosome-specific long reads. The candidate HiFi long reads were mapped the Landrace genome again to remove the autosome alignments using minimap261. Hifiasm (version 0.15.4_r343)10 was used to generate sex assembly from these reads using default parameters. After removing possible contaminants, redundant sequences were identified and removed using the Purge Haplotigs pipeline41, with the parameters -a 70. Last, we anchored the non-redundant sequences into scaffolds with Hi-C data. The protein-coding genes were predicted by miniport (https://github.com/lh3/miniprot) with genes from the Y chromosome of human, Duroc pig and goat genomes62.
Genome alignment and collinearity analysis
Landrace pig assembly was aligned to the Duroc pig genome with MUMmer (v 3.23)63 using default parameters and the genomic alignment results were extracted with the delta-filter −1 -l 10000 parameters. R (v3.5.1) was used to visualize the collinear results.
Genetic map construction and QTL mapping
Consecutive phased markers were used for recombination breakpoint detection with a sliding window approach64. The recombination map of each sperm was determined, and genotypes were determined for each 100-kb interval. Adjacent 100-kb intervals with the same genotype in each sperm were merged into a bin. The genetic map was constructed using 1481 recombination bins, and the genetic distance was calculated with the Kosambi mapping function65. Subsequently, QTL was identified using composite interval mapping (CIM) implemented in the Windows QTL Cartographer V2.566 package. A 10-cM scan window was employed, and the walking speed was set as 1 cM. LOD values and R2 were determined based on likelihood ratio tests under a hypothesis allowing both additive and dominance effects. QTLs were identified based on LOD values equal to or higher than 2.5.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The sequencing data for this project have been deposited in the NCBI Sequence Read Archive (SRA) (http://www.ncbi.nlm.nih.gov/sra) under accession number PRJNA977441. The genome sequences have been deposited into CNGB Sequence Archive (CNSA)67 of China National GeneBank DataBase (CNGBdb)68 with accession number CNP0004469. Gene annotation files were uploaded to Figshare (https://figshare.com/s/f37b58dfa53047f0b08d).
Code availability
Codes used in this study were deposited to GitHub (https://github.com/Niuyongchao/sperm_phasing).
References
Koren, S. et al. De novo assembly of haplotype-resolved genomes with trio binning. Nat. Biotechnol. 36, 1174–1182 (2018).
Hoehe, M. R. et al. Multiple haplotype-resolved genomes reveal population patterns of gene and protein diplotypes. Nat. Commun. 5, 5569 (2014).
Low, W. Y. et al. Haplotype-resolved genomes provide insights into structural variation and gene content in Angus and Brahman cattle. Nat. Commun. 11, 2071 (2020).
Lyu, R., Tsui, V., McCarthy, D. J. & Crismani, W. Personalized genome structure via single gamete sequencing. Genome Biol. 22, 112 (2021).
Bell, A. D. et al. Insights into variation in meiosis from 31,228 human sperm genomes. Nature 583, 259–264 (2020).
Chin, C. S. et al. Phased diploid genome assembly with single-molecule real-time sequencing. Nat. Methods 13, 1050–1054 (2016).
Weisenfeld, N. I., Kumar, V., Shah, P., Church, D. M. & Jaffe, D. B. Direct determination of diploid genome sequences. Genome Res 27, 757–767 (2017).
Kronenberg, Z. N. et al. Extended haplotype-phasing of long-read de novo genome assemblies using Hi-C. Nat. Commun. 12, 1935 (2021).
Liu, Y. et al. Integration of multi-omics data reveals cis-regulatory variants that are associated with phenotypic differentiation of eastern from western pigs. Genet. Select Evol. 54, 62 (2022).
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods 18, 170–175 (2021).
Garg, S. et al. Chromosome-scale, haplotype-resolved assembly of human genomes. Nat. Biotechnol. 39, 309–312 (2021).
Porubsky, D. et al. Fully phased human genome assembly without parental data using single-cell strand sequencing and long reads. Nat. Biotechnol. 39, 302–308 (2021).
Fu, Y. et al. MethPhaser: methylation-based haplotype phasing of human genomes. bioRxiv 0, 2023-05.12.540573 (2023).
Shi, D. et al. Single-pollen-cell sequencing for gamete-based phased diploid genome assembly in plants. Genome Res. 29, 1889–1899 (2019).
Kirkness, E. F. et al. Sequencing of isolated sperm cells for direct haplotyping of a human genome. Genome Res. 23, 826–832 (2013).
Groenen, M. A. M. et al. Analyses of pig genomes provide insight into porcine demography and evolution. Nature 491, 393–398 (2012).
Li, M. et al. Genomic analyses identify distinct patterns of selection in domesticated pigs and Tibetan wild boars. Nat. Genet. 45, 1431–1438 (2013).
Warr, A. et al. An improved pig reference genome sequence to enable pig genetics and genomics research. Gigascience 9, giaa051 (2020).
Zhou, R. et al. The Meishan pig genome reveals structural variation-mediated gene expression and phenotypic divergence underlying Asian pig domestication. Mol. Ecol. Resour. 21, 2077–2092 (2021).
Chen, J. H. et al. The de novo assembly of a European wild boar genome revealed unique patterns of chromosomal structural variations and segmental duplications. Anim. Genet. 53, 281–292 (2022).
Jiang, Y. F. et al. Pangenome obtained by long-read sequencing of 11 genomes reveal hidden functional structural variants in pigs. iScience 26, 106119 (2023).
Carioscia, S. A. et al. A method for low-coverage single-gamete sequence analysis demonstrates adherence to Mendel’s first law across a large sample of human sperm. Elife 11, e76383 (2022).
Li, R. et al. Inference of Chromosome-Length Haplotypes Using Genomic Data of Three or a Few More Single Gametes. Mol. Biol. Evol. 37, 3684–3698 (2020).
Lyu, R. et al. sgcocaller and comapr: personalised haplotype assembly and comparative crossover map analysis using single-gamete sequencing data. Nucleic Acids Res 50, e118 (2022).
Skaletsky, H. et al. The male-specific region of the human Y chromosome is a mosaic of discrete sequence classes. Nature 423, 825–837 (2003).
Yin, D. et al. Gapless genome assembly of East Asian finless porpoise. Sci. Data 9, 765 (2022).
Lu, S. et al. Probing meiotic recombination and aneuploidy of single sperm cells by whole-genome sequencing. Science 338, 1627–1630 (2012).
Paigen, K. & Petkov, P. Mammalian recombination hot spots: properties, control and evolution. Nat. Rev. Genet. 11, 221–233 (2010).
Hinch, A. G. et al. Factors influencing meiotic recombination revealed by whole-genome sequencing of single sperm. Science 363, eaau8861 (2019).
Zong, C., Lu, S., Chapman, A. R. & Xie, X. S. Genome-wide detection of single-nucleotide and copy-number variations of a single human cell. Science 338, 1622–1626 (2012).
Chen, S., Zhou, Y., Chen, Y. & Jia, G. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–1890 (2018).
Alonge, M. et al. RaGOO: fast and accurate reference-guided scaffolding of draft genomes. Genome Biol. 20, 224 (2019).
Walker, B. J., Abeel, T., Shea, T., Priest, M. & Earl, A. M. Pilon: An Integrated Tool for Comprehensive Microbial Variant Detection and Genome Assembly Improvement. PLoS ONE 9, e112963 (2014).
Heng, L. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv:1303.3997v2 [q-bio.GN] https://github.com/lh3/bwa (2013).
McKenna, A. et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20, 1297–1303 (2010).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinforma. (Oxf., Engl.) 25, 2078–2079 (2009).
Li, H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 27, 2987–2993 (2011).
Ruan, J. & Li, H. Fast and accurate long-read assembly with wtdbg2. Nat. Methods 17, 1–4 (2020).
Kolmogorov, M., Yuan, J., Lin, Y. & Pevzner, P. A. Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol. 37, 540–546 (2019).
Nurk, S. et al. HiCanu: Accurate assembly of segmental duplications, satellites, and allelic variants from high-fidelity long reads. Genome Res. 30, gr.263566.120 (2020).
Roach, M. J., Schmidt, S. A. & Borneman, A. R. Purge Haplotigs: allelic contig reassignment for third-gen diploid genome assemblies. BMC Bioinforma. 19, 460 (2018).
Servant, N. et al. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biol. 16, 259 (2015).
Durand, N. et al. Juicer Provides a One-Click System for Analyzing Loop-Resolution Hi-C Experiments. Cell Syst. 3, 95–98 (2016).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356, eaal3327 (2017).
Durand, N. C. et al. Juicebox Provides a Visualization System for Hi-C Contact Maps with Unlimited Zoom. Cell Syst. 3, 99–101 (2016).
Waterhouse, R. M. et al. BUSCO Applications from Quality Assessments to Gene Prediction and Phylogenomics. Mol. Biol. Evol. 35, 543–548 (2018).
Lin, Y. Z. et al. quarTeT: a telomere-to-telomere toolkit for gap-free genome assembly and centromeric repeat identification. Horticulture Res. 10, uhad127 (2023).
Jurka, J. Repbase Update: A database and an electronic journal of repetitive elements. Trends Genet. : TIG 16, 418–420 (2000).
Keilwagen, J., Hartung, F. & Grau, J. GeMoMa: Homology-Based Gene Prediction Utilizing Intron Position Conservation and RNA-seq Data. Gene Prediction: Methods Protoc. 1962, 161–177 (2019).
Kim, D., Langmead, B. & Salzberg, S. L. HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360 (2015).
Kovaka, S. et al. Transcriptome assembly from long-read RNA-seq alignments with StringTie2. Genome Biol. 20, 278 (2019).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 9, R7 (2008).
Haas, B. Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res. 31, 5654–5666 (2003).
Buchfink, B., Xie, C. & Huson, D. H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12, 59–60 (2015).
Bairoch, A. & Apweiler, R. The SWISS-PROT protein sequence data bank and its supplement TrEMBL in 1999. Nucleic Acids Res. 27, 49–54 (1999).
Kanehisa, M. & Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 28, 27–30 (2000).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990).
Jones, P. et al. InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240 (2014).
Lowe, T. M. & Eddy, S. R. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic acids Res. 25, 955–964 (1997).
Nawrocki, E. P., Kolbe, D. L. & Eddy, S. R. Infernal 1.0: inference of RNA alignments. Bioinformatics 25, 1335–1337 (2009).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100 (2018).
Xiao, C. et al. The assembly of caprine Y chromosome sequence reveals a unique paternal phylogenetic pattern and improves our understanding of the origin of domestic goat. Ecol. Evol. 11, 7779–7795 (2021).
Kurtz, S. et al. Versatile and open software for comparing large genomes. Genome Biol. 5, R12 (2004).
Huang, X. et al. High-throughput genotyping by whole-genome resequencing. Genome Res. 19, 1068–1076 (2009).
Kosambi, D.D. The estimation of map distances from recombination values. in Ann. Eugen. 172–175 (1944).
Silva Lda, C., Wang, S. & Zeng, Z. B. Composite interval mapping and multiple interval mapping: procedures and guidelines for using Windows QTL Cartographer. Methods Mol. Biol. 871, 75–119 (2012).
Guo, X.Q. et al. CNSA: a data repository for archiving omics data. Database- J Biol Databases Curation baaa055 (2020).
Chen, F. Z. et al. CNGBdb: China National GeneBank DataBase. Yi Chuan 42, 799–809 (2020).
Acknowledgements
We are grateful to Jue Ruan from the Agriculture Genomics Institute at Shenzhen, Chinese Academy of Agricultural Sciences and Guojie Zhang from Zhejiang University for discussions on bioinformatics analyses and for giving comments on this study. This study was supported by the sustainable development special project from Shenzhen (KCXFZ20201221173213037), the National Key Scientific Research Project (2023YFF1001100), the National Natural Science Foundation of China (U23A20229), the Project of Science and Technology from Guangxi (GKAB21196060), and the Agricultural Science and Technology Innovation Program (CAAS-ZDRW202006).
Author information
Authors and Affiliations
Contributions
Z.T. designed this project and coordinated research activities. Y.N., Y.Y., Jiang L., and J. L. performed bioinformatics analyses. X.F. and Y.T. conducted the experiments. L.W. and Y.Z. collected pig materials. Y.N., Z.T., and Y.Y. contributed to writing the manuscript. All authors participated in analyzing and interpreting the data.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval
The animal experiments in this study were approved by the Institutional Animal Care and Use Committee of Agricultural Genomics Institute at Shenzhen, Chinese Academy of Agricultural Sciences (AGIS-ER-2023-003). A 2-year-old male Landrace pig was used in this study. The study did not involve any wild animals. We have complied with all relevant ethical regulations for animal use.
Peer review
Peer review information
Communications Biology thanks Wai Low and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editors: John Mulley, George Inglis and Christina Karlsson Rosenthal.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Niu, Y., Fan, X., Yang, Y. et al. Haplotype-resolved assembly of a pig genome using single-sperm sequencing. Commun Biol 7, 738 (2024). https://doi.org/10.1038/s42003-024-06397-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-024-06397-x
- Springer Nature Limited