
Abstract
The hijacking of early developmental programs is a canonical feature of gliomas where neoplastic cells resemble neurodevelopmental lineages and possess mechanisms of stem cell resilience. Given these parallels, uncovering how and when in developmental time gliomagenesis intersects with normal trajectories can greatly inform our understanding of tumor biology. Here, we review how elapsing time impacts the developmental principles of astrocyte (AS) and oligodendrocyte (OL) lineages, and how these same temporal programs are replicated, distorted, or circumvented in pathological settings such as gliomas. Additionally, we discuss how normal gliogenic processes can inform our understanding of the temporal progression of gliomagenesis, including when in developmental time gliomas originate, thrive, and can be pushed towards upon therapeutic coercion.
Similar content being viewed by others
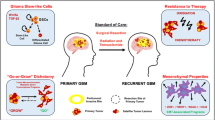

Introduction
Normal glial development is guided by a series of tightly controlled and temporally regulated lineage-determining events. Gliogenic malignancies represent a perturbation in this process, where neurodevelopmental programs are hijacked under severe genetic and environmental settings leading to aberrant cell fate decisions and pathogenic consequences. In this review, we highlight where normal and oncogenic glial differentiation paths diverge and how this information can uncover new facets of tumor biology. First, we summarize the current understanding of the timeline and instructive cues that define normal astro- and oligo-genesis. We then provide an overview of how molecular regulators of glial development and glial hierarchical organization are mimicked across glioma subtypes. Finally, we consider the ways in which normal glial differentiation can inform our understanding of how glioma cells move across developmental trajectories, including discussion on when in developmental time gliomas begin, progress to, and are capable of differentiating towards.
Molecular regulators of astrocyte and oligodendrocyte lineage commitment
Neuroectodermal development involves multipotent neural stem cells (NSC) called radial glia (RG) that give rise to three key cell populations—neurons, astrocytes (AS), and oligodendrocytes (OL). RG first undergo symmetrical divisions during early gestation to expand their pool before dividing asymmetrically towards neurogenic fates during mid-gestation, followed by gliogenic fates (astrocytes and oligodendrocytes) at later gestational and early postnatal stages1,2. This shift from neurogenesis to gliogenesis, termed the gliogenic switch, occurs around 16 gestational weeks (GW) in humans and is mediated by both intrinsic and extrinsic factors that synergize to suppress neurogenesis, release molecular brakes impeding gliogenesis, and actively promote gliogenic commitment3,4. For the purpose of this review, references to glia specifically pertain to astrocyte and oligodendrocyte macroglial populations.
During the neurogenic phase of development, premature astrogenesis is primarily prevented through inhibition of the JAK/STAT pathway and more specifically, STAT3-mediated transcription of astrocyte genes, including GFAP and S100B4,5,6,7. Pro-neuronal transcription factors (TF), such as NGN18, and the neurotrophin BDNF9, both inhibit STAT3-mediated astrogenesis while simultaneously promoting neurogenic pathways, like MEK-ERK signaling10,11. These mechanisms ensure a robust population of early immature neurons prior to the emergence of astrocytes and oligodendrocytes.
A key event that drives the shift towards gliogenesis is the remodeling of regulatory genomic regions into favorable states that promote the transcription of gliogenic genes. During astrogenesis, this occurs through synergistic activation of the JAK/STAT, BMP, and Notch signaling pathways, which modulate the landscape of DNA methylation, histone methylation, and acetylation4,12,13,14,15. The p300/CBP complex is an important component of the JAK/STAT pathway and has intrinsic acetyltransferase activity, including helping to induce H3K9 and H3K14 acetylation at the STAT3 binding site of the GFAP promoter7. Around the time of the gliogenic switch, Polycomb group (PcG) proteins silence NGN1 activity, inducing the release of p300/CBP, which forms a co-activator complex with STAT3 at the promoter of astrocyte genes to activate expression12,13. Additionally, the binding of astrocytic TFs, such as NFIA, has been shown to displace DNMT1 from astrocyte-specific promoters, helping to facilitate an active gliogenic transcriptional state16,17,18,19.
Transcription factors are powerful molecular regulators that initiate changes in cell state, differentiation, and maturation. Advancements in high-throughput sequencing coupled with new and robust methods for studying glia—such as sophisticated 2D and 3D model systems, improved glial purification methods, and more specific genetic targeting of glia20,21—helped identify several TFs that contribute to gliogenesis. Two of the first TFs that were identified as key players in the induction of astrogenesis include NFIA and SOX9. Overexpression of NFIA is sufficient to induce astrocyte formation22,23 and also drives HES5 expression, a Notch pathway effector required for the inhibition of neurogenesis24. Similarly, reduced Sox9 expression results in prolonged neurogenesis and delayed gliogenesis in vitro25. Kang and colleagues later discovered that Sox9 not only induces NFIA expression but identified that the two TFs form a complex to facilitate transcription of astrocyte genes26. Two additional Sox9 binding partners, NFIB and Zbtb20, also collectively induce cortical astrocyte differentiation in mice27,28. Several studies have subsequently identified key regulators of the SOX9-NFIA complex, including TFs PITX1, which promotes SOX9 expression29, and Brn2, which plays a key role in SOX9-induction of NFIA30. Together, this illustrates a complex network of TF activation that is required to promote the switch from neurogenesis to astrogenesis (Fig. 1). Several studies have also investigated the role of TFs at later stages of astrocyte maturation, although this developmental window remains comparatively more elusive. Work by Lattke and colleagues suggested Rorb, Dbx2, Lhx2, and Fezf2 are potential regulators of astrocyte maturation in the developing mouse cortex31. However, substantial changes in maturation were more apparent when all four TFs were simultaneously overexpressed. Most likely, these TFs, as well as yet-to-be-identified candidates, act synergistically and/or in physical complexes to promote maturation.

Schematized representation of proposed glial differentiation trajectories. RG are hypothesized to either (solid arrows) give rise to a bipotent glial intermediate progenitor that can generate both astrocytes and oligodendrocytes or (dashed arrows) directly generate astrocyte and oligodendrocyte lineages. Colored boxes indicate lineage markers and relevant TF drivers and inhibitors are listed next to respective lineage types. Outer radial glia (oRG), ventricular radial glia (vRG), glial intermediate progenitor cell (gIPC).
The gliogenic switch is a shift not only from neurogenic to astrogenic fates but also towards oligodendrocyte lineages. Several TFs are implicated in early oligodendrocyte precursor cell (OPC) development and maintenance (Fig. 1). The TF Olig2, for example, activates additional OL-lineage TFs, including Sox1032, which acts in combination with Sox9 to promote OPC maintenance and proliferation33. Co-deletion of Sox9 and Sox10 reduces the density of Olig2-positive OPCs within the developing spinal cord, and the remaining Olig2-positive OPCs are deficient in Pdgfrα, a signaling pathway that promotes OPC survival and proliferation33,34. In return, Sox10 helps maintain Olig2 expression in a positive feedback loop, together supporting the maintenance of a robust OPC population35. Additionally, OPCs express several TFs, including Sox5, Sox6, Hes5, Id2, and Id4, which prevent OPC differentiation and maturation by inhibiting Olig1/2, Sox10, and downstream transcription of key maturation genes36,37,38 (Fig. 1).
Extrinsic regulators of gliogenesis
In addition to intrinsic regulators of cell fate, multiple extrinsic cues are also important for promoting gliogenic commitment and downstream glial development39. Some of the most well-documented intrinsic factors in astrogenesis include a trio of IL-6 cytokines—cardiotrophin-1 (CT-1), leukemia inhibitory factor (LIF), and ciliary neurotrophic factor (CNTF)—that promote astrocyte formation through JAK/STAT activation40,41,42. Newborn neurons also secrete ligands Jagged 1 and Delta-like 1, which contribute to the gliogenic switch through activation of Notch signaling18. Multiple cytokines, including BMP2, BMP4, and TGF-B1, have also been implicated in astrogenesis by promoting the formation of a Smad:p300/CBP:STAT complex that facilitates the transcription of astrocyte genes43,44,45,46,47. FGF248,49 and retinoic acid (RA)50 may act more broadly to promote astrogenesis by facilitating shifts in chromatin state to elicit transcription of astrocyte genes. Additionally, synergistic activity of the ligands TGFβ2, NLGN1, TSLP, DKK1, and BMP4 act upstream of mTORC1 to promote astrocyte development, suggesting that much like TFs, astrogenesis is orchestrated by a concert of extrinsic cues51.
Extrinsic cues also play a substantial role in OL development, including PDGF-α, FGF-2, and IGF-1 signaling. PDGF- α is secreted by both neurons and astrocytes and helps maintain the OPC population by promoting proliferation and preventing precocious differentiation33,34,52,53. When the PDGF mitogen binds to and activates PDGF receptors, it triggers a reorganization of the actin filament structure, stimulating changes in cell growth and motility, a cascade that when hyperactivated can serve as an oncogenic program54. The mitogen FGF-2 helps to maintain the expression of PDGFRα and blocks oligodendrocyte differentiation by downregulating major myelin proteins55,56,57. FGF-2 and PDGFα, in combination with IGF-1, also work synergistically to promote OPC DNA synthesis and proliferation to ensure continual replenishing of OPC populations58,59.
Glial maturation
After populating the CNS, astrocytes undergo a profound maturation process, evidenced by changes in gene expression, morphology, and function. In the first month of rodent postnatal development astrocyte appearance shifts from cells with simple filopodial processes that overlap with neighboring astrocytes to dense elaborate branching where cells occupy spatially segregated non-overlapping domains, a process referred to as tiling60,61,62,63. Likewise, recent studies using mouse models31 and primary human fetal tissue samples64,65 have identified thousands of differentially expressed genes (DEGs) between prenatal astrocyte precursor cells and postnatal astrocytes, highlighting differences in physiology and function between these two maturation states. For instance, immature astrocytes express high levels of proliferation genes TOP2A and MKI67, consistent with a developmental window when these cells are populating the CNS. During this time, immature astrocytes promote neuron migration and axon pathfinding66,67, as well as guide synapse formation68,69,70 and elimination71,72. While immature astrocytes help guide CNS construction, mature astrocyte functions shift towards supporting a homeostatic state. This is evidenced by the upregulation of gap junction (GJA1 and GJB6) and water channel (AQP4) genes in mature astrocytes, which are important for mediating neuronal signaling and meeting the fluctuating metabolic demands of the CNS73,74,75.
Similar to astrocytes, the OL lineage also demonstrates morphological, transcriptomic, and functional changes throughout maturation. Structurally, OPCs closely resemble NPCs, with a bipolar morphology and a small number of processes that emanate from opposing regions of the soma76,77. As OPCs differentiate into postmitotic pre-OLs and pre-myelinating OLs, they expand their total surface area by engaging with neighboring axons, losing their bipolarity, and acquiring filamentous myelin outgrowths77,78. This change in morphology coincides with a cascade of TFs binding to regulatory sites of myelination-promoting genes79. For instance, during early differentiation, Olig2 is recruited to Sox10 and myelin regulatory factor (Myrf) enhancers, activating their expression80,81,82. The activation of Myrf and additional TFs, including Nkx2-2, Olig1, Ascl1, YY1, Zfhx1b, and Sox10, is necessary for proper OL differentiation into mature myelinating cells78. Later in OL development, Olig2 and Brg1 are recruited to the enhancers of cell morphogenesis regulators, such as Cdc42 and Rac1, guiding cytoskeleton reorganization, an important step in the progression toward myelinating OLs83. During this shift from pre-myelinating OLs to mature myelinating OLs, myelin structural proteins, including proteolipid protein (PLP), myelin-associated glycoprotein (MAG), and myelin basic protein (MBP), are upregulated, coinciding with increased myelin ensheathment of axons84,85.
Tipping the scales: making astrocytes vs oligodendrocytes
Greater access to primary human tissue specimens has revealed new and diverse progenitor populations, including some that are uniquely hominid86,87,88. Recently, several single-cell RNA-seq papers suggested the presence of a bipotent glial progenitor in the developing human brain that is EGFR+/OLIG2+/OLIG1+/ASCL1+89,90,91. In both the cortex and spinal cord EGFR positive cells are split into two groups—those enriched for astrocyte markers (SOX9 and AQP4) and a separate population expressing canonical oligodendrocyte markers (SOX10, PDGFRA, and PCDH15). These data suggest that at some point, this bipotent glial precursor may diverge towards either an astrocyte or oligodendrocyte trajectory89,91 (Fig. 1). Notably, there also appears to be a population of EGFR-negative multipotent intermediate progenitor cells (mIPC), which are enriched for both neuronal and radial glial markers (RBFOX1, ADGRV1, and NRG). Thus, EGFR may serve as a marker for progenitors committed specifically to the glial fate89.
Assuming astrocytes and OPCs emanate from a shared progenitor, it would be critical that molecular regulators are positioned at the right time and place to assure appropriate proportions and developmental timing of each glial lineage. Transcription factors are one such class of lineage fate determinants that can simultaneously promote one lineage trajectory and repress another. This is evident during gliogenesis when there is strong overlap in the molecular programs that drive AS and OL lineages; however, these shared drivers of development behave in unique and in some cases opposing ways to promote one cellular fate over another.
This concept is perhaps most evident when evaluating the role of SOX9 and its binding partners in determining glial fate specification. SOX9 appears to be an important component of both astrocyte and oligodendrocyte development25,26,33,92 as Sox9 knockout in the developing spinal cord inhibits both astrogenesis and oligogenesis25. However, it serves contrasting roles in each lineage because of differences in when, where, and with which partners it binds. Studies in the developing rodent spinal cord indicate that glial genes are prebound by Sox3 in NSCs. During the initial wave of astrogenesis, genomic sites marked by Sox3 are targeted by Sox9, specifically at regions enriched for Nfi binding motifs93. Together, Sox9 and Nfi facilitate the transcription of astrocyte genes to drive early astrogenesis26 (Fig. 1). In oligogenesis, Sox9 is prebound at multiple oligodendrocyte genes, which are then targeted by Sox10 to facilitate oligodendrocyte development25,93 (Fig. 1). Unlike in astrocyte development, Sox9 expression appears to peak during the OPC lineage commitment phase of OL development but then drops off during later stages of maturation94, suggesting that it serves different roles in astrocyte and oligodendrocyte developmental progression.
Not only does SOX9 display differential binding and functional properties in AS and OL lineages, but there is also evidence that the binding partners of SOX9 in one lineage may directly antagonize SOX9 binding partners of a diverging lineage. Work by Glasgow et al. in chick and mouse models demonstrates that NFIA and Sox10 exhibit antagonizing effects on each other. Expression of SOX10 impedes NFIA-induced expression of AS genes and reciprocally, NFIA inhibits SOX10-induction of OL genes95. The same study provided evidence suggesting that Olig2 may play a key role in the NFIA/SOX10 dynamic by reinforcing the interaction between SOX10 and NFIA, promoting a lineage-fate-decision stage95. This suggests that while NFIA and SOX10 promote their respective lineages by interacting with SOX9, they also suppress competing lineages by interfering with each other’s ability to transcribe specific glial gene sets, thereby tipping the scales toward a specific glial lineage (Fig. 1).
The prospect of a shared AS/OL precursor cell and a precarious scale of AS/OL fate has important implications for glioma research, where malignant cells resemble AS- and OL- like cell types and in some cases, have the capacity to differentiate between the two glial fates.
Gliomas echo glial development
Cancer echoes many early developmental principles, including rapid cell proliferation, the activation of nascent developmental signaling pathways, a high degree of cellular plasticity, and susceptibility to local environmental cues. Brain tumors in particular are a prime example of this developmental mimicry. Advancements in single-cell sequencing datasets confirm that brain tumors, especially glioblastomas, exhibit cellular heterogeneity comprised of hierarchies reflective of early neurodevelopment (Fig. 2). This mirroring of early glial lineages may be explained by aberrant activation of developmental regulatory programs, including key TFs, a frequent phenomenon in gliomas96,97,98,99,100,101. Additionally, functional studies in Drosophila and rodent models implicate key fundamental neurodevelopmental signaling cascades, such as Wnt, Notch, and Hedgehog pathways in tumorigenesis102,103,104,105,106.

Schematic depicting the normal neurodevelopmental cell hierarchy and how these cell states are (over)represented across glioma subtypes. The glioma subtypes are arranged in order of relative aggressiveness, with the most aggressive and dedifferentiated (glioblastoma) on the far left. Relative enrichment of neurodevelopmental cell states are represented by arrow thickness. Relevant single-cell publications that support these findings are listed below respective tumor types.
Stem-like populations that are abundant in neurodevelopment have also been identified in most primary brain malignancies, including various glioma subtypes, where they are referred to as glioma stem cells (GSCs). While it remains unclear what type of cell(s) these represent and if a pan-GSC marker exists, GSCs exhibit high expression of embryonic stem cell genes and self-renewal capabilities107,108,109. GSCs also demonstrate the ability to self-renew, adapt to the tumor microenvironment, and differentiate into multiple lineage types, reminiscent of the NSC population within the embryonic brain107,108,109. This population is believed to be the source of tumor propagation108,109 and is capable of evading immune surveillance and therapeutic interventions such as chemotherapy and radiation110,111,112. Essentially, brain tumors recycle early developmental blueprints for generating and maintaining progenitor populations102,103,104,105,106.
Glioblastoma
Glioblastoma (GBM) is classified by the World Health Organization (WHO) as a grade IV glioma. These tumors are the most aggressive and common primary CNS malignancy, accounting for approximately 16% of all primary CNS neoplasms113. For primary (de novo) GBMs, which account for 80% of all GBMs, the median age of diagnosis is 62114. Secondary GBMs, which develop from lower-grade astrocytomas or oligodendrogliomas, are more frequent in younger adults (mean age 45 years)114,115. The typical treatment course for patients with GBM consists of maximal safe surgical resection followed by radiotherapy and temozolomide (TMZ) chemotherapy116. Unfortunately, due to the diffuse, heterogeneous, and resilient nature of GBM, these tumors are nearly impossible to entirely irradicate and the prognosis remains bleak with a median survival of 15 months117,118.
The first GBM datasets included in The Cancer Genome Atlas highlighted inter-tumoral transcriptional heterogeneity across tumors, partitioning them into four transcriptional subtypes—proneural, neural, mesenchymal, and classical—where each exhibits unique cell type-specific gene signatures and oncogenic events119,120. However, subsequent studies incorporating multi-region sampling across individual GBM tumors demonstrated that many transcriptional subtypes exist within different regions of the same tumor121. This finding was confirmed and delineated by a series of GBM single-cell transcriptomic studies, which uniquely afforded the ability to distinguish between neoplastic and non-neoplastic cells in the tumor bulk using predicted copy number variations for each individual cell. Implementing this approach revealed that the neural tumor signature was likely an artifact of non-cancerous neuronal populations122 and allowed for more nuanced transcriptional classification of neoplastic cells. Neftel et al. and other groups demonstrated that GBM malignant tumor cells align to neurodevelopmental lineages and generally fall into four transcriptional subtypes that reflect- (1) neural-progenitor-like (NPC-like), (2) oligodendrocyte-progenitor-like (OPC-like), (3) astrocyte-like (AC-like), and (4) mesenchymal-like (MES-like) states, where any given tumor possesses varying ratios of cells that exist in all of these states123,124,125. Further, pseudotime analysis suggests that these cells exist along a stemness hierarchy, with a small population of malignant tumor cells that resemble multipotent NSCs at the apex, and the remaining majority of neoplastic cells existing along the four cellular differentiation trajectories123 (Fig. 2).
It is important to note that these transcriptomic analyses capture cell states at a single moment in time. Functional studies where cells of a specific GSC population have been engrafted into patient-derived xenografts demonstrate that GSC state is anything but stagnant, and that regardless of the cell population used to initiate the xenograft—AC-like, NPC-like, or MES-like—resulting tumors present all three cell states in comparable frequencies123,126,127. Cell state fluctuation may result from endogenous tumor microenvironmental (TME) niches, which have been shown to influence tumor cell biology, including the perivascular128,129,130,131, hypoxic132,133,134,135, and invasive edge136 niches. Additionally, evidence suggests that therapeutic intervention, itself, can induce a shift in GSC state to a phenotype more conducive for evading harsh treatment strategies137,138. However, intrinsic molecular landscape also plays a role in determining GSC state. Neftel et al. found that frequencies of each transcriptional state are associated with genetic alterations in CDK4, PDGFRA, EGFR, and NF1 that appear to bias cell identity towards a particular state123. Thus, while GSCs and normal developmental cell types share the capacity to respond to environmental cues, it is the combination of oncogenic mutations, genomic instability, and disruption of chromatin regulators that permits GSCs to override normal systems of checks and balances.
Many of the genetic aberrations in GBM occur in genes that play critical roles in normal glial development. Genetically engineered mouse models (GEMMs) and genetic manipulation of primary glial cell populations illustrate how loss-of-function of GBM-associated tumor suppressor genes (TP53, PTEN, NF1) or gain-of-function of oncogenes (EGFR, PDGFR, RAS, AKT) induce dedifferentiation of quiescent glia139,140,141,142,143, restrict progenitors to an immature state144,145, and may even promote the inter-conversion between glial types146. Likewise, neurodevelopmental TFs, such as ASCL1, POU3F2, SOX2, SALL2, and OLIG2 can act as oncogenes by inappropriately activating developmental programs that push differentiated GBM cells into tumor propagating GSCs96,98,147,148,149. Olig2 has also demonstrated the capacity to dictate GSC subtype, as a loss of Olig2 causes a shift from a proneural transcriptional subtype towards a more astrocytic phenotype, including downregulation of PDGFR and concomitant upregulation of EGFR150. Recent studies have also demonstrated reciprocal binding of ASCL1 and OLIG2 in part determines the cell types and degree of migration of tumor cells151. When ASCL1 levels are greater than OLIG2, tumors are biased towards astrocyte/NPC-like lineages, whereas the converse scenario pushes cells towards oligodendrocyte fates151.
Another clear example of the convergence between developmental and oncogenic programs is the redundancy of EGFR activity in development and gliomagenesis. EGFR activity is essential during normal gliogenesis90,152,153,154 and both EGFR amplification or constitutively activating mutations (EGFRvIII) are among the most common molecular features of GBM, occurring in about 50% of all cases155,156. The tumor biology of EGFR signaling is highly nuanced. Liu et al. demonstrated that the most common EGFR mutation, EGFRvIII, remodels the enhancer regulatory landscape of GBM to induce two key TFs that regulate astrocyte development, SOX9, and FOXG1. Together, these EGFR-dependent TFs work collaboratively to induce oncogenic programs, including c-MYC target genes and EGFR-regulated genes157. Interestingly, a recent study from the Deneen group showed that one of these EGFR targets, SOX9, has divergent roles in varying brain tumor subtypes, which each exhibit unique epigenomic states158. Thus, while the activation of developmental programs is a shared biological phenomenon across gliomas, individual molecular perturbations can induce opposing outcomes within different cellular contexts.
Adult-type diffuse gliomas
Adult-type diffuse gliomas consist of astrocytomas (WHO grades II, III, and IV) and oligodendrogliomas (WHO grades II and III). Unlike GBM, most of the tumors in this class exhibit IDH1/2 mutations, and oligodendrogliomas are further distinguished by the common chromosomal 1p/19 co-deletion159,160. IDH-mutant oligodendrogliomas and astrocytomas demonstrate a recycling of early glial differentiation programs to fuel immature developmental cell states. For instance, the regulatory chromatin architecture that is present in normal gliogenesis and the binding of astrocytic TFs like SOX9, NFIA, and BRN2, is shared by models of diffuse glioma and promoted tumorigenesis30. Several of these potent glial fate determinants even demonstrate the capacity to regulate glioma subtype specification reminiscent of the early developmental decision to bias towards AS versus OL lineages. This was perhaps most clearly demonstrated by experiments overexpressing NFIA in a mouse model of oligodendroglioma, which shifted tumor histopathology to more closely reflect astrocytomas95. In addition to intrinsic regulators, extrinsic cues also play a role in driving glioma phenotype. PDGF, a potent mitogen involved in generating and maintaining OPCs in the developing brain, induces tumors that reflect oligodendroglioma biology161,162.
Although diffuse astrocytomas and oligodendrogliomas are characterized by unique histological features, genomic perturbations, and markers of gliogenic regulation, neurodevelopmental lineages are reflected quite consistently between the two tumor types. Single-cell transcriptomic work by Venteicher et al. highlights the similarities between the two glioma subtypes, demonstrating that both harbor three main groups of malignant tumor cells—a relatively small proliferative NSC-like population, and two populations of nonproliferating cells that resemble AS and OL lineages163 (Fig. 2). Interestingly, the primary differences between astrocytomas and oligodendrogliomas are related to genetic events and tumor microenvironmental niches163. When focusing on the cellular heterogeneity within oligodendrogliomas, Tirosh et al. found that CNV-subclones within these tumors span all three transcriptional states—NSC-like, OL-like, and AS-like—suggesting that factors beyond genetic events contribute to the observed developmental hierarchy164. This finding is supported by experiments where PDGF exposure yields an inconsistent tumor phenotype between WHO grade II oligodendrogliomas and a mixed oligoastrocytoma profile that expresses both GFAP and Vimentin161,162.
Pediatric-type diffuse midline gliomas
H3 K27-altered diffuse midline gliomas (K27M-DMGs) are a primarily pediatric and extremely aggressive glioma subtype with a median survival of about one-year post-diagnosis165. These tumors are regionally specific to midline structures occurring in the thalamus, midbrain, cerebellum, or pons; the latter of which are designated as diffuse intrinsic pontine gliomas (DIPG)166. A major breakthrough in the tumor biology of DMGs was the finding that many of these neoplasms contain a lysine27-to-methionine (K27M) mutation in histone 3 (H3). In H3K27-altered DMGs, H3K27M suppresses EZH2, the catalytic subunit of polycomb repressive complex 2 (PRC2). Polycomb activity is involved in a variety of epigenetic regulatory processes, including trimethylation of Lys-27 on histone 3 (H3K27me3)167,168, which leads to genome-wide dysregulation of gene repression and cell differentiation168,169.
Single-cell transcriptomic profiling of these tumors has uncovered a similar developmental hierarchy in K27M-DMG to other diffuse gliomas; however, there are several noteworthy differences170. K27M-DMGs contain a substantially larger pool of undifferentiated cells, consistent with the more aggressive nature of this tumor subtype170. Additionally, undifferentiated cells in K27M-DMGs most closely resemble OPC lineages170, unlike the putative GSCs in IDH-mutant diffuse gliomas that reflect an NSC identity (Fig. 2).
More recent work from Jessa et al. and Liu et al. implemented a barrage of single-cell genomic, epigenomic, and chromatin profiling approaches to dissect region- and age-related developmental signatures in K27-altered DMG. Jessa et al. profiled cells across DMGs that harbor the H3K27M mutation in different histone variants (H3.1 and H3.3) and demonstrated that while K27M-DMGs appear to maintain a developmentally conserved OPC chromatin signature, differences between H3.1 and H3.3 samples point to distinct OPC developmental origins171. Specifically, the molecular profiles of H3.1K27M ACVR1-mutant pontine gliomas resemble early ventral NKX6-1+/SHH-dependent brainstem OPCs, whereas the H3.3K27M signature is more closely aligned with later dorsal PAX3+/BMP-dependent progenitors171. These results are supported by work from Michelle Monje and collaborators, which showed that H3.3K27M and H3.1K27M DIPG demonstrate variant-specific PRC2 regulation of developmental gene sets and cell signaling programs172. In a similar study, Liu et al. observed the presence of a stem-like OPC population across all H3K27M-DMGs, regardless of age or tumor location173. Remarkably, the team identified location-specific OPC subpopulations, where pontine tumors were enriched for a more immature pre-OPC-like signature in comparison with thalamic tumor OPC signatures, corroborating the findings that pontine K27M-DMGs may arise from an OPC population of earlier origins173. Recent functional studies also suggests that OPC-like tumor cells play a critical role in fueling K27M-DMG tumor survival by synapsing with surrounding neurons, catalyzing neuronal signaling-induced tumor growth174,175,176. Together, these findings suggest that K27-altered DMGs arising in different brain regions may descend from distinct cells of origin, but likely undergo similar developmental pressures that shape a shared OPC-enriched cellular hierarchy.
Liu et al. further leveraged their age- and region-matched K27M-DMG samples to investigate how patient age, brain tumor location, and mutational status contribute to the global cellular makeup of K27M-DMG tumors. This dataset revealed an enrichment of MES-like cells (a state also observed in adult GBM) in K27M-DMGs from adult patients, compared to region-matched K27M-DMGs from pediatric patients173. The authors postulate that this difference may be influenced by endogenous developmental shifts in brain myeloid cell composition, whereby older patients display an enrichment of macrophages compared to pediatric patients who exhibit higher proportions of microglia173. When comparing K27M-DMG to age- and region- matched IDH-mutant midline gliomas, it appears that the K27M mutation may skew tumor cells toward a glial/OPC-like cell fate173. Lastly, Liu et al. assessed the spatial distribution of cell states in K27M-DMGs identifying physical niches of proliferative OPC-like and OC-like cells, accompanied by larger portions of nonproliferative diffusely-dispersed AC-like cells173. This discrepancy between the spatial transcriptomic and existing scRNA-seq datasets may be due to a loss of vulnerable cell populations during tissue dissociation and suggests that spatial transcriptomic approaches, which utilize in-tact tissue sections, might help mitigate these biases in future studies.
Circumscribed pilocytic astrocytomas
Unlike grade II and III astrocytomas, pilocytic astrocytomas (PAs) are circumscribed astrocytic gliomas, they do not progress to higher-grade tumors, and most commonly arise in the optic pathway, brainstem, and cerebellum177. In comparison to higher-grade diffuse gliomas, PAs have less complex genetics, with most exhibiting only a single-driver alteration activating the MAPK pathway177. PAs in the cerebellum commonly develop sporadically and display a somatic rearrangement where the BRAF gene kinase domain is fused to the KIAA1549 gene (referred to as KIAA1549:BRAF)177. An additional PA subtype is present in children with Neurofibromatosis type 1 (NF1) who typically experience tumors in optic pathways177.
Akin to other gliomas, the developmental origins of PAs remain largely unknown, although, there is some evidence implicating OPC and astrocyte populations, specifically. For instance, NF1-deficient astrocytes display hyperactive mTOR signaling and a greater proliferative capacity, a phenotype that is observed in patient NF1 PA tumors178,179. Conversely, limited evidence suggests that ectopic expression of the KIAA1549:BRAF fusion protein does increase proliferation of NSCs in vitro and in vivo; however, there is more uncertainty about the cell of origin in KIAA1549:BRAF fusion PAs as a result of limited experimental models180,181. Recent single-cell RNA-seq studies have begun unraveling the cellular hierarchies within these tumors, providing new evidence that an OPC-like progenitor population enriched for MAPK signaling may give rise to a much larger group of AC-like cells with diminished MAPK signaling activity182,183 (Fig. 2). In comparison to GBM and other diffuse astrocytomas/oligodendrogliomas, the NSC signature is noticeably absent from PA cells, suggesting that PAs may be driven by a more developmentally committed OPC-like cell182,183 (Fig. 2).
How do glioma cells move across developmental time?
While early glial cell types and developmental hierarchies are recapitulated in most gliomas, we are still learning how to use normal developmental trajectories to better understand glioma biology and potential therapeutic interventions. More detailed and complete glial maturation atlases can provide insight into the initial, present, and future developmental stages that glioma cells progress through. This can be thought of as (initial) when in developmental time do gliomas begin (i.e., cell of origin); (present) when in developmental time glioma cells reside during tumor progression; and (future) when in developmental time glioma cells are capable of moving towards with intervention (Fig. 3).

Schematized summary of how normal glial differentiation trajectories (generated from transcriptomic, functional, and morphological information) can inform how glioma cells progress through developmental time—when in developmental time do tumor cells (1) begin, (2) thrive during tumorigenesis, and (3) are capable of being coerced to? Citations are listed for primary evidence related to each respective question.
When in developmental time do gliomas begin?
The first of these questions is a long-standing enigma: which cell types have the capacity to give rise to gliomas? An important note here is that there is certainly a difference between cells with gliomagenic capacity and the reality of which cells originate tumors in vivo. There are two prevailing theories addressing glioma cellular origin(s). The first is a scenario where a differentiated somatic cell stochastically gains a combination of oncogenic and/or tumor suppressor mutations, through a variety of possible mechanisms including replication errors or DNA damage, transforming quiescent cells into a stem-like state (Fig. 3). Critics of this theory argue that it is unlikely that a mature non-proliferative cell with a limited lifespan could accumulate the perfect combination of mutations to induce such tumorigenic potential. However, recent work by Simpson Ragdale et al. demonstrates that p53 knockout in reactive astrocytes destabilizes astrocyte fate, such that adult astrocytes are capable of dedifferentiating after chronic injury paradigms140. Reacquisition of a stem-like program was largely mediated through age-exacerbated inflammation coupled with EGF secretion from periwound astrocytes, which induces mTOR-dependent reacquisition of early neurodevelopmental TF programs, including Sox2, Olig2, and Ascl1 activation140. This suggests that p53 mutation may lift a restraint on fate commitment, while chronic inflammation could serve as a second hit to induce dedifferentiation at later time points.
The second cell-of-origin theory proposes that GSCs arise when an endogenous quiescent stem cell in the brain acquires oncogenic mutations (Fig. 3). Importantly, the exact identity of this stem-like cell is still debated and likely varies across glioma subtypes. Given the cellular heterogeneity of GBM, many hypothesize that the GSC origin in GBM is adult NSCs, which developmentally have the capacity to generate each of the transcriptomic subtypes documented by Neftel et al. (AS-like, OPC-like, and NPC-like). Indeed, a multitude of evidence supports this theory. For instance, GBMs are thought to arise in the SVZ, where quiescent NSCs reside. Additionally, GBM tumor cells share many properties with NSCs, including high expression of NSC markers184,185; they can form neurospheres that have a similar structure to those derived from adult human subventricular zone cells186,187; and Nestin-positive tumor cells are critical for tumor growth and chemotherapy resistance110. Alternatively, others hypothesize that a lineage-committed precursor, such as an OPC or astrocyte precursor cell, is a more likely culprit. This is because GSCs also express abundant markers for these cell types188,189,190,191 and there is evidence that both lineages possess tumor propagating potential141,188,192,193. OPCs vastly outnumber NSCs, which are restricted to ventricular zone niches and the dentate gyrus194,195. Additionally, while there is some evidence of adult neurogenesis in the rodent hippocampus, this phenomenon is more controversial in the adult human brain, making OPCs the major proliferative cell population in the adult human CNS176. Far less evidence exists on whether astrocyte progenitor cells exist in the adult brain and if so, exhibit the same proliferative potential as OPCs in the adult CNS196. It is also conceivable, and highly likely, that there are discrete cells of origin for different GSC subtypes or for the same GSC subtype across different brain regions. The former of these ideas is strongly supported by work from Parada and colleagues, which has demonstrated that the same genetic drivers in different adult lineage-committed progenitors give rise to molecularly and phenotypically distinct GBM subtypes197.
When in developmental time do glioma cells reside and thrive during tumor progression?
While it is evident that gliomas reflect multiple early developmental cell types, it is unclear if there are specific maturation stages of glial development reflected in glioma tumors (Fig. 3). This is largely due to our fragmented understanding of glial maturation, which in humans primarily occurs between the third trimester of gestation and the first postnatal month, a brief but critical period when access to primary human tissue samples is greatly restricted. Much of what we do know about glial development and maturation is derived from murine model systems and a limited number of second trimester primary fetal tissue samples. While informative, there are major temporal gaps during this developmental window in human samples, which exhibit many neurodevelopmental differences from rodents64,198,199,200,201. Curating more comprehensive developmental timelines of glial lineages will help inform whether glioma cells are stalled at particular developmental stages and which molecular programs could be leveraged to coerce maturation towards a quiescent state.
One viable option for building comprehensive timelines of human glial maturation is to leverage human in vitro model systems, such as human brain organoids. There are now numerous robust protocols for forming and culturing human stem cell-induced 3D organoids that are patterned to reflect various regions of the CNS, including forebrain202,203, midbrain204,205, hindbrain206,207, and spinal cord208,209. This platform recapitulates many key features of human neurodevelopment including complex cellular composition, intricate tissue architecture, and functionally active neurons203,204,205,210,211,212,213,214. Additionally, long-term culture of human brain organoids depicts maturing astrocyte215,216 and oligodendrocyte217,218,219 lineages with transcriptomic profiles that reflect pre- and postnatal stages of human brain development. Altogether, this makes organoids an ideal system for chronicling elusive windows of development at a high temporal resolution to capture all phases of glial maturation220.
The human brain organoid model is also well-suited to investigate glioma-glia and glioma-neuron interactions. There are several organoid-based approaches that inherently incorporate both malignant and normal differentiated cell types. These include neoplastic cerebral organoids (neoCOR), whereby specific genetic alterations are introduced at the stem cell stage prior to 3D formation to induce spontaneous tumor formation221,222, and organoid-glioma coculture models, in which patient-derived tumor cells are engrafted into healthy organoids127,223,224,225,226. While the neoCOR approach serves as a reductionist system ideal for questions that pertain to specific oncogenic molecular events, the organoid-glioma coculture approach is well-suited for those who wish to account for the diverse and complex molecular nature of human gliomas. Using these models, several groups have demonstrated that GSCs readily invade organoids without losing their proliferative capacity222,223,224, form tumor microtubes with host organoid cells223,225, and maintain a transcriptional profile that closely reflects the parent tumor127,226. Given the amenability of organoids to a variety of functional and high-throughput sequencing assays, these models would be optimal for investigating the relationships between malignant tumor cells and the surrounding host tissue.
When in developmental time are glioma cells capable of moving towards with intervention?
Given the parallels between neurodevelopment and glioma biology, one might reasonably hypothesize that malignant glioma cells are susceptible to the same maturation cues that coerce quiescence in normal glial development (Fig. 3). This is the rationale behind differentiation therapy, which explores therapeutic options to coerce tumor cells through developmental time by exploiting extrinsic and intrinsic factors that regulate cell differentiation and maturation. Of course, a major challenge remains in identifying the optimal glial maturation cues to target in gliomas.
One avenue under active investigation is targeting signaling pathways that are critical in initiating gliogenesis (BMP, Wnt, Notch, STAT3, MAPK/ERK, and TGF-B) and that are frequently hijacked in glioma progression227. Several of these pathways appear to have particularly potent impacts on GSC growth, proliferation, and differentiation when targeted through BMP4 and RA treatment228,229,230,231, both of which are important in early astrogenesis4 and are currently being tested in clinical trials232. Another approach is to inhibit glial development TFs that are inappropriately activated in glioma97,232,233,234. For example, STAT3, a key player in the gliogenic switch, is highly upregulated in gliomas, is associated with glioma EGFR amplification, and contributes to GSC proliferation and migration, thus making it a high-priority target for inhibition233,235. Multiple groups have identified approaches for suppressing STAT3 activity in GSCs, resulting in increased GSC sensitivity to subsequent chemo and radiation therapy235,236,237.
While there is accumulating evidence that malignant glioma cells are receptive to glial developmental cues, there are several technical challenges and caveats to differentiation therapies that must be considered. As illustrated through rigorous testing of BMP4 treatment, it is extremely challenging to identify targetable molecular programs that overcome inter- and intra- tumoral heterogeneity boundaries238,239. Not only do tumor cells from different patients exhibit varying genetic backgrounds that respond inconsistently to BMP4 treatment, but there is evidence that GSCs exposed to separate TME niches may also respond uniquely to treatment240. Another obstacle is defining the benchmarks for successful GSC differentiation. As demonstrated by in vitro experiments overexpressing TFs to induce normal glial development31, different TFs will likely induce unique epigenetic and transcriptomic changes. Deciphering which set(s) of changes indicate sufficient maturation will be especially challenging without more detailed molecular maps of normal maturation in glial lineages. Lastly, even if GSCs respond to differentiation cues to progress through developmental time, these changes may be transient. In fact, Caren et al. demonstrated that GSCs are capable of reverting to a stem-like state following BMP-treatment as a result of incomplete chromatin accessibility changes that permit aberrant SOX TF binding241. This suggests that the most effective differentiation method will need to induce large-scale chromatin architecture shifts that are comparable to what occurs in normal glial maturation234.
Conclusion
Neurodevelpomental hierarchies are reflected across glioma subtypes, where the programs that drive normal lineage specification and maturation are aberrantly activated to promote tumorigenesis. Utilizing blueprints of normal glial trajectories has revealed new information about glioma occurrence, growth, and resilience. However, there are discontinuities in our current timelines of glial development, particularly between the third trimester and into the first few postnatal months, a critical window of time when immature precursor populations give rise to mature quiescent counterparts. Rapidly evolving model systems, lineage tracing methods, and sequencing platforms will be fundamental for filling in these gaps, and understanding how these elusive developmental stages are represented across gliomas and contribute to tumor progression.
References
Takouda, J., Katada, S. & Nakashima, K. Emerging mechanisms underlying astrogenesis in the developing mammalian brain. Proc. Jpn Acad. Ser. B Phys. Biol. Sci. 93, 386–398 (2017).
Zeng, B. et al. The single-cell and spatial transcriptional landscape of human gastrulation and early brain development. Cell Stem Cell 30, 851–866 e857 (2023).
Desai, A. R. & McConnell, S. K. Progressive restriction in fate potential by neural progenitors during cerebral cortical development. Development 127, 2863–2872 (2000).
Kanski, R., van Strien, M. E., van Tijn, P. & Hol, E. M. A star is born: new insights into the mechanism of astrogenesis. Cell Mol. Life Sci. 71, 433–447 (2014).
Derouet, D. et al. Neuropoietin, a new IL-6-related cytokine signaling through the ciliary neurotrophic factor receptor. Proc. Natl Acad. Sci. USA 101, 4827–4832 (2004).
Uemura, A. et al. Cardiotrophin-like cytokine induces astrocyte differentiation of fetal neuroepithelial cells via activation of STAT3. Cytokine 18, 1–7 (2002).
Cheng, P. Y. et al. Interplay between SIN3A and STAT3 mediates chromatin conformational changes and GFAP expression during cellular differentiation. PLoS One 6, e22018 (2011).
He, F. et al. A positive autoregulatory loop of Jak-STAT signaling controls the onset of astrogliogenesis. Nat. Neurosci. 8, 616–625 (2005).
Menard, C. et al. An essential role for a MEK-C/EBP pathway during growth factor-regulated cortical neurogenesis. Neuron 36, 597–610 (2002).
Gauthier, A. S. et al. Control of CNS cell-fate decisions by SHP-2 and its dysregulation in Noonan syndrome. Neuron 54, 245–262 (2007).
Ernst, M. & Jenkins, B. J. Acquiring signalling specificity from the cytokine receptor gp130. Trends Genet. 20, 23–32 (2004).
Hirabayashi, Y. et al. Polycomb limits the neurogenic competence of neural precursor cells to promote astrogenic fate transition. Neuron 63, 600–613 (2009).
Yuan, Z. L., Guan, Y. J., Chatterjee, D. & Chin, Y. E. Stat3 dimerization regulated by reversible acetylation of a single lysine residue. Science 307, 269–273 (2005).
Nakashima, K. et al. Synergistic signaling in fetal brain by STAT3-Smad1 complex bridged by p300. Science 284, 479–482 (1999).
Kamakura, S. et al. Hes binding to STAT3 mediates crosstalk between Notch and JAK-STAT signalling. Nat. Cell Biol. 6, 547–554 (2004).
Wilczynska, K. M. et al. Nuclear factor I isoforms regulate gene expression during the differentiation of human neural progenitors to astrocytes. Stem Cells 27, 1173–1181 (2009).
Cebolla, B. & Vallejo, M. Nuclear factor-I regulates glial fibrillary acidic protein gene expression in astrocytes differentiated from cortical precursor cells. J. Neurochem 97, 1057–1070 (2006).
Namihira, M. et al. Committed neuronal precursors confer astrocytic potential on residual neural precursor cells. Dev. Cell 16, 245–255 (2009).
das Neves, L. et al. Disruption of the murine nuclear factor I-A gene (Nfia) results in perinatal lethality, hydrocephalus, and agenesis of the corpus callosum. Proc. Natl Acad. Sci. USA 96, 11946–11951 (1999).
Yu, X., Nagai, J. & Khakh, B. S. Improved tools to study astrocytes. Nat. Rev. Neurosci. 21, 121–138 (2020).
Lanjewar, S. N. & Sloan, S. A. Growing Glia: Cultivating human stem cell models of gliogenesis in health and disease. Front Cell Dev. Biol. 9, 649538 (2021).
Tchieu, J. et al. NFIA is a gliogenic switch enabling rapid derivation of functional human astrocytes from pluripotent stem cells. Nat. Biotechnol. 37, 267–275 (2019).
Tiwari, N. et al. Stage-specific transcription factors drive astrogliogenesis by remodeling gene regulatory landscapes. Cell Stem Cell 23, 557–571.e558 (2018).
Deneen, B. et al. The transcription factor NFIA controls the onset of gliogenesis in the developing spinal cord. Neuron 52, 953–968 (2006).
Stolt, C. C. et al. The Sox9 transcription factor determines glial fate choice in the developing spinal cord. Genes Dev. 17, 1677–1689 (2003).
Kang, P. et al. Sox9 and NFIA coordinate a transcriptional regulatory cascade during the initiation of gliogenesis. Neuron 74, 79–94 (2012).
Nagao, M., Ogata, T., Sawada, Y. & Gotoh, Y. Zbtb20 promotes astrocytogenesis during neocortical development. Nat. Commun. 7, 11102 (2016).
Yeon, G. B. et al. NFIB induces functional astrocytes from human pluripotent stem cell-derived neural precursor cells mimicking in vivo astrogliogenesis. J. Cell Physiol. 236, 7625–7641 (2021).
Byun, J. S. et al. The transcription factor PITX1 drives astrocyte differentiation by regulating the SOX9 gene. J. Biol. Chem. 295, 13677–13690 (2020).
Glasgow, S. M. et al. Glia-specific enhancers and chromatin structure regulate NFIA expression and glioma tumorigenesis. Nat. Neurosci. 20, 1520–1528 (2017).
Lattke, M. et al. Extensive transcriptional and chromatin changes underlie astrocyte maturation in vivo and in culture. Nat. Commun. 12, 4335 (2021).
Liu, Z. et al. Induction of oligodendrocyte differentiation by Olig2 and Sox10: evidence for reciprocal interactions and dosage-dependent mechanisms. Dev. Biol. 302, 683–693 (2007).
Finzsch, M., Stolt, C. C., Lommes, P. & Wegner, M. Sox9 and Sox10 influence survival and migration of oligodendrocyte precursors in the spinal cord by regulating PDGF receptor alpha expression. Development 135, 637–646 (2008).
Watzlawik, J. O., Warrington, A. E. & Rodriguez, M. PDGF is required for remyelination-promoting IgM stimulation of oligodendrocyte progenitor cell proliferation. PLoS One 8, e55149 (2013).
Weider, M. et al. Elevated in vivo levels of a single transcription factor directly convert satellite glia into oligodendrocyte-like cells. PLoS Genet 11, e1005008 (2015).
Stolt, C. C. et al. SoxD proteins influence multiple stages of oligodendrocyte development and modulate SoxE protein function. Dev. Cell 11, 697–709 (2006).
Samanta, J. & Kessler, J. A. Interactions between ID and OLIG proteins mediate the inhibitory effects of BMP4 on oligodendroglial differentiation. Development 131, 4131–4142 (2004).
Liu, A. et al. A molecular insight of Hes5-dependent inhibition of myelin gene expression: old partners and new players. EMBO J. 25, 4833–4842 (2006).
Morrow, T., Song, M. R. & Ghosh, A. Sequential specification of neurons and glia by developmentally regulated extracellular factors. Development 128, 3585–3594 (2001).
Barnabe-Heider, F. et al. Evidence that embryonic neurons regulate the onset of cortical gliogenesis via cardiotrophin-1. Neuron 48, 253–265 (2005).
Bonni, A. et al. Regulation of gliogenesis in the central nervous system by the JAK-STAT signaling pathway. Science 278, 477–483 (1997).
Bugga, L., Gadient, R. A., Kwan, K., Stewart, C. L. & Patterson, P. H. Analysis of neuronal and glial phenotypes in brains of mice deficient in leukemia inhibitory factor. J. Neurobiol. 36, 509–524 (1998).
Nakashima, K. et al. BMP2-mediated alteration in the developmental pathway of fetal mouse brain cells from neurogenesis to astrocytogenesis. Proc. Natl Acad. Sci. USA 98, 5868–5873 (2001).
Nakashima, K., Yanagisawa, M., Arakawa, H. & Taga, T. Astrocyte differentiation mediated by LIF in cooperation with BMP2. FEBS Lett. 457, 43–46 (1999).
Bonaguidi, M. A. et al. LIF and BMP signaling generate separate and discrete types of GFAP-expressing cells. Development 132, 5503–5514 (2005).
Gross, R. E. et al. Bone morphogenetic proteins promote astroglial lineage commitment by mammalian subventricular zone progenitor cells. Neuron 17, 595–606 (1996).
Miller, M. W. Expression of transforming growth factor-beta in developing rat cerebral cortex: effects of prenatal exposure to ethanol. J. Comp. Neurol. 460, 410–424 (2003).
Song, M. R. & Ghosh, A. FGF2-induced chromatin remodeling regulates CNTF-mediated gene expression and astrocyte differentiation. Nat. Neurosci. 7, 229–235 (2004).
Irmady, K., Zechel, S. & Unsicker, K. Fibroblast growth factor 2 regulates astrocyte differentiation in a region-specific manner in the hindbrain. Glia 59, 708–719 (2011).
Asano, H. et al. Astrocyte differentiation of neural precursor cells is enhanced by retinoic acid through a change in epigenetic modification. Stem Cells 27, 2744–2752 (2009).
Voss, A. et al. Computational Identification of Ligand-Receptor Pairs that Drive Human Astrocyte Development. bioRxiv, 2022.2005.2031.491513, https://doi.org/10.1101/2022.05.31.491513 (2022).
Noble, M., Murray, K., Stroobant, P., Waterfield, M. D. & Riddle, P. Platelet-derived growth factor promotes division and motility and inhibits premature differentiation of the oligodendrocyte/type-2 astrocyte progenitor cell. Nature 333, 560–562 (1988).
Raff, M. C., Lillien, L. E., Richardson, W. D., Burne, J. F. & Noble, M. D. Platelet-derived growth factor from astrocytes drives the clock that times oligodendrocyte development in culture. Nature 333, 562–565 (1988).
Heldin, C. H. & Westermark, B. Mechanism of action and in vivo role of platelet-derived growth factor. Physiol. Rev. 79, 1283–1316 (1999).
Goddard, D. R., Berry, M., Kirvell, S. L. & Butt, A. M. Fibroblast growth factor-2 inhibits myelin production by oligodendrocytes in vivo. Mol. Cell Neurosci. 18, 557–569 (2001).
Fressinaud, C., Vallat, J. M. & Labourdette, G. Basic fibroblast growth factor down-regulates myelin basic protein gene expression and alters myelin compaction of mature oligodendrocytes in vitro. J. Neurosci. Res 40, 285–293 (1995).
Bansal, R. & Pfeiffer, S. E. Inhibition of protein and lipid sulfation in oligodendrocytes blocks biological responses to FGF-2 and retards cytoarchitectural maturation, but not developmental lineage progression. Dev. Biol. 162, 511–524 (1994).
Jiang, F., Frederick, T. J. & Wood, T. L. IGF-I synergizes with FGF-2 to stimulate oligodendrocyte progenitor entry into the cell cycle. Dev. Biol. 232, 414–423 (2001).
McMorris, F. A., Furlanetto, R. W., Mozell, R. L., Carson, M. J. & Raible, D. W. Regulation of oligodendrocyte development by insulin-like growth factors and cyclic nucleotides. Ann. N. Y Acad. Sci. 605, 101–109 (1990).
Bushong, E. A., Martone, M. E. & Ellisman, M. H. Maturation of astrocyte morphology and the establishment of astrocyte domains during postnatal hippocampal development. Int J. Dev. Neurosci. 22, 73–86 (2004).
Bushong, E. A., Martone, M. E., Jones, Y. Z. & Ellisman, M. H. Protoplasmic astrocytes in CA1 stratum radiatum occupy separate anatomical domains. J. Neurosci. 22, 183–192 (2002).
Halassa, M. M., Fellin, T., Takano, H., Dong, J. H. & Haydon, P. G. Synaptic islands defined by the territory of a single astrocyte. J. Neurosci. 27, 6473–6477 (2007).
Ogata, K. & Kosaka, T. Structural and quantitative analysis of astrocytes in the mouse hippocampus. Neuroscience 113, 221–233 (2002).
Zhang, Y. et al. Purification and characterization of progenitor and mature human astrocytes reveals transcriptional and functional differences with mouse. Neuron 89, 37–53 (2016).
Krawczyk, M. C. et al. Human astrocytes exhibit tumor microenvironment-, age-, and sex-related transcriptomic signatures. J. Neurosci. 42, 1587–1603 (2022).
Le Roux, P. D. & Reh, T. A. Independent regulation of primary dendritic and axonal growth by maturing astrocytes in vitro. Neurosci Lett. 198, 5–8 (1995).
Le, R. & Esquenazi, S. Astrocytes mediate cerebral cortical neuronal axon and dendrite growth, in part, by release of fibroblast growth factor. Neurol Res. 24, 81–92 (2002).
Allen, N. J. et al. Astrocyte glypicans 4 and 6 promote formation of excitatory synapses via GluA1 AMPA receptors. Nature 486, 410–414 (2012).
Christopherson, K. S. et al. Thrombospondins are astrocyte-secreted proteins that promote CNS synaptogenesis. Cell 120, 421–433 (2005).
Eroglu, C. et al. Gabapentin receptor alpha2delta-1 is a neuronal thrombospondin receptor responsible for excitatory CNS synaptogenesis. Cell 139, 380–392 (2009).
Chung, W. S. et al. Astrocytes mediate synapse elimination through MEGF10 and MERTK pathways. Nature 504, 394–400 (2013).
Stevens, B. et al. The classical complement cascade mediates CNS synapse elimination. Cell 131, 1164–1178 (2007).
Hosli, L. et al. Decoupling astrocytes in adult mice impairs synaptic plasticity and spatial learning. Cell Rep. 38, 110484 (2022).
Szu, J. I. & Binder, D. K. The role of astrocytic Aquaporin-4 in synaptic plasticity and learning and memory. Front Integr. Neurosci. 10, 8 (2016).
Cooper, M. L. et al. Redistribution of metabolic resources through astrocyte networks mitigates neurodegenerative stress. Proc. Natl Acad. Sci. USA 117, 18810–18821 (2020).
Chittajallu, R., Aguirre, A. & Gallo, V. NG2-positive cells in the mouse white and grey matter display distinct physiological properties. J. Physiol. 561, 109–122 (2004).
Michalski, J. P. & Kothary, R. Oligodendrocytes in a Nutshell. Front Cell Neurosci. 9, 340 (2015).
Emery, B. & Lu, Q. R. Transcriptional and epigenetic regulation of oligodendrocyte development and myelination in the central nervous system. Cold Spring Harb. Perspect. Biol. 7, a020461 (2015).
Gokhan, S. et al. Combinatorial profiles of oligodendrocyte-selective classes of transcriptional regulators differentially modulate myelin basic protein gene expression. J. Neurosci. 25, 8311–8321 (2005).
Hornig, J. et al. The transcription factors Sox10 and Myrf define an essential regulatory network module in differentiating oligodendrocytes. PLoS Genet 9, e1003907 (2013).
Yu, Y. et al. Olig2 targets chromatin remodelers to enhancers to initiate oligodendrocyte differentiation. Cell 152, 248–261 (2013).
Bujalka, H. et al. MYRF is a membrane-associated transcription factor that autoproteolytically cleaves to directly activate myelin genes. PLoS Biol. 11, e1001625 (2013).
Thurnherr, T. et al. Cdc42 and Rac1 signaling are both required for and act synergistically in the correct formation of myelin sheaths in the CNS. J. Neurosci. 26, 10110–10119 (2006).
Zhang, S. C. Defining glial cells during CNS development. Nat. Rev. Neurosci. 2, 840–843 (2001).
Scolding, N. J. et al. Myelin-oligodendrocyte glycoprotein (MOG) is a surface marker of oligodendrocyte maturation. J. Neuroimmunol. 22, 169–176 (1989).
Bedner, P., Jabs, R. & Steinhauser, C. Properties of human astrocytes and NG2 glia. Glia 68, 756–767 (2020).
Hansen, D. V., Lui, J. H., Parker, P. R. & Kriegstein, A. R. Neurogenic radial glia in the outer subventricular zone of human neocortex. Nature 464, 554–561 (2010).
Eze, U. C., Bhaduri, A., Haeussler, M., Nowakowski, T. J. & Kriegstein, A. R. Single-cell atlas of early human brain development highlights heterogeneity of human neuroepithelial cells and early radial glia. Nat. Neurosci. 24, 584–594 (2021).
Ramos, S. I. et al. An atlas of late prenatal human neurodevelopment resolved by single-nucleus transcriptomics. Nat. Commun. 13, 7671 (2022).
Yang, L., Li, Z., Liu, G., Li, X. & Yang, Z. Developmental origins of human cortical oligodendrocytes and astrocytes. Neurosci. Bull. 38, 47–68 (2022).
Andersen, J. et al. Single-cell transcriptomic landscape of the developing human spinal cord. Nat. Neurosci. 26, 902–914 (2023).
Pozniak, C. D. et al. Sox10 directs neural stem cells toward the oligodendrocyte lineage by decreasing Suppressor of Fused expression. Proc. Natl Acad. Sci. USA 107, 21795–21800 (2010).
Klum, S. et al. Sequentially acting SOX proteins orchestrate astrocyte- and oligodendrocyte-specific gene expression. EMBO Rep 19, https://doi.org/10.15252/embr.201846635 (2018).
Hassel, L. A. et al. Differential activity of transcription factor Sox9 in early and adult oligodendroglial progenitor cells. Glia 71, 1890–1905 (2023).
Glasgow, S. M. et al. Mutual antagonism between Sox10 and NFIA regulates diversification of glial lineages and glioma subtypes. Nat. Neurosci. 17, 1322–1329 (2014).
Rheinbay, E. et al. An aberrant transcription factor network essential for Wnt signaling and stem cell maintenance in glioblastoma. Cell Rep. 3, 1567–1579 (2013).
Vue, T. Y. et al. ASCL1 regulates neurodevelopmental transcription factors and cell cycle genes in brain tumors of glioma mouse models. Glia 68, 2613–2630 (2020).
Suva, M. L. et al. Reconstructing and reprogramming the tumor-propagating potential of glioblastoma stem-like cells. Cell 157, 580–594 (2014).
Ignatova, T. N. et al. Gliomagenesis is orchestrated by the Oct3/4 regulatory network. J. Neurosurg. Sci. https://doi.org/10.23736/S0390-5616.21.05437-0 (2021).
Kobayashi, K. et al. Oct-3/4 promotes migration and invasion of glioblastoma cells. J. Cell Biochem 113, 508–517 (2012).
Song, W. S. et al. Sox2, a stemness gene, regulates tumor-initiating and drug-resistant properties in CD133-positive glioblastoma stem cells. J. Chin. Med. Assoc. 79, 538–545 (2016).
Li, S. et al. Signaling pathways in brain tumors and therapeutic interventions. Signal Transduct. Target Ther. 8, 8 (2023).
Reya, T. & Clevers, H. Wnt signalling in stem cells and cancer. Nature 434, 843–850 (2005).
Teodorczyk, M. & Schmidt, M. H. H. Notching on cancer’s door: notch signaling in brain tumors. Front Oncol. 4, 341 (2014).
Yu, Z., Pestell, T. G., Lisanti, M. P. & Pestell, R. G. Cancer stem cells. Int J. Biochem Cell Biol. 44, 2144–2151 (2012).
Curry, R. N. & Glasgow, S. M. The role of neurodevelopmental pathways in brain tumors. Front Cell Dev. Biol. 9, 659055 (2021).
Lathia, J. D., Mack, S. C., Mulkearns-Hubert, E. E., Valentim, C. L. & Rich, J. N. Cancer stem cells in glioblastoma. Genes Dev. 29, 1203–1217 (2015).
Gimple, R. C., Bhargava, S., Dixit, D. & Rich, J. N. Glioblastoma stem cells: lessons from the tumor hierarchy in a lethal cancer. Genes Dev. 33, 591–609 (2019).
Kreso, A. & Dick, J. E. Evolution of the cancer stem cell model. Cell Stem Cell 14, 275–291 (2014).
Chen, J. et al. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature 488, 522–526 (2012).
Bao, S. et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 444, 756–760 (2006).
Bao, B., Ahmad, A., Azmi, A. S., Ali, S. & Sarkar, F. H. Overview of cancer stem cells (CSCs) and mechanisms of their regulation: implications for cancer therapy. Curr Protoc Pharmacol Chapter 14, Unit 14 25, https://doi.org/10.1002/0471141755.ph1425s61 (2013).
Ostrom, Q. T. et al. CBTRUS statistical report: Primary brain and central nervous system tumors diagnosed in the United States in 2006-2010. Neuro Oncol. 15, ii1–56, (2013).
Thakkar, J. P. et al. Epidemiologic and molecular prognostic review of glioblastoma. Cancer Epidemiol. Biomark. Prev. 23, 1985–1996 (2014).
Mansouri, A., Karamchandani, J. & Das, S. in Glioblastoma (ed S. De Vleeschouwer) (2017).
Stupp, R. et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 352, 987–996 (2005).
Tran, B. & Rosenthal, M. A. Survival comparison between glioblastoma multiforme and other incurable cancers. J. Clin. Neurosci. 17, 417–421 (2010).
Koshy, M. et al. Improved survival time trends for glioblastoma using the SEER 17 population-based registries. J. Neuro Oncol. 107, 207–212 (2012).
Cancer Genome Atlas Research, N. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 455, 1061–1068 (2008).
Verhaak, R. G. et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 17, 98–110 (2010).
Sottoriva, A. et al. Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proc. Natl Acad. Sci. USA 110, 4009–4014 (2013).
Wang, Q. et al. Tumor evolution of glioma-intrinsic gene expression subtypes associates with immunological changes in the microenvironment. Cancer Cell 32, 42–56.e46 (2017).
Neftel, C. et al. An integrative model of cellular states, plasticity, and genetics for glioblastoma. Cell 178, 835–849.e821 (2019).
Bhaduri, A. et al. Outer radial glia-like cancer stem cells contribute to heterogeneity of glioblastoma. Cell Stem Cell 26, 48–63.e46 (2020).
Couturier, C. P. et al. Single-cell RNA-seq reveals that glioblastoma recapitulates a normal neurodevelopmental hierarchy. Nat. Commun. 11, 3406 (2020).
Dirkse, A. et al. Stem cell-associated heterogeneity in Glioblastoma results from intrinsic tumor plasticity shaped by the microenvironment. Nat. Commun. 10, 1787 (2019).
Pine, A. R. et al. Tumor microenvironment is critical for the maintenance of cellular states found in primary glioblastomas. Cancer Discov. 10, 964–979 (2020).
Cheng, L. et al. Glioblastoma stem cells generate vascular pericytes to support vessel function and tumor growth. Cell 153, 139–152 (2013).
Charles, N. et al. Perivascular nitric oxide activates notch signaling and promotes stem-like character in PDGF-induced glioma cells. Cell Stem Cell 6, 141–152 (2010).
Brooks, M. D., Sengupta, R., Snyder, S. C. & Rubin, J. B. Hitting them where they live: targeting the glioblastoma perivascular stem cell niche. Curr. Pathobiol. Rep. 1, 101–110 (2013).
Sharma, A. & Shiras, A. Cancer stem cell-vascular endothelial cell interactions in glioblastoma. Biochem Biophys. Res Commun. 473, 688–692 (2016).
Covello, K. L. et al. HIF-2alpha regulates Oct-4: effects of hypoxia on stem cell function, embryonic development, and tumor growth. Genes Dev. 20, 557–570 (2006).
Heddleston, J. M., Li, Z., McLendon, R. E., Hjelmeland, A. B. & Rich, J. N. The hypoxic microenvironment maintains glioblastoma stem cells and promotes reprogramming towards a cancer stem cell phenotype. Cell Cycle 8, 3274–3284 (2009).
Li, Z. et al. Hypoxia-inducible factors regulate tumorigenic capacity of glioma stem cells. Cancer Cell 15, 501–513 (2009).
Seidel, S. et al. A hypoxic niche regulates glioblastoma stem cells through hypoxia inducible factor 2 alpha. Brain 133, 983–995 (2010).
Ortensi, B., Setti, M., Osti, D. & Pelicci, G. Cancer stem cell contribution to glioblastoma invasiveness. Stem Cell Res Ther. 4, 18 (2013).
Wang, Z., Zhang, H., Xu, S., Liu, Z. & Cheng, Q. The adaptive transition of glioblastoma stem cells and its implications on treatments. Signal Transduct. Target Ther. 6, 124 (2021).
Liau, B. B. et al. Adaptive Chromatin remodeling drives glioblastoma stem cell plasticity and drug tolerance. Cell Stem Cell 20, 233–246.e237 (2017).
Friedmann-Morvinski, D. et al. Dedifferentiation of neurons and astrocytes by oncogenes can induce gliomas in mice. Science 338, 1080–1084 (2012).
Simpson Ragdale, H. et al. Injury primes mutation-bearing astrocytes for dedifferentiation in later life. Curr. Biol. 33, 1082–1098.e1088 (2023).
Bachoo, R. M. et al. Epidermal growth factor receptor and Ink4a/Arf: convergent mechanisms governing terminal differentiation and transformation along the neural stem cell to astrocyte axis. Cancer Cell 1, 269–277 (2002).
Friedmann-Morvinski, D. & Verma, I. M. Dedifferentiation and reprogramming: origins of cancer stem cells. EMBO Rep. 15, 244–253 (2014).
Liu, B. & Neufeld, A. H. Activation of epidermal growth factor receptors in astrocytes: from development to neural injury. J. Neurosci. Res. 85, 3523–3529 (2007).
Zhu, Q. et al. Genetic evidence that Nkx2.2 and Pdgfra are major determinants of the timing of oligodendrocyte differentiation in the developing CNS. Development 141, 548–555 (2014).
Calver, A. R. et al. Oligodendrocyte population dynamics and the role of PDGF in vivo. Neuron 20, 869–882 (1998).
Liu, X. et al. EGF signaling promotes the lineage conversion of astrocytes into oligodendrocytes. Mol. Med. 28, 50 (2022).
Suva, M. L., Riggi, N. & Bernstein, B. E. Epigenetic reprogramming in cancer. Science 339, 1567–1570 (2013).
Ligon, K. L. et al. The oligodendroglial lineage marker OLIG2 is universally expressed in diffuse gliomas. J. Neuropathol. Exp. Neurol. 63, 499–509 (2004).
Kosty, J., Lu, F., Kupp, R., Mehta, S. & Lu, Q. R. Harnessing OLIG2 function in tumorigenicity and plasticity to target malignant gliomas. Cell Cycle 16, 1654–1660 (2017).
Lu, F. et al. Olig2-dependent reciprocal shift in PDGF and EGF receptor signaling regulates tumor phenotype and mitotic growth in malignant glioma. Cancer Cell 29, 669–683 (2016).
Myers, B. L. et al. Glioblastoma initiation, migration, and cell types are regulated by core bHLH transcription factors ASCL1 and OLIG2. bioRxiv, 2023.2009.2030.560206, https://doi.org/10.1101/2023.09.30.560206 (2023).
Galvez-Contreras, A. Y., Quinones-Hinojosa, A. & Gonzalez-Perez, O. The role of EGFR and ErbB family related proteins in the oligodendrocyte specification in germinal niches of the adult mammalian brain. Front Cell Neurosci. 7, 258 (2013).
Fu, Y. et al. Heterogeneity of glial progenitor cells during the neurogenesis-to-gliogenesis switch in the developing human cerebral cortex. Cell Rep. 34, 108788 (2021).
Zhang, X. et al. Bulk and mosaic deletions of Egfr reveal regionally defined gliogenesis in the developing mouse forebrain. iScience 26, 106242 (2023).
Furnari, F. B., Cloughesy, T. F., Cavenee, W. K. & Mischel, P. S. Heterogeneity of epidermal growth factor receptor signalling networks in glioblastoma. Nat. Rev. Cancer 15, 302–310 (2015).
Lassman, A. B. et al. Epidermal growth factor receptor (EGFR) amplification rates observed in screening patients for randomized trials in glioblastoma. J. Neuro Oncol 144, 205–210 (2019).
Liu, F. et al. EGFR mutation promotes glioblastoma through epigenome and transcription factor network remodeling. Mol. Cell 60, 307–318 (2015).
Sardar, D. et al. Sox9 directs divergent epigenomic states in brain tumor subtypes. Proc. Natl Acad. Sci. USA 119, e2202015119 (2022).
Louis, D. N. et al. The 2021 WHO Classification of Tumors of the Central Nervous System: a summary. Neuro Oncol. 23, 1231–1251 (2021).
Whitfield, B. T. & Huse, J. T. Classification of adult-type diffuse gliomas: Impact of the World Health Organization 2021 update. Brain Pathol. 32, e13062 (2022).
Lindberg, N., Kastemar, M., Olofsson, T., Smits, A. & Uhrbom, L. Oligodendrocyte progenitor cells can act as cell of origin for experimental glioma. Oncogene 28, 2266–2275 (2009).
Dai, C. et al. PDGF autocrine stimulation dedifferentiates cultured astrocytes and induces oligodendrogliomas and oligoastrocytomas from neural progenitors and astrocytes in vivo. Genes Dev. 15, 1913–1925 (2001).
Venteicher, A. S. et al. Decoupling genetics, lineages, and microenvironment in IDH-mutant gliomas by single-cell RNA-seq. Science 355, https://doi.org/10.1126/science.aai8478 (2017).
Tirosh, I. et al. Single-cell RNA-seq supports a developmental hierarchy in human oligodendroglioma. Nature 539, 309–313 (2016).
Pachocki, C. J. & Hol, E. M. Current perspectives on diffuse midline glioma and a different role for the immune microenvironment compared to glioblastoma. J. Neuroinflammation 19, 276 (2022).
Di Ruscio, V. et al. Pediatric Diffuse Midline Gliomas: An Unfinished Puzzle. Diagnostics 12, https://doi.org/10.3390/diagnostics12092064 (2022).
Duan, R., Du, W. & Guo, W. EZH2: a novel target for cancer treatment. J. Hematol. Oncol. 13, 104 (2020).
Lewis, P. W. et al. Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science 340, 857–861 (2013).
Funato, K., Major, T., Lewis, P. W., Allis, C. D. & Tabar, V. Use of human embryonic stem cells to model pediatric gliomas with H3.3K27M histone mutation. Science 346, 1529–1533 (2014).
Filbin, M. G. et al. Developmental and oncogenic programs in H3K27M gliomas dissected by single-cell RNA-seq. Science 360, 331–335 (2018).
Jessa, S. et al. K27M in canonical and noncanonical H3 variants occurs in distinct oligodendroglial cell lineages in brain midline gliomas. Nat. Genet 54, 1865–1880 (2022).
Nagaraja, S. et al. Histone variant and cell context determine H3K27M reprogramming of the enhancer landscape and oncogenic state. Mol. Cell 76, 965–980 e912 (2019).
Liu, I. et al. The landscape of tumor cell states and spatial organization in H3-K27M mutant diffuse midline glioma across age and location. Nat. Genet. 54, 1881–1894 (2022).
Venkatesh, H. S. et al. Electrical and synaptic integration of glioma into neural circuits. Nature, https://doi.org/10.1038/s41586-019-1563-y (2019).
Venkatesh, H. S. et al. Neuronal activity promotes glioma growth through Neuroligin-3 secretion. Cell 161, 803–816 (2015).
Taylor, K. R. et al. Glioma synapses recruit mechanisms of adaptive plasticity. Nature 623, 366–374 (2023).
Chen, Y. H. & Gutmann, D. H. The molecular and cell biology of pediatric low-grade gliomas. Oncogene 33, 2019–2026 (2014).
Lee, D. Y., Yeh, T. H., Emnett, R. J., White, C. R. & Gutmann, D. H. Neurofibromatosis-1 regulates neuroglial progenitor proliferation and glial differentiation in a brain region-specific manner. Genes Dev. 24, 2317–2329 (2010).
Dasgupta, B., Yi, Y., Chen, D. Y., Weber, J. D. & Gutmann, D. H. Proteomic analysis reveals hyperactivation of the mammalian target of rapamycin pathway in neurofibromatosis 1-associated human and mouse brain tumors. Cancer Res. 65, 2755–2760 (2005).
Kaul, A., Chen, Y. H., Emnett, R. J., Dahiya, S. & Gutmann, D. H. Pediatric glioma-associated KIAA1549:BRAF expression regulates neuroglial cell growth in a cell type-specific and mTOR-dependent manner. Genes Dev. 26, 2561–2566 (2012).
Kaul, A., Chen, Y. H., Emnett, R. J., Gianino, S. M. & Gutmann, D. H. Conditional KIAA1549:BRAF mice reveal brain region- and cell type-specific effects. Genesis 51, 708–716 (2013).
Lammers, J. A. S. et al. Lgg-15. Single-nucleus and bulk RNA sequencing of pediatric pilocytic astrocytomas reveals cns location-associated tumor cell and microenvironmental heterogeneity. Neuro-Oncol. 25, i58–i59 (2023).
Reitman, Z. J. et al. Mitogenic and progenitor gene programmes in single pilocytic astrocytoma cells. Nat. Commun. 10, 3731 (2019).
Neradil, J. & Veselska, R. Nestin as a marker of cancer stem cells. Cancer Sci. 106, 803–811 (2015).
Hassn Mesrati, M., Behrooz, A. B., A., Y. A. & Syahir, A. Understanding Glioblastoma biomarkers: knocking a mountain with a hammer. Cells 9, https://doi.org/10.3390/cells9051236 (2020).
Ignatova, T. N. et al. Human cortical glial tumors contain neural stem-like cells expressing astroglial and neuronal markers in vitro. Glia 39, 193–206 (2002).
Vik-Mo, E. O. et al. A comparative study of the structural organization of spheres derived from the adult human subventricular zone and glioblastoma biopsies. Exp. Cell Res. 317, 1049–1059 (2011).
Liu, C. et al. Mosaic analysis with double markers reveals tumor cell of origin in glioma. Cell 146, 209–221 (2011).
Bouchart, C., Trepant, A. L., Hein, M., Van Gestel, D. & Demetter, P. Prognostic impact of glioblastoma stem cell markers OLIG2 and CCND2. Cancer Med 9, 1069–1078 (2020).
Jeong, H. Y. et al. High expression of RFX4 is associated with tumor progression and poor prognosis in patients with glioblastoma. Int J. Neurosci. 131, 7–14 (2021).
Uceda-Castro, R. et al. GFAP splice variants fine-tune glioma cell invasion and tumour dynamics by modulating migration persistence. Sci. Rep. 12, 424 (2022).
Ghazi, S. O. et al. Cell of origin determines tumor phenotype in an oncogenic Ras/p53 knockout transgenic model of high-grade glioma. J. Neuropathol. Exp. Neurol. 71, 729–740 (2012).
Radke, J., Bortolussi, G. & Pagenstecher, A. Akt and c-Myc induce stem-cell markers in mature primary p53(-)/(-) astrocytes and render these cells gliomagenic in the brain of immunocompetent mice. PLoS One 8, e56691 (2013).
Kondo, T. & Raff, M. Oligodendrocyte precursor cells reprogrammed to become multipotential CNS stem cells. Science 289, 1754–1757 (2000).
Dawson, M. R., Polito, A., Levine, J. M. & Reynolds, R. NG2-expressing glial progenitor cells: an abundant and widespread population of cycling cells in the adult rat CNS. Mol. Cell Neurosci. 24, 476–488 (2003).
Bardehle, S. et al. Live imaging of astrocyte responses to acute injury reveals selective juxtavascular proliferation. Nat. Neurosci. 16, 580–586 (2013).
Alcantara Llaguno, S. R. et al. Adult lineage-restricted CNS progenitors specify distinct glioblastoma subtypes. Cancer Cell 28, 429–440 (2015).
Beauchamp, A. et al. Whole-brain comparison of rodent and human brains using spatial transcriptomics. Elife 11, https://doi.org/10.7554/eLife.79418 (2022).
Zhao, X. & Bhattacharyya, A. Human Models Are Needed for Studying Human Neurodevelopmental Disorders. Am. J. Hum. Genet 103, 829–857 (2018).
Semple, B. D., Blomgren, K., Gimlin, K., Ferriero, D. M. & Noble-Haeusslein, L. J. Brain development in rodents and humans: Identifying benchmarks of maturation and vulnerability to injury across species. Prog. Neurobiol. 106-107, 1–16 (2013).
Zeiss, C. J. Comparative Milestones in Rodent and Human Postnatal Central Nervous System Development. Toxicol. Pathol. 49, 1368–1373 (2021).
Sloan, S. A., Andersen, J., Pasca, A. M., Birey, F. & Pasca, S. P. Generation and assembly of human brain region-specific three-dimensional cultures. Nat. Protoc. 13, 2062–2085 (2018).
Birey, F. et al. Assembly of functionally integrated human forebrain spheroids. Nature 545, 54–59 (2017).
Jo, J. et al. Midbrain-like organoids from human pluripotent stem cells contain functional dopaminergic and neuromelanin-producing neurons. Cell Stem Cell 19, 248–257 (2016).
Zagare, A., Gobin, M., Monzel, A. S. & Schwamborn, J. C. A robust protocol for the generation of human midbrain organoids. STAR Protoc. 2, 100524 (2021).
Muguruma, K., Nishiyama, A., Kawakami, H., Hashimoto, K. & Sasai, Y. Self-organization of polarized cerebellar tissue in 3D culture of human pluripotent stem cells. Cell Rep. 10, 537–550 (2015).
Valiulahi, P. et al. Generation of caudal-type serotonin neurons and hindbrain-fate organoids from hPSCs. Stem Cell Rep. 16, 1938–1952 (2021).
Ogura, T., Sakaguchi, H., Miyamoto, S. & Takahashi, J. Three-dimensional induction of dorsal, intermediate and ventral spinal cord tissues from human pluripotent stem cells. Development 145, https://doi.org/10.1242/dev.162214 (2018).
Duval, N. et al. BMP4 patterns Smad activity and generates stereotyped cell fate organization in spinal organoids. Development 146, https://doi.org/10.1242/dev.175430 (2019).
Pasca, S. P. Assembling human brain organoids. Science 363, 126–127 (2019).
Chiaradia, I. & Lancaster, M. A. Brain organoids for the study of human neurobiology at the interface of in vitro and in vivo. Nat. Neurosci. 23, 1496–1508 (2020).
Pasca, S. P. The rise of three-dimensional human brain cultures. Nature 553, 437–445 (2018).
Fiorenzano, A. et al. Single-cell transcriptomics captures features of human midbrain development and dopamine neuron diversity in brain organoids. Nat. Commun. 12, 7302 (2021).
He, Z. et al. An integrated transcriptomic cell atlas of human neural organoids. bioRxiv, 2023.2010.2005.561097, https://doi.org/10.1101/2023.10.05.561097 (2023).
Sloan, S. A. et al. Human Astrocyte Maturation Captured in 3D Cerebral Cortical Spheroids Derived from Pluripotent Stem Cells. Neuron 95, 779–790 e776 (2017).
Trevino, A. E. et al. Chromatin accessibility dynamics in a model of human forebrain development. Science 367, https://doi.org/10.1126/science.aay1645 (2020).
Marton, R. M. et al. Differentiation and maturation of oligodendrocytes in human three-dimensional neural cultures. Nat. Neurosci. 22, 484–491 (2019).
Madhavan, M. et al. Induction of myelinating oligodendrocytes in human cortical spheroids. Nat. Methods 15, 700–706 (2018).
Garcia-Leon, J. A. et al. Generation of oligodendrocytes and establishment of an all-human myelinating platform from human pluripotent stem cells. Nat. Protoc. 15, 3716–3744 (2020).
Gordon, A. et al. Long-term maturation of human cortical organoids matches key early postnatal transitions. Nat. Neurosci. 24, 331–342 (2021).
Bian, S. et al. Genetically engineered cerebral organoids model brain tumor formation. Nat. Methods 15, 631–639 (2018).
Ogawa, J., Pao, G. M., Shokhirev, M. N. & Verma, I. M. Glioblastoma Model using human cerebral organoids. Cell Rep. 23, 1220–1229 (2018).
Linkous, A. et al. Modeling patient-derived glioblastoma with cerebral organoids. Cell Rep. 26, 3203–3211.e3205 (2019).
da Silva, B., Mathew, R. K., Polson, E. S., Williams, J. & Wurdak, H. Spontaneous Glioblastoma spheroid infiltration of early-stage cerebral organoids models brain tumor invasion. SLAS Discov. 23, 862–868 (2018).
Goranci-Buzhala, G. et al. Rapid and efficient invasion assay of glioblastoma in human brain organoids. Cell Rep. 31, 107738 (2020).
Pine, A. R. et al. Microenvironment-driven dynamic chromatin changes in glioblastoma recapitulate early neural development at single-cell resolution. Cancer Res 83, 1581–1595 (2023).
Jin, J., Grigore, F., Chen, C. C. & Li, M. Self‑renewal signaling pathways and differentiation therapies of glioblastoma stem cells (Review). Int. J. Oncol. 59, https://doi.org/10.3892/ijo.2021.5225 (2021).
Wang, R. & Liu, C. All-trans retinoic acid therapy induces asymmetric division of glioma stem cells from the U87MG cell line. Oncol. Lett. 18, 3646–3654 (2019).
Ying, M. et al. Regulation of glioblastoma stem cells by retinoic acid: role for Notch pathway inhibition. Oncogene 30, 3454–3467 (2011).
Koguchi, M. et al. BMP4 induces asymmetric cell division in human glioma stem-like cells. Oncol. Lett. 19, 1247–1254 (2020).
Nayak, S. et al. Bone Morphogenetic Protein 4 targeting glioma stem-like cells for malignant glioma treatment: latest advances and implications for clinical application. Cancers 12, https://doi.org/10.3390/cancers12020516 (2020).
Swartling, F. J., Cancer, M., Frantz, A., Weishaupt, H. & Persson, A. I. Deregulated proliferation and differentiation in brain tumors. Cell Tissue Res. 359, 225–254 (2015).
Giannopoulou, A. I., Kanakoglou, D. S. & Piperi, C. Transcription Factors with targeting potential in Gliomas. Int. J. Mol. Sci. 23, https://doi.org/10.3390/ijms23073720 (2022).
Giannopoulou, A. I., Kanakoglou, D. S., Papavassiliou, A. G. & Piperi, C. Insights into the multi-faceted role of Pioneer transcription factors in glioma formation and progression with targeting options. Biochim Biophys. Acta Rev. Cancer 1877, 188801 (2022).
Lo, H. W., Cao, X., Zhu, H. & Ali-Osman, F. Constitutively activated STAT3 frequently coexpresses with epidermal growth factor receptor in high-grade gliomas and targeting STAT3 sensitizes them to Iressa and alkylators. Clin. Cancer Res. 14, 6042–6054 (2008).
Shi, Y. et al. Ibrutinib inactivates BMX-STAT3 in glioma stem cells to impair malignant growth and radioresistance. Sci. Transl. Med. 10, https://doi.org/10.1126/scitranslmed.aah6816 (2018).
Fuh, B. et al. LLL-3 inhibits STAT3 activity, suppresses glioblastoma cell growth and prolongs survival in a mouse glioblastoma model. Br. J. Cancer 100, 106–112 (2009).
Videla Richardson, G. A. et al. Specific preferences in lineage choice and phenotypic plasticity of glioma stem cells under BMP4 and Noggin influence. Brain Pathol. 26, 43–61 (2016).
Caren, H., Beck, S. & Pollard, S. M. Differentiation therapy for glioblastoma - too many obstacles? Mol. Cell Oncol. 3, e1124174 (2016).
Pistollato, F. et al. Hypoxia and HIF1alpha repress the differentiative effects of BMPs in high-grade glioma. Stem Cells 27, 7–17 (2009).
Caren, H. et al. Glioblastoma stem cells respond to differentiation cues but fail to undergo commitment and terminal cell-cycle arrest. Stem Cell Rep. 5, 829–842 (2015).
Acknowledgements
We would like to thank Jimena Andersen for helpful discussions. The authors apologize to colleagues whose relevant studies were not cited due to limited space. The research in the author’s laboratory was supported by grants from the National Institutes of Health (NIMH R01 MH125956 and NINDS R01 NS123562).
Author information
Authors and Affiliations
Contributions
C.S. composed the manuscript and C.S. and S.A.S. edited and finalized the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks the anonymous reviewers for their contribution to the peer review of this work. Primary Handling Editor: Manuel Breuer. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sojka, C., Sloan, S.A. Gliomas: a reflection of temporal gliogenic principles. Commun Biol 7, 156 (2024). https://doi.org/10.1038/s42003-024-05833-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-024-05833-2
- Springer Nature Limited