
Abstract
Despite regulatory elements such as long non - coding RNAs representing most of the transcriptome, the functional understanding of long non - coding RNAs in relation to major health conditions including bovine mastitis is limited. This study examined the milk somatic cell transcriptome from udder quarters of 6 Holstein dairy cows to identify differentially expressed long non - coding RNAs using RNA - Sequencing. Ninety - four differentially expressed long non - coding RNAs are identified, 5 of which are previously annotated for gene name and length, 11 are annotated for gene name and 78 are novel, having no gene name or length previously annotated. Significant inflammatory response and regulation of immune response pathways (false discovery rate < 0.05) are associated with the differentially expressed long non - coding RNAs. QTL annotation analysis revealed 31 QTL previously annotated in the genomic regions of the 94 differentially expressed long non - coding RNAs, and the majority are associated with milk traits. This research provides a better understanding of long non - coding RNAs regulatory elements in milk somatic cells, which may enhance current breeding strategies for more adaptable or high mastitis resistant cattle.
Similar content being viewed by others

Introduction
Bovine mastitis is a highly prevalent disease of lactating dairy cows that results in milk yield reduction, discarded milk, and early culling. Mastitis can be classified as clinical or subclinical, with clinical status defined as changes in the milk, such as flakes and clotting or inflammation in the udder1; and subclinical, status defined as an elevated level of milk somatic cells (SC) which consist of macrophages, polymorphonuclear leukocytes, lymphocytes, and epithelial cells2,3. Although abundant research exists on cattle mastitis, it remains a major economic challenge for dairy producers due to the complexity of the trait.
With the advent of RNA-sequencing (RNA-Seq), the highly dynamic host transcriptome can be studied at a high-throughput level to identify key molecular differences between healthy and mastitic animals, such as genes, transcripts, and regulatory elements. The host transcriptome consists of both protein-coding and non-coding genes. Messenger RNAs are protein-coding RNAs and well-studied; however, they account for <2% of the total genomic sequence, illustrating that most transcripts are non-coding4. More specifically, non-coding transcripts such as long non-coding RNAs (lncRNAs), defined as transcripts longer than 200 nucleotides in length with limited to no coding capacity5, implement diverse cellular and biological functions through a multiplicity of biochemical activities, including transcriptional and post-transcriptional processing6. The lncRNA is classified as cis-acting, where they recognize complementary sequences of genes in close proximity (i.e., neighboring genes) and carry out their functions there, or trans-acting, where the lncRNA is transcribed, processed, and then vacate their sites of transcription to exert their functions elsewhere7. Regardless of functional region, it is crucial that lncRNA recognize complementary sequences of their targets to allow specific interactions to occur, such as editing, translation, degradation, splicing, and transport8.
Previous research using RNA-Seq aimed to identify and annotate novel lncRNAs in the transcriptome of 18 bovine tissues from a single animal, including lung, liver, kidney, white blood cells, and mammary gland9. The latter study revealed that kidney, liver, and lung tissue have large proportion of unknown transcripts (53%, 52%, and 48%, respectively), whereas the mammary gland has only 13.13% unknown transcripts. However, lncRNA expression is highly tissue and time-specific, and many are only detected under stress conditions10. Studying the lncRNA in the bovine mammary gland under stress conditions has revealed their involvement in many biological functions including susceptibility to clinical mastitis5. Wang et al.11 studied novel lncRNA expressed in bovine tissues and found that the novel lncRNA-TUB was expressed at higher concentrations in mammary epithelial cells that received a pro-inflammatory stimulus in comparison to normal cells. Therefore, if the lncRNAs have a fundamental role in immune regulation due to their interactions with transcripts in close proximity, this may provide key insights regarding mastitis resistance, which will aid in the development of breeding programs for mastitis.
The objectives of this study were to: (1) detect lncRNAs present in the bovine milk SC transcriptome of 6 healthy and 6 mastitic samples collected from 6 Holstein dairy cows using RNA-Seq; (2) identify lncRNAs that are differentially expressed (DE) between healthy and mastitic samples; (3) classify previously annotated lncRNAs and identify novel lncRNAs having no previously annotated gene name or length; (4) perform functional analysis to determine if these lncRNAs acting in close proximity to mRNA, miRNA etc. are associated with the host immune response or mastitis resistance; (5) perform QTL annotation and QTL enrichment analysis using the genomic regions of the DE lncRNAs to find additional positional evidence of their involvement in immune response or mastitis resistance.
This study identified 94 DE lncRNAs between healthy and mastitic samples, and these DE lncRNAs were significantly involved in functional metabolic pathways (false discovery rate (FDR) < 0.05) such as inflammatory response and regulation of immune response. It was also found that 31 QTL were annotated in the genomic regions of the 94 DE lncRNAs and the majority of these were associated with milk traits. This research provides a better understanding of lncRNA regulatory elements in the transcriptome of bovine milk SC, which may help to improve the selection of cows better able to adapt or be more resistant to mastitis.
Results and discussion
Differential lncRNA expression analysis and classification
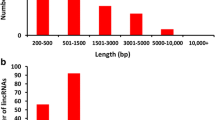
A total of 182 million single-end reads were generated from the 12 milk SC samples. RNA - Sequencing analysis revealed that 90% (87% healthy; 93% mastitic) of these reads were uniquely mapped to the bovine reference genome (ARS - UCD1.2.1) using a Large Gap Read Mapping (LGRM) approach (Supplementary Table 1). As shown in Supplementary Table 1, differences in the total mapped reads between the samples was observed. This may be due to the different cell types present in mastitic vs healthy udder quarters, as isolation of different cell types is a common limitation to transcriptomic studies12,13,14. Although the bovine reference genome currently has 27,607 annotated transcripts, our analysis revealed a total of 31,870 and 29,154 transcripts in healthy and mastitic samples, respectively (reads per kilobase per million mapped reads (RPKM) ≥ 0.2). This difference accounts for the identification of novel transcripts not yet annotated. In total, 659 transcripts were DE between healthy and mastitic samples (FDR < 0.05 and fold-change ( | FC | ) > 2). After filtering these transcripts for only DE lncRNA, 94 DE lncRNAs between healthy and mastitic samples were identified. These lncRNAs were then further categorized based on previous annotation in the bovine reference genome; 5 being previously annotated for gene name and length, 11 being previously annotated for gene name, whereas the majority (N = 78) were novel with no annotated gene name or length.
Annotation of long non-coding RNA
The lncRNA were categorized as previously annotated when they had both a specific Ensembl identifier (e.g., ENSBTAG00000013417) and the length corresponded to or was different from that reported in the ARS_UCD1.2.1 bovine reference genome. Of the 94 DE lncRNAs, only 5 of the identified DE lncRNAs were previously annotated for both gene name and length in the bovine reference genome (Table 1), whereas 11 DE lncRNAs had an associated gene name, but the length was different than that reported in the bovine reference genome (Table 2). Therefore, there were 16 DE lncRNAs classified as previously annotated; of these, 14 were over-expressed and 2 were under-expressed in the mastitic samples compared to the healthy samples. Although these 16 DE lncRNAs were previously annotated, they do not currently have a gene symbol assigned to them; therefore, all functional analysis was completed using their Ensembl identifier. The most over-expressed DE lncRNA in the mastitic samples compared to the healthy samples was ENSBTAG00000070418_2 (FC = 52.90, FDR = 1.01E-02). This lncRNA was annotated for gene name only as the length is different than what is reported in the bovine reference genome. This lncRNA predicted target is phosphoinositide 3-kinase regulatory subunit 4 (PIK3R4) based on its downstream proximity in the bovine reference genome (Table 2). This interaction was significantly negatively correlated (r = −0.82, P-value = 0.05; Supplementary Table 2) with the specific mRNA isoform of PIK3R4_3 in the healthy group. This gene family (PI3K) is involved in many aspects of immune cell development including regulatory T cells which are important in regulating and suppressing other cells in the immune system15. Therefore, if the lncRNA/gene interaction is altered, this could negatively impact the host’s immune response. Alternatively, the most under-expressed lncRNA (annotated for gene name only) in the mastitic samples was ENSBTAG00000082333 (FC = −39.49, FDR = 4.34E-02). This lncRNA predicted target interaction is with kappa casein (CSN3), which is located upstream of the lncRNA in the bovine reference genome. This specific casein constitutes about 25% of the casein fraction in bovine milk16. It also determines the size of casein micelles and initiates micelle aggregation for cheese production17. Thus, if the lncRNA / gene interaction is altered, this could in turn negatively affect the CSN3 protein that is formed. However, it was found that in our dataset, the correlation between the expression levels of these isoforms was not significant.
Identification of novel long non-coding RNA
Approximately 83% of the DE lncRNAs (78/94 lncRNAs) identified in this study were classified as novel as they were not previously annotated for gene name or length in the bovine reference genome (Table 3). We identified the potential transcript interaction of these novel lncRNAs relative to their position to other transcripts in the genome. For example, transcripts that were close to an annotated mRNA or overlapped with an annotated mRNA were inferred as the target of the lncRNA in relation to its functionality within the genome. As shown in Table 3, most of the novel DE lncRNAs were found to be intergenic (71.79%), whereas 28.21% were genic. Previous research has demonstrated that bovine lncRNA are mostly located in intergenic regions; however, this is commonly found in muscle tissues9,18,19.
Intergenic non-coding RNA
As mentioned, 71.79% of the novel DE lncRNAs identified in this study were long intergenic non-coding RNA; (lincRNA; N = 56), meaning they are located upstream or downstream between genes20. The most under-expressed novel DE lincRNA was lincRNA_64.1 (FC = − 41.23, FDR = 3.69E-03). Based on its downstream position in the bovine genome, it is predicted to interact with cordon - bleu WH2 repeat protein like 1 (COBLL1). The expression levels between linc_64.1 and COBLLI are significantly negatively correlated (r = −0.86, P-value = 0.03) for the specific mRNA isoform COBLLI_3 in the healthy group (Supplementary Table 2). The COBLL1 gene has been linked with metritis, which is an inflammatory condition of the uterus, generally caused by a bacterial infection after calving when the cows have a suppressed immune system and are more vulnerable to bacterial infection21,22. This is interesting to consider, as some post-calving diseases also indicate an animal’s susceptibility to mastitis. Research by Thompson-Crispi et al.23 demonstrated high immune responding cows had less mastitis, along with metritis, retained placenta, and displaced abomasum23. Additionally, a positive genetic correlation (0.19–0.49) between mastitis and other diseases such as milk fever, ketosis, and retained placenta exist24. Therefore, if the focus is breeding on immune responsiveness, this could decrease the overall disease occurrence25. As the lincRNA that potentially interacts with this COBLL1 gene was under-expressed, this could impact the functionality of the mRNA, making the animal more susceptible to metritis and in turn mastitis. Alternatively, the most over-expressed (FC = 683.48, FDR = 4.57E-02) novel lncRNA in the mastitic samples compared to the healthy samples was lincRNA_2159.2. This novel lncRNA in cattle was predicted to interact with C - X - C motif chemokine receptor 3 (CXCR3), which is located downstream from the lncRNA. The correlation between expression values was not significant in our dataset. Chemokines are a large family of cytokines and their receptors play an important role in the recruitment, activation, and differentiation of immune cells26. Previous research identified 3 polymorphisms associated with subclinical mastitis within the chemokine receptor C - X - C motif chemokine receptor 2 (CXCR2)27. Based on the current literature, no polymorphisms associated with mastitis have been found in CXCR3. Therefore, further research is needed to determine if polymorphisms in this gene might impact the lncRNA / gene relationship and how this could potentially impact mastitis resistance.
Genic long non-coding RNA
Alternatively, 28.21% of the novel DE lncRNAs were genic (N = 22), meaning they are found overlapping a gene. The most under-expressed genic lncRNA (FC = −170.31, FDR = 4.85E-02) in the mastitic samples compared to the healthy samples is the novel lncRNA_68.2. The lncRNA_68.2 is predicted to interact with ENSBTAG00000016785 as the lncRNA overlaps an exonic region. This gene does not currently have a gene symbol assigned to it for bovine; however, in mice it is the gene 5 - hydroxytryptamine receptor 5B (Htr5b) gene. This gene is conserved across chimpanzee, cow, and rat; however, through evolution, the conservation has been lost in humans28. The Htr5b gene product acts as a receptor for serotonin which is a neurotransmitter with vital roles in neural activities29. In bovine, serotonin is a potent regulator of calcium homeostasis, energy homeostasis and energy balance during lactation30,31. This energy balance is critical to ensure that the cow can meet the demand for milk production. Research has shown that the highest rates of mastitis occur during early lactation when most cows experience a negative energy balance32,33,34. As this lncRNA was found to overlap an exonic region of Htr5b, it is hypothesized that it could play a key role in regulation of this receptor. Since lncRNA_68.2 was under-expressed in the mastitic samples, this could be a key lncRNA to target for future analysis to minimize the impact of the negative energy balance. Alternatively, the most over-expressed genic lncRNA in the mastitic samples compared to the healthy samples, was lncRNA_1505.1 (FC = 496.05, FDR = 1.20E-02) and it was predicted to interact with U6 spliceosomal RNA (U6) due to it overlapping an intronic region of the gene. This transcript is the most highly conserved of the five spliceosomal RNAs and plays a catalytic role in the spliceosome and undergoes extensive structural rearrangements35,36. At the present time, the potential role of U6 on bovine mastitis is unknown. Therefore, further research is needed to better understand the functionality of lncRNA_1505.1 and its potential interaction with U6 in relation to mastitis or the host immune response.
Functional analysis of differentially expressed lncRNAs
Functional analysis was performed to deeply investigate the functional regulatory elements and their impact on the host’s response to mastitis causing agents. If the lncRNA is suppressing or enhancing the ability of its target (e.g., mRNA, miRNA), this could negatively impact the cow’s immune response, making her more susceptible to mastitis and prevent her from efficiently eliminating the threat. As the DE lncRNAs were split into 1) previously annotated; 2) novel intergenic and: 3) novel genic, the functional analysis follows this structure.
Functional analysis of previously annotated lncRNAs for both gene name and length
As mentioned previously, 5 of the annotated lncRNA were annotated previously for both gene name and length (Table 1). To determine their predicted target(s), the genomic coordinates of the lncRNA and Genome Data Viewer was used (https://www.ncbi.nlm.nih.gov/genome/gdv/). Based on the proximal location of other mRNA, miRNA etc. the predicted interaction of each of these lncRNA was identified (Table 1).
The first annotated lncRNA ENSBTAG00000050065_2 was 35 × under-expressed in the mastitic samples compared to the healthy samples (FC = −35.43, FDR = 4.80E-02; Table 1). The Iroquois homeobox 2 (IRX2) gene is located downstream from out DE lncRNA and therefore this is its predicted target. This family of genes has been reported to affect tumor growth, invasion and metastasis and have been closely linked to tumor progression and prognosis37. The potential effect of this lncRNA / gene interaction would have on mastitis, or the immune system is unknown, thus further research is needed. As shown in Supplementary Table 2, there were 2 potential mRNA isoforms of IRX2 that demonstrated significant, positive interactions in the mastitic (IRX2_1 (r = 0.93, P-value = 0.01); and IRX2_2 (r = 0.99, P-value = 0.00) and healthy groups (IRX2_2 (r = 0.98, P-value = 0.00).
The annotated lncRNA ENSBTAG00000054150_1 was over-expressed in the mastitic samples compared to the healthy samples (FC = 13.61, FDR = 4.94E-02; Table 1). It is predicted to interact with X – C motif chemokine receptor 1 (XCR1) as the lncRNA overlaps with this gene. This gene is expressed on a subset of dendritic cells which are involved in antigen cross-presentation38. Antigen presentation is critical to ensure the cow can fight off the mastitis causing pathogens, such as Staphylococcus aureus (S. aureus). As this mRNA plays a key role in activating this part of the immune system, this lncRNA could help in the regulation of this mRNA.
The next annotated lncRNA ENSBTAG00000051221_3 is predicted to interact with tribbles - 1 (TRIB1), based on the gene’s downstream proximity (Table 1). The correlation between these two was significantly positive (r = 0.90, P-value = 0.01; Supplementary Table 2). This gene has been linked to the regulation of anti-inflammatory macrophage polarization and inflammatory responses39,40,41,42. It also has important roles in controlling inflammatory cytokines, including the pro-inflammatory cytokine interleukin 8 (IL-8)41. Therefore, due to its critical roles in inflammatory responses, this is of key interest in terms of mastitis. The lncRNA, which is predicted to interact with this gene is over-expressed in the mastitic samples compared to the healthy samples (FC = 16.08, FDR = 4.66E-02) and, therefore, its over expression could act to suppress the functions of this key gene and therefore impact the hosts’ ability to regulate the inflammatory response, making the mastitis incidence more severe.
Next, the over-expressed (FC = 16.31, FDR = 4.53E-02) lncRNA, which is annotated for both gene name and length is ENSBTAG00000052589_1 (Table 1). This lncRNA is predicted to interact with the downstream gene PIK3R4. As mentioned earlier, this family of genes are involved in in many aspects of immune cell development15. Therefore, since this lncRNA is over-expressed in the mastitic samples, it could impact the genes expression and thus the cow’s ability to mount a proper immune response to the mastitis causing agent.
Lastly, ENSBTAG00000050727_2 is over-expressed in the mastitic samples (FC = 19.02, FDR = 3.28E-02; Table 1). This lncRNA has two potential target interactions based on their upstream proximity to the lncRNA. The first potential interaction is with bta-mir-10163. This miRNA has been previously found in the corpus luteum of pregnant animals and has numerous targets including retinoic acid receptor RXR - alpha (RXRA) which is involved in cell proliferation and apoptosis43,44. As lncRNA can act as miRNA sponges by binding miRNAs and preventing their interaction with their target, this could inhibit the miRNA regulatory function45,46. In relation to mastitis, the relationship between our DE lncRNA ENSBTAG00000050727_2 and the miRNA bta-mir-10163 is unclear, but further research is necessary to investigate this. Additionally, this lncRNA has another potential interaction with the upstream gene embigin (EMB). Embigin is a transmembrane glycoprotein which belongs to the immunoglobulin superfamily47. It has been reported to be expressed in a variety of prostate and mammary cancer cell lines48. Currently, the potential impact of this gene on bovine mastitis is unknown, but as it has been expressed in mammary cancer cell lines, this is something that could be relevant to bovine mastitis. The correlation between the lncRNA and both of these predicted targets was not significant in our dataset.
Functional analysis of previously annotated lncRNAs for gene name
In addition, there were 11 DE lncRNAs that were annotated for gene name, but length was different than the length reported in the ARS_UCD1.2.1 bovine reference genome (Table 2).
The first lncRNA, ENSBTAT00000072182 is over-expressed in the mastitic samples (FC = 5.93, FDR = 1.77E-02) and has two predicted target interactions (Table 2). The first predicted target is located upstream and is family with sequence similarity 111 member B (FAM111B). This gene has been linked to multiple malignancies as it is an oncoprotein. The expression of FAM111B in breast cancer tissues is higher than observed in healthy tissues in humans49. Therefore, future research should confirm if this lncRNA / mRNA interaction could have a negative effect on the bovine mammary gland. This lncRNA is also predicted to interact with the downstream gene deltex E3 ubiquitin ligase 4 (DTX4), which has roles in the IFN-1 signaling pathway in innate immunity50. Innate immunity is the cows first line of defence against mastitis-causing agents. Therefore, if the over-expression of this lncRNA impacts the ability of the mRNA protein to form, this could negatively impact the cow’s initial response to mastitis.
Next, lncRNA ENSBTAT00000084749 has two predicted targets due to their proximity to the lncRNA (Table 2). The first predicted target which is located upstream is solute carrier organic anion transporter family member 4A1 (SLCO4A1). This gene is highly expressed in colon cancers and promotes the cancers’ proliferation51. The second predicted target is the miRNA bta-mir-133a-1 and is located downstream. This miRNA has been linked to human cardiac remodeling52, as well as in muscle development in beef cattle53. As shown in Table 2, this lncRNA is over-expressed in the mastitic samples (FC = 6.60, FDR = 1.43E-02); however, the relationship between this lncRNA and its predicted targets is unknown, so further research should look at the potential impact its expression may have on SLCO4A1 and bta-mir-133a-1. The correlation between expression levels of the lncRNA and both of these predicted targets was observed to be not significant in our dataset.
The lncRNA ENSBTAT00000086278 overlaps with toll-like receptor 8 (TLR8) and therefore this is its predicted target. No significant correlation between the two was found. The family of toll-like receptors (TLRs) are a family of highly conserved pattern-recognition receptors which are essential for host immune response54 and triggering the onset of the inflammatory cascade55. The lncRNA is over-expressed in the mastitic samples (FC = 8.91, FDR = 3.27E-02), therefore, over-expression of TLR8 could impact its ability to carry out important roles in the host immune response. Therefore, future research should investigate the relationship between this lncRNA / mRNA to determine the impact on the host’s immune response.
The lncRNA ENSBTAT00000084307 is predicted to interact with the upstream gene serum / glucocorticoid regulated kinase 1 (SGK1) which has roles in promoting glucose metabolism56. The role of SGK1 in bovine mastitis is not currently known; however, previous research conducting gene expression and GWAS have shown that it is commonly DE between mastitic and healthy tissue conditions57. In our study, the lncRNA that is predicted to interact with SGK1 is over-expressed in the mastitic samples (FC = 45.79, FDR = 2.16E-03) and thus, further research is warranted to better understand the relationship of this lncRNA/ mRNA in relation to mastitis or immune response. The correlation between the lncRNA and the isoform of SGK1 (SGKI_2) expression levels was positive (r = 0.82) and significant (P-value = 0.05).
Lastly, ENSBTAT00000070418_2 and ENSBTAT00000082333 and their predicted targets were both previously discussed as they were the most over-expressed and under-expressed annotated lncRNA and therefore, not be further mentioned. Additionally, 5 of the lncRNAs in Table 2 have the same predicted targets of other DE lncRNA previously discussed throughout the manuscript, therefore, their functions will not be discussed. The first lncRNA, ENSBTAT00000068605 is overlapping and predicted to interact with TRIB1 (FC = 19.82, FDR = 2.37E-02). The second lncRNA ENSBTAT00000069074 is predicted to interact with both bta-mir-10163 and EMB, which are both located upstream from the lncRNA (FC = 25.06, FDR = 4.09E-03). Only the interaction between the lncRNA and EMB was significantly positively correlated (r = 0.94, P-value = 0.00) in the healthy group. Lastly, the lncRNAs ENSBTAT00000070418_1, ENSBTAT00000070418_3 and ENSBTAT00000070418_4 were predicted to interact with the downstream gene PIK3R4 and are all over-expressed in the mastitic samples compared to the healthy samples (FC = 25.41, FDR = 3.06E-03; FC = 46.89, FDR = 4.37E-02; FC = 27.38, FDR = 3.14E-03, respectively). When looking at the correlations, there were significant interactions between ENSBTAT00000070418_1 and PIK3R4_1 (r = 0.83; P-value = 0.04) in the healthy group, ENSBTAT00000070418_1 and PIK3R4_3 (r = 0.94; P-value = 0.01) in the mastitic group and ENSBTAT00000070418_2 in the healthy group (r = −0.82; P-value = 0.05; Supplementary Table 2).
Functional analysis of novel differentially expressed lncRNA
As mentioned earlier, novel lncRNA were split into intergenic and genic. Therefore, functional analysis was completed separately for both groups using the platform NetworkAnalyst. It was hypothesized that some of the lincRNAs and genic lncRNAs act in close proximity to mRNAs involved in immune system pathways. As such, if the lncRNA is acting to suppress or enhance the mRNA, this could negatively impact the cow’s immune response and increase susceptibility to mastitis infection, preventing her from being able to efficiently eliminate the threat.
Functional analysis of novel differentially expressed long intergenic non-coding RNA
Using the list of transcript interactions associated with the DE lincRNAs (N = 56; Table 3) to perform the functional analysis, 44 significantly enriched metabolic pathways were identified (FDR < 0.05; Table 4). The significant metabolic pathways were associated with immune mechanisms such as positive regulation of immune response, regulation of cytokine biosynthetic process and T-cell differentiation, among others. Two of the lincRNAs (lincRNA_2411.6 and lincRNA_1450.2) act in close proximity to two genes, tumor necrosis factor receptor superfamily member 1 A (TNFRSF1A) and baculoviral IAP repeat containing 3 (BIRC3), which explain the majority of the topology of the network (Fig. 1).

The two red circles represent two significantly enriched genes, which explain the majority of the topology of the network. Both tumor necrosis factor receptor superfamily member 1A (TNFRSF1A) and baculoviral IAP repeat containing 3 (BIRC3) are located in close proximity to two of the identified DE lincRNA. The purple circles represent genes used to construct the network.
The lincRNA_2411.6 was 11 × over-expressed in the mastitic samples compared to the healthy samples (FC = 11.31, FDR = 3.19E-02) and is predicted to interact with TNFRSF1A which is located upstream. This receptor encodes the type 1 receptor for tumor necrosis factor - α (TNFα)58, which is a pro-inflammatory cytokine secreted by inflammatory cells59. Numerous studies have investigated TNFα in relation to mastitis and have found that concentrations increase during mastitis caused by E. coli and its endotoxin60. Other studies have identified polymorphisms in TNFα that caused an amino acid sequence change and made animals more susceptible to mastitis61. Tumor necrosis factor receptor superfamily member 1 A TNFRSF1A is also a central gene connecting numerous other immune genes such as inhibitor of nuclear factor kappa B kinase subunit beta (IKBKB), which regulates multiple aspects of the innate and adaptive immune system62 and cluster of differentiation 40 (CD40) that is induced by proinflammatory stimuli63. Thus, the close connection of this lincRNA (lincRNA_2411.6) with TNFRSF1A may potentially affect the functionality of this gene and other genes connected to this central node. We observed a significantly strong positive correlation between lincRNA_2411.6 and the isoform TNFRSF1A_2 (r = 0.92, P-value = 0.01; Supplementary Table 2).
The baculoviral IAP repeat containing 3 (BIRC3) gene also explained the majority of the topology of the network and is located upstream of the novel lincRNA lincRNA_1450.2. This lincRNA is 6.88 × over-expressed in the mastitic samples compared to the healthy samples (FC = 6.88, FDR = 1.60E-02). This gene is also commonly known as cellular inhibitor of apoptosis 2 (cIAP2). The inhibitors of apoptosis family of proteins have numerous biological functions such as cell proliferation, cell migration, apoptosis, and regulation of innate immunity and inflammation64. In an intramammary infection, one aspect of the host’s innate immunity are neutrophils and neutrophils act as the first line of defense to protect the mammary gland through phagocytosis and intracellular killing of bacterial pathogens. Once the mammary gland has cleared the pathogens, apoptosis of the neutrophils is critical to limit the inflammation in the udder and return it back to normal to prevent permanent scarring in the mammary gland, which results in a loss of milk production65,66. This central gene is also connected to numerous caspase (CASP) genes including CASP3, CASP7, and CASP9, which function mainly in programmed cell death67. Programmed cell death is important to ensure that pathogen - infected cells actively initiate cell death to prevent the pathogens, such as S. aureus, from multiplying and spreading, causing the mastitis infection to worsen68. Therefore, due to the central role of BIRC3 with the immune system, further research is required to confirm if lincRNA_1450.2 has an impact on the BIRC3 gene functionality. However, the correlation between them was not significant.
Functional analysis of novel differentially expressed genic long non-coding RNA
Using the list of mRNAs associated with 22 DE genic lncRNAs (Table 3), 65 significant metabolic pathways were identified such as inflammatory response and regulation of cytokine biosynthetic process (FDR < 0.05; Table 5). Three genes explain the majority of the topology of the network analysis, however only hypoxia-inducible factor 1 - alpha (HIF1A or HIF - 1α) and Plexin - A2 (PLXNA2) are in close proximity with DE genic lncRNAs (Fig. 2).

The three red circles represent three significantly enriched genes that explain the majority of the topology of the network. Both Plexin - A2 (PLXNA2) and hypoxia-inducible factor 1 - alpha (HIF1A) are located in close proximity to three of the identified DE genic lncRNA. The purple circles represent genes used to construct the network.
The HIF - 1α gene has been detected in almost all innate and adaptive immune populations and the HIF transcription factors are key elements in immune cell metabolism and function69. Previous studies have shown that HIF - 1α are induced by pro-inflammatory cytokines such as TNF - α and IL - 1β70 and the expression of both increase during cases of mastitis60,71. As illustrated in Fig. 2 this gene is a central node for 22 other genes, some of which are immune genes (e.g., vascular endothelial growth factor A (VEGFA), b - cell lymphoma 2 (BCL2)) and some of which are involved in regulating normal cell function (e.g., mediator complex subunit 1 (MED1), retinoic acid receptor - related orphan receptor alpha (RORA)). Given the variety of functions these central genes have, it is important to consider how the DE genic lncRNAs associated with them act to enhance or impact their expression. In our analysis the HIF - 1α gene was associated with two DE lncRNAs (lncRNA_577.1 and lncRNA_577.4) which overlap with the exonic region of this gene. Both of these lncRNA were over-expressed in the mastitic samples in comparison to the healthy samples (FC = 6.93, FDR = 4.55E-02; and FC = 5.59, FDR = 4.47E-02, respectively). These transcripts may be a potential regulator of HIF - 1α gene, but further research is needed to confirm this. Additionally, in our dataset, the correlation between HIF - 1α and both lncRNA_577.1 and lncRNA_577.4 was not significant.
Genic lncRNA_2901.1 (FC = 7.12, FDR = 4.81E-02) was over-expressed in the mastitic samples compared to the healthy samples and is predicted to interact with PLXNA2 as it is nested in an intronic region of the gene. This central node (Fig. 2) is connected to 11 other genes which are mainly involved in normal cell function. Plexins are a family of proteins that act as receptors for semaphorins, which are extracellular signaling proteins, essential in the development and maintenance of organs and tissues72. Plexins and semaphorins have also been found to mediate critical processes to the immune system including cytokine secretion, migration and cell - cell contact73. However, no studies have directly linked PLXNA2 with mastitis, so further research into this specific plexin is needed and how the DE lncRNA could impact its expression.
QTL annotation and enrichment analysis
The current cattle QTL database has 159,844 QTL reported, relating to 653 different traits (release 42; https://www.animalgenome.org/cgi-bin/QTLdb/index)74. Previous research by Tong et al.5 reported that several QTLs in regions of lncRNAs affect clinical mastitis, milk quality or production. In our study, 31 QTL were previously annotated within the regions of the 94 DE lncRNAs (Table 6). These QTL were associated with milk (65%), reproduction (24%), production (7%), health (2%) and meat/carcass (2%; Fig. 3). The majority of QTL within the lncRNA regions were associated with milk, milk kappa - casein percentage and milk protein percentage (Supplementary Fig. 1) which is to be expected as the majority of QTL reported in the cattle QTL database are associated with production traits. However, when the QTL associated with health trait was plotted, the only QTL annotated was for ketosis and the lincRNA annotated in this region is lincRNA_2322.5 (3:104443741-104466628). Ketosis is another highly prevalent disease cows face in early lactation when energy expenditure is higher than the dietary intake75,76. Some studies have studied the effects of ketosis on mastitis and other diseases, for example Uyarlar et al. found that the incidence of mastitis, metritis and the coexistence of both infections was significantly (p < 0.01) higher in subclinically and clinically ketotic cows75. Therefore, this QTL could make the cows more susceptible to ketosis and in turn, other diseases common in the negative energy balance, including mastitis.

The pie chart represents the percentages of each QTL type from the annotation analysis. The slices represent as follows: purple = milk, blue = reproduction, red = production, green = health and yellow = meat and carcass.
Additionally, QTL enrichment analysis was performed to correct for the large proportion of existing studies which have evaluated milk traits in dairy cattle77,78. As shown in the bubble plot in Fig. 4, the milk production traits were still the most enriched traits, as the number of associated studies for a specific trait can directly influence the enrichment results (Supplementary Table 3). Although QTL directly related to mastitis (e.g., somatic cell count, somatic cell score, clinical mastitis) were not found, previous research has shown genetic and phenotypic correlations between mastitis and milk production related traits. Milk yield is a trait with a genetically unfavorable correlation with clinical mastitis79,80. Additionally, the abundance of numerous milk proteins changes during cases of mastitis, which is unfavorable for the producer81. As there are numerous QTL within the lncRNAs genomic regions associated with milk production traits, careful consideration must be made before targeting specific genomic regions for breeding purposes. However, studying these regions could provide a deeper insight into the potential functionality of the lncRNAs identified in this study, in relation to both mastitis resistance and milk production traits.

The area of the bubbles represents the number of observed QTL for that QTL class, and the color represents the P-value scale (darker color = smaller P-value). The richness factor for each QTL represents the ratio of the number of QTL and the expected number of QTL.
Conclusions
RNA - Sequencing analysis was used to identify DE lncRNAs between 6 healthy and 6 mastitic milk somatic cell samples. The previously annotated lncRNAs identified in this study were predicted to interact with mRNA and miRNA that are involved in immune pathways and could impact the host’s immune response to mastitis. Functional candidate novel lncRNAs were identified due to their involvement with the immune system and acted as central nodes in immune pathways, specifically those interacting with hypoxia - inducible factor 1 - alpha (HIF - 1α) which is detected in most innate and adaptive immune populations, plexin - A2 (PLXNA2) which mediates critical processes to the immune system, tumor necrosis factor receptor superfamily member 1 A (TNFRSF1A) which is involved with numerous other immune genes, and lastly baculoviral IAP repeat containing 3 (BIRC3) which helps regulate innate immunity and inflammation. The QTLs in the lncRNA genomic regions are associated with numerous milk traits, such as milk protein percentage, as well as in the QTL region associated with ketosis. Therefore, studying the transcriptome at a high - throughput level using RNA – Seq enabled the identification of lncRNA regulatory elements that have a potential functional role in immune response to mastitis in Holstein dairy cows. In turn, this research could aid in the development of breeding programs to select animals that are more resistant to mastitis infections.
Methods
Animal material and sample collection
This study was approved by the UC Davis Institutional Animal Care and Use Committee (IACUC). Sample collections and procedures were performed in accordance with the approved guidelines of UC Davis IACUC. As described in Asselstine et al., 6 Holstein dairy cows, ranging from first to third lactation from the University of California - Davis were used in this study and all 6 of these animals had natural cases of mastitis82. The natural cases of mastitis were diagnosed by the California mastitis test which detects early presence of mastitis in the milk based on the SC count3,83. The California mastitis test can be performed quickly cow-side and works by using a reagent that disrupts the cell membrane of somatic cells present in the milk sample. The DNA in the cells then reacts with the test reagent to form a gel, and the degree of gelling is positively associated with SCC in the sample83. Generally, this test cannot detect SCC below 350,000 cells/mL. Two different samples were taken from each cow, one sample from the mastitic quarter (N = 6) and the other sample taken diagonally away from the mastitic quarter and classified as healthy (N = 6) which was verified based on having a somatic cell count <100,000 cells/mL (N = 12)82. Using examination gloves, the same technician for all the samples cleaned the cow’s teats was with gauze and damped in 70% isopropanol and 50-mL of milk sample was taken from each quarter using a 3 - cm plastic cannula (Genesis Industries Inc., Elmwood, WI) to ensure no external bacteria contaminated the sample. Milk was kept on ice and immediately processed for RNA extraction. After the milk samples were collected, the cow was immediately treated to mediate the mammary infection.
After being stored on ice, the technician then separated the pellet of SC from the upper milk fat globule membrane and the pellet was washed using RNase - free Phosphate Buffered Saline (PBS) and EDTA (50 mL PBS + 50 μL EDTA) following protocol outlined by Cánovas et al.84. Total RNA was purified following the Trizol protocol (Invitrogen, Carlsbad, CA); and the RNA was quantified by an ND - 1000 Nanodrop Spectrometer (Thermo Scientific, Pittsburgh, PA)85. All samples passed the RNA integrity number (range of 8.0–9.0), indicating good RNA quality85. As described in Cánovas et al.86,87 library construction was performed using the TruSeq RNA sample preparation kit (Illumina, San Diego, CA). Sequencing was completed at the same time by a facility technician with an Illumina HiSeq 2000 analyzer that yielded 100 - bp single - read sequences.
The data discussed in this publication have been deposited in NCBI’s Gene Expression Omnibus and are accessible through Gene Expression Omnibus series accession number GSE131607 (https://www.ncbi.nlm.nih.gov/geo/ query/acc.cgi?acc = GSE131607).
RNA - sequencing analysis
Read trimming and quality control
The raw sequence data was trimmed using the automatic trimmer function of CLC Genomics Workbench (CLC Bio, Aarhus, Denmark) using a quality trimming score = 0.05. After the reads were trimmed, quality control was performed using the NGS quality control tool of CLC Genomics Workbench as described by Cánovas et al.86. All samples passed the quality control analysis based on GC content, Phred score and over-represented sequence parameters to name a few82,86.
Sequence assembly
The Large Gap Read Mapping (LGRM) tool in CLC Genomics Workbench was used to map the reads to the bovine reference genome (ARS_UCD1.2.1; ftp.ensembl.org/pub/release-100/fasta/bos_taurus)88. The LGRM tool can map sequence reads that span introns without requiring prior transcript annotation89. Assembly was conducted with a length fraction of 0.7 and a similarity of 0.8 to exclude paralogous sequence variants and the settings were as follows: a mismatch cost = 2, deletion cost = 3, insert cost = 3, minimum contig length = 200 base pairs (bp) were allowed88.
Using CLC Genomics Workbench, transcript discovery was performed to identify transcripts in each group individually. Starting with the healthy group, the transcript discovery used: 1) the bovine reference genome and 2) the LGRM assembly for the healthy group. Parameters for filtering include gene merging distance = 50, minimum reads in gene = 10 and minimum predicted gene length ≥ 200 bp88. For the mastitic transcript discovery, the predicted RNA and gene tracks generated from the healthy group as well as the annotated bovine reference genome were used. Thus, the predicted RNA file (.gtf) contains predicted information from both groups of samples (healthy and mastitis), in addition to the annotated genome information.
Long non-coding RNA identification
To identify lncRNAs, three different FIExible Extraction of LncRNAs (FEELnc) pipelines were used (FEELnc filter, FEELnc codpot and FEELnc classifier; https://github.com/tderrien/FEELnc)90. The FEELnc filter pipeline was used to filter and remove protein-coding, pseudogene, miRNA etc and capture transcripts with a minimal size of 200 bp. The FEELnc codpot pipeline was applied to compute the coding potential score (CPS; [0–1]) for each of the candidate transcripts in the predicted RNA file (.gtf) generated in the previous step. From this, the mRNA vs lncRNA can be separated based on their maximized specificity (Sp) and sensitivity (Sn). The CPS calculation was based on three parameters: the k - mer frequencies, which were left as default with values of: 1, 2, 3, 6, 9 and 12 mers, the Open Reading Frame (ORF) coverage and the mRNA size91. The CPS cut - off for the samples was 0.92 of both sensitivity and specificity (Supplementary Fig. 2). Lastly, FEELnc classifier pipeline was used to identify the best partner interaction of each lncRNA. This is determined based on different aspects including: the lncRNAs type, subtype and location. To classify the lncRNA type, lncRNA can either be ‘genic’ where they overlap with a gene, or they can be classified as ‘intergenic’ when they are located between genes (either upstream or downstream) and these are commonly referred to as (long intergenic non-coding RNA; lincRNA). For the subtype category, there are different classifications; genic lncRNA can be classified as: 1) ‘nested’ in which the lncRNA is contained in the RNA partner transcript, or (2) ‘overlapping’ meaning the lncRNA partly overlaps with the RNA partner transcript. For lincRNA, they can be classified as (1) ‘same_strand’ in which the lncRNA is transcribed in the same orientation with its RNA partner, (2) ‘divergent’ in which the lncRNA is transcribed in head-to-head orientation with the RNA partner, or (3) ‘convergent’ where the lncRNA is orientated in tail to tail with its RNA partner. Next, the location can be defined according to the orientation of the interactions and the localization of the interactions (genic can be overlapping an intronic or exonic region of the partner RNA; lincRNA can be located upstream or downstream). Next, to determine the best partner RNA, for genic lncRNA, the best RNA partner is by rule of priority exonic, then intronic, then containing. Whereas for lincRNA, the best RNA partner is the closest to the lincRNA. For both genic and lincRNA, the best partner interaction is assigned a numerical value of 1 = best identified match for the lncRNA or 0 = indicating there is a better match. Once this is determined, functional annotation and relationships between lncRNA and their predicted target interactions, e.g., mRNA or miRNA is identified90.
From the previous step, a file is generated containing the identified lncRNA, which was then combined with the annotated reference genome (.gtf file). Using this combined file, the trimmed reads were then aligned to the bovine reference genome using CLC Genomics Workbench (CLC Bio, Aarhus, Denmark). RNA - Sequencing analysis was then performed in CLC Genomics Workbench using mapping criteria as mentioned prior (mismatch = 2, insertion = 3, deletion costs = 3). We also used the same criteria for the length and similarity fractions (0.7 and 0.8, respectively). Expression values for the lncRNA were on a count - based model which were then transformed and normalized.
Differential lncRNA expression analysis and classification
Differential expression analysis on the lncRNAs was performed between healthy (N = 6) and mastitic (N = 6) samples by Empirical analysis of differential gene expression tool (CLC Genomics Workbench). Transcripts were classified as DE between healthy and mastitic samples when FDR < 0.05 and a fold change (|FC|) > 2. Among the DE transcripts, only those annotated as lncRNA using the FEELnc software were used for further analysis. We were interested in looking not only at the lncRNA as a whole, but also looking at if they were 1) previously annotated for both gene name and length in the ARS_UCD1.2.1 bovine reference genome, 2) previously annotated for gene name in the ARS_UCD1.2.1 bovine reference genome and 3) novel lncRNAs for which no gene name or gene length was previously annotated in the ARS_UCD1.2.1 bovine reference genome.
Functional analysis
Functional analysis including metabolic pathway analysis and gene networks analysis was performed on the partner interactions of the DE lncRNAs identified in this study. To determine the functions of the DE lncRNAs, the lncRNAs were split into 1) previously annotated, including those annotated for both gene name and length and those annotated for gene name, but length was different than that reported in the bovine reference genome (ARS.UCD 1.2.1) and 2) novel lncRNAs with neither gene name nor length reported in the bovine reference genome (ARS.UCD 1.2.1).
For the previously annotated DE lncRNAs, the genomic position (e.g., chr: start - end region) of each DE lncRNA was used to identify proximal RNA transcripts through Genome Data Viewer (https://www.ncbi.nlm.nih.gov/genome/gdv/?org=bos-taurus). These proximal RNA transcripts were then considered to be the predicted target of the DE lncRNA and functional analysis was performed on a lncRNA – by – lncRNA basis.
For the novel lncRNA, as previously mentioned the FEELnc classifier pipeline was used to determine the predicted target interaction. After this predicted target was identified, the NetworkAnalyst platform was used to perform the gene network analysis (http://www.networkanalyst.ca), using the list of predicted target interactions associated with the DE lncRNAs identified as the input. This software performs meta - analysis on gene expression data sets, to determine important features, patterns, functions and connections between genes92,93,94,95,96.
Significant (P-value < 0.05) correlations between the lncRNA and its predicted interaction within all categories (previously annotated (genes + length), previously annotated (genes), novel (genic) and novel (intergenic)) are shown in Supplementary Table 2. Within each category of lncRNA, a Pearson Correlation analysis was performed in SAS (SAS version 9.4) for each lncRNA and its predicted interaction to provide further insight on the relationship between their expression level patterns. The RPKM values of the predicted interactions was downloaded from the raw expression analysis in CLC genomics workbench. Each predicted interaction could have had multiple mRNA isoforms present, so the correlation analysis was performed for every potential mRNA isoform of the predicted interaction. Only statistically significant (P-value < 0.05) interactions were discussed in the manuscript where relevant.
QTL annotation and enrichment analysis
Lastly, QTL annotation was performed using the R package: Genomic functional Annotation in Livestock for positional candidate LOci (GALLO)77. The genome coordinates of the DE lncRNAs was used, as well as the QTL gff annotation file retrieved from the cattle QTL Database (https://www.animalgenome.org/cgi-bin/QTLdb/index)74. Intervals of 1000 bp was used to account for 1000 bp upstream and 1000 bp downstream of each DE lncRNA coordinates97. Additionally, to evaluate if the QTL classes and traits identified around the selected DE lncRNA were significantly overrepresented, QTL enrichment qtl_enrich() function from GALLO was performed using the output obtained from the QTL annotation step78,98.
Statistics and reproducibility
No statistical method was used to determine the minimum sample size in this study. There was no data excluded from the analysis. The experiment was not randomized and the investigators were not blinded during the experiment and outcome assessment. For comparisons between two groups (healthy and mastitic) differential expression analysis on the lncRNAs was performed using the Empirical analysis of differential gene expression tool of CLC Genomics Workbench. The program FEELnc was used to identify lncRNA from the dataset. The R package GALLO was used to perform the QTL annotation and enrichment analysis.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The datasets generated and/or analyzed during the current study are available in the NCBI’s Gene Expression Omnibus repository, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc. Accession ID = GSE131607.
References
Heikkilä, A.-M., Liski, E., Pyörälä, S. & Taponen, S. Pathogen-specific production losses in bovine mastitis. J. Dairy Sci. 101, 9493–9504 (2018).
Li, N., Richoux, R., Boutinaud, M., Martin, P. & Gagnaire, V. Role of somatic cells on dairy processes and products: a review. Dairy Sci. Technol. 94, 517–538 (2014).
Guerrero, A. et al. Peptidomic analysis of healthy and subclinically mastitic bovine milk. Int. Dairy J. 46, 46–52 (2015).
Sun, X. et al. The developmental transcriptome sequencing of bovine skeletal muscle reveals a long noncoding RNA, lncMD, promotes muscle differentiation by sponging miR-125b. Biochim. Biophys. Acta (BBA) - Mol. Cell Res. 1863, 2835–2845 (2016).
Tong, C. et al. Identification and characterization of long intergenic noncoding RNAs in bovine mammary glands. BMC Genomics 18, 468 (2017).
Ulitsky, I. Evolution to the rescue: using comparative genomics to understand long non-coding RNAs. Nat. Rev. Genet. 17, 601–614 (2016).
Gil, N. & Ulitsky, I. Regulation of gene expression by cis-acting long non-coding RNAs. Nat. Rev. Genet. 21, 102–117 (2020).
Kukurba, K. R. & Montgomery, S. B. RNA sequencing and analysis. Cold Spring Harb. Protoc. 2015, pdb.top084970 (2015).
Koufariotis, L. T., Chen, Y.-P. P., Chamberlain, A., Vander Jagt, C. & Hayes, B. J. A catalogue of novel bovine long noncoding RNA across 18 tissues. PLoS ONE 10, e0141225 (2015).
Diamantopoulos, M. A., Tsiakanikas, P. & Scorilas, A. Non-coding RNAs: the riddle of the transcriptome and their perspectives in cancer. Ann. Transl. Med. 6, 241–241 (2018).
Wang, H. et al. A novel long non‐coding RNA regulates the immune response in MAC ‐T cells and contributes to bovine mastitis. FEBS J. 286, 1780–1795 (2019).
Becker, D., Weikard, R., Hadlich, F. & Kühn, C. Single-cell RNA sequencing of freshly isolated bovine milk cells and cultured primary mammary epithelial cells. Sci. Data 8, 177 (2021).
Niedziela, D. A., Cormican, P., Foucras, G., Leonard, F. C. & Keane, O. M. Bovine milk somatic cell transcriptomic response to Staphylococcus aureus is dependent on strain genotype. BMC Genomics 22, 796 (2021).
Twigger, A.-J. et al. Transcriptional changes in the mammary gland during lactation revealed by single cell sequencing of cells from human milk. Nat. Commun. 13, 562 (2022).
Pompura, S. L. & Dominguez-Villar, M. The PI3K/AKT signaling pathway in regulatory T-cell development, stability, and function. J. Leukoc. Biol. 103, 1065–1076 (2018).
Patel, J. B. & Chauhan, J. B. Computational analysis of non-synonymous single nucleotide polymorphism in the bovine cattle kappa-casein (CSN3) gene. Meta Gene 15, 1–9 (2018).
Threadgill, D. W. & Womack, J. E. Genomic analysis of the major bovine milk protein genes. Nucleic Acids Res. 18, 6935–6942 (1990).
Qu, Z. & Adelson, D. L. Bovine ncRNAs are abundant, primarily intergenic, conserved and associated with regulatory genes. PLoS ONE 7, e42638 (2012).
Huang, W., Long, N. & Khatib, H. Genome-wide identification and initial characterization of bovine long non-coding RNAs from EST data. Anim. Genet. 43, 674–682 (2012).
Colinas, J., Schmidler, S. C., Bohrer, G., Iordanov, B. & Benfey, P. N. Intergenic and genic sequence lengths have opposite relationships with respect to gene expression. PLoS ONE 3, e3670 (2008).
Bartlett, C. P., Kirk, J. H., Wilke, M. A., Kaneene, J. B. & Mather, E. C. Metritis complex in Michigan Holstein-Friesian cattle: incidence, descriptive epidemiology and estimated economic impact. Prev. Vet. Med. 4, 235–248 (1986).
Freebern, E. et al. GWAS and fine-mapping of livability and six disease traits in Holstein cattle. BMC Genomics 21, 41 (2020).
Thompson-Crispi, K. A., Hine, B., Quinton, M., Miglior, F., & Mallard, B. A. Short communication: Association of disease incidence and adaptive immune response in Holstein dairy cows. J. Dairy Sci. 95, 3888–3893 (2012).
Koeck, A., Miglior, F., Kelton, D. F. & Schenkel, F. S. Health recording in Canadian Holsteins: data and genetic parameters. J. Dairy Sci. 95, 4099–4108 (2012).
Thompson-Crispi, K. A., Miglior, F., & Mallard, B. A. Incidence rates of clinical mastitis among canadian holsteins classified as high, average, or low immune responders. Clin. Vaccine Immunol. 20, 106–112 (2013).
Kuo, P. T. et al. The role of CXCR3 and its chemokine ligands in skin disease and cancer. Front. Med. 5, 271 (2018).
Youngerman, S. M., Saxton, A. M., Oliver, S. P. & Pighetti, G. M. Association of CXCR2 polymorphisms with subclinical and clinical mastitis in dairy cattle. J. Dairy Sci. 87, 2442–2448 (2004).
Grailhe, R., Grabtree, G. W. & Hen, R. Human 5-HT5 receptors: the 5-HT5A receptor is functional but the 5-HT5B receptor was lost during mammalian evolution. Eur. J. Pharmacol. 418, 157–167 (2001).
Lv, J. & Liu, F. The role of serotonin beyond the central nervous system during embryogenesis. Front. Cell. Neurosci. 11, 74 (2017).
Donovan, M. H. & Tecott, L. H. Serotonin and the regulation of mammalian energy balance. Front. Neurosci. 7, 36 (2013).
Weaver, S. R. et al. Elevating serotonin pre-partum alters the Holstein dairy cow hepatic adaptation to lactation. PLoS ONE 12, e0184939 (2017).
Suriyasathaporn, W. et al. β-hydroxybutyrate levels in peripheral blood and ketone bodies supplemented in culture media affect the in vitro chemotaxis of bovine leukocytes. Vet. Immunol. Immunopathol. 68, 177–186 (1999).
Perkins, K. H. et al. Clinical responses to intramammary endotoxin infusion in dairy cows subjected to feed restriction. J. Dairy Sci. 85, 1724–1731 (2002).
Moyes, K. M. et al. Mammary gene expression profiles during an intramammary challenge reveal potential mechanisms linking negative energy balance with impaired immune response. Physiol. Genomics 41, 161–170 (2010).
Madhani, H. D., Bordonne, R. & Guthrie, C. Multiple roles for U6 snRNA in the splicing pathway. Genes Dev. 4, 2264–2277 (1990).
Didychuk, A. L., Butcher, S. E. & Brow, D. A. The life of U6 small nuclear RNA, from cradle to grave. RNA 24, 437–460 (2018).
Zhu, Q. et al. IRX5 promotes colorectal cancer metastasis by negatively regulating the core components of the RHOA pathway. Mol. Carcinog. 58, 2065–2076 (2019).
Kroczek, R. A. & Henn, V. The role of XCR1 and its ligand XCL1 in antigen cross-presentation by murine and human dendritic cells. Front. Immunol. 3, 14 (2012).
Ostertag, A. et al. Control of adipose tissue inflammation through TRB1. Diabetes 59, 1991–2000 (2010).
Satoh, T. et al. Critical role of Trib1 in differentiation of tissue-resident M2-like macrophages. Nature 495, 524–528 (2013).
Niespolo, C. et al. Tribbles-1 expression and its function to control inflammatory cytokines, including interleukin-8 levels are regulated by miRNAs in macrophages and prostate cancer cells. Front. Immunol. 11, 574046 (2020).
Arndt, L. et al. Tribbles homolog 1 deficiency modulates function and polarization of murine bone marrow–derived macrophages. J. Biol. Chem. 293, 11527–11536 (2018).
Liu, B. et al. Direct functional interactions between insulin-like growth factor-binding protein-3 and retinoid X receptor-α regulate transcriptional signaling and apoptosis. J. Biol. Chem. 275, 33607–33613 (2000).
Maalouf, S. W., Liu, W.-S., Albert, I. & Pate, J. L. Regulating life or death: potential role of microRNA in rescue of the corpus luteum. Mol. Cell. Endocrinol. 398, 78–88 (2014).
Salmena, L., Poliseno, L., Tay, Y., Kats, L. & Pandolfi, P. P. A ceRNA hypothesis: the rosetta stone of a hidden RNA language? Cell 146, 353–358 (2011).
López-Urrutia, E., Bustamante Montes, L. P., Ladrón de Guevara Cervantes, D., Pérez-Plasencia, C. & Campos-Parra, A. D. Crosstalk between long non-coding RNAs, Micro-RNAs and mRNAs: deciphering molecular mechanisms of master regulators in cancer. Front. Oncol. 9, 669 (2019).
Chao, F. et al. Embigin, regulated by HOXC8, plays a suppressive role in breast tumorigenesis. Oncotarget 6, 23496–23509 (2015).
Guenette, R. S. et al. Embigin, a developmentally expressed member of the immunoglobulin super family, is also expressed during regression of prostate and mammary gland. Dev. Genet. 21, 268–278 (1997).
Li, W., Hu, S., Han, Z. & Jiang, X. YY1-induced transcriptional activation of FAM111B contributes to the malignancy of breast cancer. Clin. Breast Cancer. 22, E417–E425 (2021).
Wang, L., Sun, X., He, J. & Liu, Z. Functions and molecular mechanisms of deltex family ubiquitin E3 ligases in development and disease. Front. Cell Dev. Biol. 9, 706997 (2021).
Wu, K. et al. LncRNA SLCO4A1-AS1 modulates colon cancer stem cell properties by binding to miR-150-3p and positively regulating SLCO4A1. Lab Invest. 101, 908–920 (2021).
Li, N., Zhou, H. & Tang, Q. miR-133: a suppressor of cardiac remodeling? Front. Pharmacol. 9, 903 (2018).
Sun, J. et al. Identification and profiling of conserved and novel microRNAs from Chinese Qinchuan bovine longissimus thoracis. BMC Genomics 14, 42 (2013).
Zhu, J. et al. Characterization of bovine Toll-like receptor 8: Ligand specificity, signaling essential sites and dimerization. Mol. Immunol. 46, 978–990 (2009).
Sordillo, L. M. Mammary gland immunobiology and resistance to mastitis. Vet. Clin. North Am.: Food Anim. Pract. 34, 507–523 (2018).
Mason, J. A. et al. SGK1 signaling promotes glucose metabolism and survival in extracellular matrix detached cells. Cell Rep. 34, 108821 (2021).
Naserkheil, M. et al. Multi-omics integration and network analysis reveal potential hub genes and genetic mechanisms regulating bovine mastitis. Curr. Issues Mol. Biol. 44, 309–328 (2022).
Korytina, G. F. et al. Association of CRP, CD14, pro-inflammatory cytokines and their receptors (TNFA, LTA, TNFRSF1A, TNFRSF1B, IL1B, and IL6) genes with chronic obstructive pulmonary disease development. Russ. J. Genet. 56, 972–981 (2020).
Wang, X. & Lin, Y. Tumor necrosis factor and cancer, buddies or foes? Acta Pharm. Sin. 29, 1275–1288 (2008).
Hoeben, D. et al. Role of endotoxin and TNF-α in the pathogenesis of experimentally induced coliform mastitis in periparturient cows. J. Dairy Res. 67, 503–514 (2000).
Sattar, H. et al. Genetic association of bovine TNF-α gene polymorphism with clinical and sub-clinical mastitis in Sahiwal Cows. Pak. J. Zool. 51, 1–4 (2019).
Liu, T., Zhang, L., Joo, D. & Sun, S.-C. NF-κB signaling in inflammation. Sig. Transduct. Target Ther. 2, 17023 (2017).
Antoniades, C., Bakogiannis, C., Tousoulis, D., Antonopoulos, A. S. & Stefanadis, C. The CD40/CD40 ligand system. J. Am. Coll. Cardiol. 54, 669–677 (2009).
Berthelet, J. & Dubrez, L. Regulation of apoptosis by inhibitors of apoptosis (IAPs). Cells 2, 163–187 (2013).
Tanji-Matsuba, K. et al. Functional changes in aging polymorphonuclear leukocytes. Circulation 97, 91–98 (1998).
Van Oostveldt, K., Tomita, G. M., Paape, M. J., Capuco, A. V. & Burvenich, C. Apoptosis of bovine neutrophils during mastitis experimentally induced with Escherichia coli or endotoxin. Am. J. Vet. Res. 63, 448–453 (2002).
Van Ba, H. & Hwang, I. Role of caspase-9 in the effector caspases and genome expressions, and growth of bovine skeletal myoblasts. Dev. Growth Differ. 56, 131–142 (2014).
Nagata, S. & Tanaka, M. Programmed cell death and the immune system. Nat. Rev. Immunol. 17, 333–340 (2017).
Palazon, A., Goldrath, A. W., Nizet, V. & Johnson, R. S. HIF Transcription Factors, Inflammation, and Immunity. Immunity 41, 518–528 (2014).
Imtiyaz, H. Z. & Simon, M. C. Diverse Effects of Hypoxia on Tumor Progression (ed. Simon, M. C.) vol. 345. p. 105–120 (Springer Berlin Heidelberg, 2010).
Lazzari, A. M. et al. Produção de interleucina-1beta e severidade da mastite pós-inoculação de Staphylococcus aureus na glândula mamária de bovinos e bubalinos. Cienc. Rural 44, 1816–1822 (2014).
Alto, L. T. & Terman, J. R. Semaphorin Signaling (ed. Terman, J. R.) vol. 1493. p. 1–25 (Springer New York, 2017).
Roney, K., Holl, E. & Ting, J. Immune plexins and semaphorins: old proteins, new immune functions. Protein Cell 4, 17–26 (2013).
Hu, Z.-L., Park, C. A. & Reecy, J. M. Bringing the animal QTLdb and CorrDB into the future: meeting new challenges and providing updated services. Nucleic Acids Res. 50, D956–D961 (2022).
Uyarlar, C., Cetingul, S., Gültepe, E. E., Sial, A. R. & Bayram, İ. Süt i̇neklerinde görülen subklinik ve kinik ketozisin bazi hematolojik parametreler, mastitis, metritis i̇nsidensleri ile sürü dişi kalma oranina etkileri. Kocatepe Vet. J. https://doi.org/10.30607/kvj.419839 (2018).
Soares, R. A. N. et al. Differential gene expression in dairy cows under negative energy balance and ketosis: a systematic review and meta-analysis. J. Dairy Sci. 104, 602–615 (2021).
Fonseca, P. A. S., Suárez-Vega, A., Marras, G. & Cánovas, Á. GALLO: an R package for genomic annotation and integration of multiple data sources in livestock for positional candidate loci. GigaScience 9, giaa149 (2020).
Lam, S. et al. Identification of functional candidate variants and genes for feed efficiency in Holstein and Jersey cattle breeds using RNA-sequencing. J. Dairy Sci. 104, 1928–1950 (2021).
Carlén, E., Strandberg, E. & Roth, A. Genetic parameters for clinical mastitis, somatic cell score, and production in the first three lactations of Swedish Holstein Cows. J. Dairy Sci. 87, 3062–3070 (2004).
Martin, P., Barkema, H. W., Brito, L. F., Narayana, S. G. & Miglior, F. Symposium review: Novel strategies to genetically improve mastitis resistance in dairy cattle. J. Dairy Sci. 101, 2724–2736 (2018).
Turk, R. et al. Milk and serum proteomes in subclinical and clinical mastitis in Simmental cows. J. Proteom. 244, 104277 (2021).
Asselstine, V. et al. Genetic mechanisms regulating the host response during mastitis. J. Dairy Sci. 102, 9043–9059 (2019).
Godden, S. M., Royster, E., Timmerman, J., Rapnicki, P. & Green, H. Evaluation of an automated milk leukocyte differential test and the California Mastitis Test for detecting intramammary infection in early- and late-lactation quarters and cows. J. Dairy Sci. 100, 6527–6544 (2017).
Cánovas, A., Rincon, G., Islas-Trejo, A., Wickramasinghe, S. & Medrano, J. F. SNP discovery in the bovine milk transcriptome using RNA-Seq technology. Mamm. Genome 21, 592–598 (2010).
Cánovas, A. et al. RNA sequencing to study gene expression and single nucleotide polymorphism variation associated with citrate content in cow milk. J. Dairy Sci. 96, 2637–2648 (2013).
Cánovas, A. et al. Multi-Tissue Omics Analyses Reveal Molecular Regulatory Networks for Puberty in Composite Beef Cattle. PLoS ONE 9, e102551 (2014a).
Cánovas, A. et al. Comparison of five different RNA sources to examine the lactating bovine mammary gland transcriptome using RNA-Sequencing. Sci. Rep. 4, 5297 (2014b).
Muniz, M. M. M. et al. Identification of novel mRNA isoforms associated with meat tenderness using RNA sequencing data in beef cattle. Meat Sci. 173, 108378 (2021).
Cardoso, T. F. et al. Differential expression of mRNA isoforms in the skeletal muscle of pigs with distinct growth and fatness profiles. BMC Genomics 19, 145 (2018).
Wucher, V. et al. FEELnc: a tool for long non-coding RNA annotation and its application to the dog transcriptome. Nucleic Acids Res. 45, e57, https://doi.org/10.1093/nar/gkw1306 (2017).
Muret, K. et al. Long noncoding RNA repertoire in chicken liver and adipose tissue. Genet Sel. Evol. 49, 6 (2017).
Xia, J. et al. INMEX—a web-based tool for integrative meta-analysis of expression data. Nucleic Acids Res. 41, W63–W70 (2013).
Xia, J., Lyle, N. H., Mayer, M. L., Pena, O. M. & Hancock, R. E. W. INVEX—a web-based tool for integrative visualization of expression data. Bioinformatics 29, 3232–3234 (2013).
Xia, J., Benner, M. J. & Hancock, R. E. W. NetworkAnalyst - integrative approaches for protein–protein interaction network analysis and visual exploration. Nucleic Acids Res. 42, W167–W174 (2014).
Xia, J., Gill, E. E. & Hancock, R. E. W. NetworkAnalyst for statistical, visual and network-based meta-analysis of gene expression data. Nat. Protoc. 10, 823–844 (2015).
Zhou, G. et al. NetworkAnalyst 3.0: a visual analytics platform for comprehensive gene expression profiling and meta-analysis. Nucleic Acids Res. 47, W234–W241 (2019).
Sweett, H. et al. Genome-wide association study to identify genomic regions and positional candidate genes associated with male fertility in beef cattle. Sci. Rep. 10, 20102 (2020).
Fonseca, P. A. S., Schenkel, F. S. & Cánovas, A. Genome-wide association study using haplotype libraries and repeated-measures model to identify candidate genomic regions for stillbirth in Holstein cattle. J. Dairy Sci. 105, 1314–1326 (2022).
Acknowledgements
This study was funded by the Ontario Agri - Food Innovation Alliance [Ontario Ministry of Agriculture, Food and Rural Affairs (OMAFRA), Guelph, Ontario, Canada] and Natural Sciences and Engineering Research Council of Canada (Ottawa, Ontario, Canada). This paper is also a contribution to the Food from Thought research program supported by the Canada First Research Excellence Fund. Victoria Asselstine’s PhD program was funded by the OMAFRA - HQP program. We also sincerely thank Douglas Gisi at the University of California Dairy, University of California – Davis, for collecting all of the milk samples used in this study. We also sincerely thank Alma Islas - Trejo for her work on the construction of the sequencing libraries.
Author information
Authors and Affiliations
Contributions
A.C. and J.F.M. were responsible for the conceptualization and experimental design of the study and the RNA - Sequencing project. A.C. was the responsible for the RNA - Sequencing bioinformatic pipeline execution and theoretical discussions. V.A. analyzed the data and performed the functional analysis. M.M.M.M. provided support on data analysis. N.A.K., A.C., and B.A.M. contributed to interpreting the biological results. A.C. was responsible for funding acquisition. All authors contributed to writing the manuscript and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks the anonymous reviewers for their contribution to the peer review of this work. Primary Handling Editors: Caitlin Karniski and Luke R. Grinham.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Asselstine, V., Medrano, J.F., Muniz, M.M.M. et al. Novel lncRNA regulatory elements in milk somatic cells of Holstein dairy cows associated with mastitis. Commun Biol 7, 98 (2024). https://doi.org/10.1038/s42003-024-05764-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-024-05764-y
- Springer Nature Limited