
Abstract
Phenome-wide association studies identified numerous loci associated with traits and diseases. To help interpret these associations, we constructed a phenome-wide network map of colocalized genes and phenotypes. We generated colocalized signals using the Genotype-Tissue Expression data and genome-wide association results in UK Biobank. We identified 9151 colocalized genes for 1411 phenotypes across 48 tissues. Then, we constructed bipartite networks using the colocalized signals in each tissue, and showed that the majority of links were observed in a single tissue. We applied the biLouvain clustering algorithm in each tissue-specific network to identify co-clusters of genes and phenotypes. We observed significant enrichments of these co-clusters with known biological and functional gene classes. Overall, the phenome-wide map provides links between genes, phenotypes and tissues, and can yield biological and clinical discoveries.
Similar content being viewed by others


Introduction
Electronic health records (EHR)-linked biobanks coupled with genome-wide genotyping and sequencing data allows for the study of the impact of genetic variation on thousands of medical phenotypes simultaneously. Phenome-wide association analyses have been conducted in several EHR-linked biobanks and genome-wide association study (GWAS) summary statistics have been made publicly available for large biobanks such as the UK Biobank and FinnGen study. For example, GWAS summary statistics from a phenome-wide scan of the UK Biobank (UKBB)—a prospective cohort with deep genetic and rich phenotypic data collected on approximately 500,000 middle-aged individuals (aged between 40 and 69 years old) recruited from across the United Kingdom1—now exists and is a rich resource in the human genetics community.
Large-scale EHR-linked biobanks with available genetic data, such as the UKBB, permit the study of the relationship of tens of thousands of genes and phenotypes simultaneously. However, a major challenge is an interpretation due in large part to the complexity and heterogeneity of this wealth of data. Furthermore, there is a general lack of statistical methods available for such high-throughput analysis. Hence, only a few efforts have systematically characterized disease relationships in EHR data2,3.
In this study, we sought to enhance our understanding of the complex relationship of genes and phenotypes in the medical phenome by constructing a tissue-level phenome-wide network map of colocalized genes and phenotypes. Our main motivation was to create a tool for researchers to evaluate shared links between colocalized genes and a wide array of phenotypes. The approach is an extension of the PheWAS approach but instead of simply uncovering cross-phenotype associations4, the approach directly generates links between specific colocalized genes and phenotypes in specific tissues and identifies clusters within these shared links that reflect meaningful common causal mechanisms and/or pleiotropic genetic effects. To construct the phenome-wide map, we first generated tens of thousands of colocalized expression quantitative trait loci (eQTL) from 48 tissues of the Genotype-Tissue Expression (GTEx) v7 project5,6,7, and from ~3800 GWAS of biological and medical phenotypes from the UKBB. We then applied a bipartite (or two-mode) network approach8,9 followed by the biLouvain clustering method10, to identify networks of genes and phenotypes that co-cluster together in different tissues, giving us broad insight into the biological structure of genes, phenotypes, and tissues. Finally, we demonstrate the functionality of the phenome-wide map by highlighting co-clusters that are biologically relevant.
Results
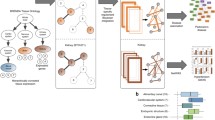
We performed three steps to generate the phenome-wide network map of genes and phenotypes: (1) identification of colocalization signals of eQTLs and GWAS loci for various continuous and binary phenotypes in 48 tissues from the GTEx project v7; (2) construction of a bipartite network using the colocalization signals to establish links between genes and phenotypes in each tissue; and (3) identification of co-clusters of colocalized genes and phenotypes in each bipartite network using the biLouvain clustering algorithm. A graphical flowchart of the study is shown in Fig. 1.

The flowchart illustrates all the different steps of our study. GWAS genome-wide association study, GTEx Genotype-Tissue Expression, eQTL expression quantitative trait locus.
Identification of colocalization signals of eQTL and GWAS loci in multiple tissues
We used coloc211, along with GWAS summary association statistics available for 3822 phenotypes in UKBB and eQTL data to identify colocalization signals in 48 tissues from the GTEx project. Before running coloc2, we performed stringent quality control (QC) on the phenotypes in UKBB. We removed phenotypes related to cause of death and case–control phenotypes with less than 1250 cases (or controls), except phenotypes showing prior gene/locus association as reported in the NHGRI-EBI GWAS catalog. In Neale GWAS data, we retained coloc2 results for 496 continuous and binary phenotypes. In SAIGE data, we excluded case–control phenotypes with less than 200 cases, retaining coloc2 results for 915 case–control (PheCode) phenotypes. We selected variants with minor allele frequency (MAF) > 0.1% in both Neale and SAIGE datasets; in GTEX, we included cis-eQTL variants with MAF >1% from Analysis V7. We restricted our study to the list of 48 tissues (from 620 donors) having a sample size of at least 80. In total, after QC (see “Methods”), we identified 9151 unique colocalized genes for 1411 unique phenotypes across the 48 selected tissues. Colocalization results for each tissue are reported in Supplementary Data 1. Unsurprisingly, the number of colocalized genes and phenotypes increases with respect to the GTEx tissue sample size (from n = 80 for brain–substantia nigra to n = 491 for muscle − skeletal), reflecting the enhanced statistical power of the method to uncover colocalized genes (see Supplementary Fig. 1).
Construction of tissue-level bipartite networks
Using the colocalized data of 9151 genes and 1411 phenotypes, we next created a bipartite network for each tissue. In brief, a bipartite network—also called a two-mode network—is a network in which nodes of one mode (i.e., type) are only connected to nodes of the other mode, as opposed to a unipartite (or one-mode) network commonly found in the network literature. In our colocalization results, phenotypes are not directly connected to other phenotypes, but could only be indirectly connected to each other through genes they share, while genes are indirectly connected to other genes if they appear in the same phenotype. In Supplementary Fig. 2a, a typical graphical representation of a bipartite network is displayed, comprised of seven phenotypes and six genes. Associations between genes and phenotypes are indicated by links (or edges) between them.
For each tissue, Supplementary Data 2 displays the number of unique colocalized genes and phenotypes, along with the number of links between the two sets. When all 48 tissues are aggregated, there are 9151 unique colocalized genes and 1411 unique phenotypes, with 25,710 unique links between the two sets. We aggregated the tissues using an unweighted approach which means that if a link between a gene and a phenotype was found in more than one tissue, we counted this link only once. In fact, we observed that the majority of links between a given gene and a given phenotype are observed in one single tissue, but a few links are present in all 48 tissues (see Supplementary Fig. 3 and Supplementary Data 3).
To characterize and compare colocalization results across tissues, we computed the average degree of colocalized genes and phenotypes in each tissue. The degree of a given gene (respectively, phenotype) is simply the number of unique phenotypes (respectively, genes) connected to it in the network, the average being taken over the total number of genes (respectively, phenotypes)12. The average degree for both genes and phenotypes does not vary much across tissues (see Supplementary Data 2), although it increases with larger tissue sample size. When aggregating all 48 tissues, each gene is connected to an average of ~2.8 phenotypes, while each phenotype is connected to an average of ~18.2 genes. The fact that the majority of gene and phenotype links are observed in a single tissue and not across all tissues, but the average degree of genes and phenotypes does not vary across tissues, suggests an architecture where tissue-specific gene regulatory mechanisms drive GWAS loci, and the size and structure of these mechanisms are largely similar across different tissues.
Identification of co-clusters in tissue-level bipartite networks
To identify structure within the phenome-wide map, we applied a clustering algorithm, called biLouvain, which extends the well-known unipartite Louvain clustering algorithm10. This algorithm efficiently identifies co-clusters of non-overlapping genes and phenotypes by maximizing a bipartite modularity measure (see “Methods” for details). Supplementary Fig. 2b illustrates co-clusters identified by the biLouvain algorithm in the bipartite network of Supplementary Fig. 2a.
We applied the biLouvain algorithm to identify co-clusters in each of the tissue-level bipartite networks. We identified a large number of co-clusters, ranging from 218 co-clusters for tissue brain-anterior cingulate cortex (BA24) to 314 co-clusters for adipose–subcutaneous. Across all bipartite networks, we observed that the majority of co-clusters had a small number of genes and phenotypes, on one hand, whereas a few co-clusters had a large number of genes and phenotypes, on the other hand (Fig. 2a, Supplementary Fig. 4 and Supplementary Data 4). Across the 48 tissues, the vast majority of co-clusters (8389/9472 = 88.6%) were found in only one tissue (Fig. 2b). Hence, the structure of the phenome-wide map involves hundreds of isolated tissue-specific subnetworks comprised of a small number of interrelated genes and phenotypes. Large co-clusters were also identified, although these are the exception rather than the norm. The complete list of genes and phenotypes per co-cluster in each tissue is provided in Supplementary Data 4.

a Number of genes and phenotypes per co-cluster identified by the biLouvain algorithm. Diamonds are proportional to the frequency of co-cluster size across all 48 tissues. Both axes are displayed on the log scale. b Number of unique co-clusters and how many times they appear in a given number of tissues. y axis is displayed in log scale.
Enrichment analysis of co-clusters with biological and functional gene classes
To demonstrate the functionality of the phenome-wide map, we tested if the identified biLouvain co-clusters were enriched with known biological and functional gene classes. We selected 183 co-clusters consisting of 10 genes or more, and performed enrichment analysis using PANTHER13,14 on four different annotation types: Biological process (2064 gene ontology (GO) terms), Cellular component (520 GO terms), Molecular function (532 GO terms), and 164 different Pathways. For each co-cluster and each annotation type, we selected the minimal P value of all Fisher overrepresentation tests, and plotted it against the expected minimal P value under the null hypothesis of no enrichment (see “Methods” for details). All four annotation types demonstrated significant enrichment (Fig. 3). We observed enrichment in GO terms related to (i) antibody-mediated immune response, upregulation response to biotic stimulus, glutathione metabolism, zymogen activation, downregulation of blood pressure, and cellular nitrogen compound metabolism in seven co-clusters in the Biological process annotation; (ii) outer surface of cytoplasmic membrane, and obsolete intracellular part in two co-clusters in the Cellular component annotation; (iii) signaling receptor binding, metallopeptidase activity, zinc ion binding, NADH-dependent glyoxylate reductase, and phosphatase activity in five co-clusters in the Molecular function annotation; and (iv) toll-like receptor signaling pathway, muscarinic acetylcholine receptor 2 and 4 signaling pathway, serine and glycine biosynthesis, and heterotrimeric G-protein signaling pathway-Gq alpha and Go alpha mediated pathway in six co-clusters in the Pathway annotation.

Each panel represents a PANTHER annotation type: (a) Biological process; (b) Cellular component; (c) Molecular function; and (d) Pathway. In each panel, the observed minimal P value across all GO terms is plotted against the expected minimal P value under the null hypothesis of no enrichment for all 183 co-clusters selected. Some plots (a–c) show a breakdown in the y axis to help display very small P values.
As an example, the most significant pathway detected by PANTHER is a toll-like receptor signaling pathway for co-cluster 111 comprised of hypothyroidism/myxedema and levothyroxine sodium medication, and genes TLR1, TLR6, and TLR10 in the cells—EBV-transformed lymphocytes tissue (Fig. 3a, Table 1, and Supplementary Fig. 5). Toll-like receptor 1 (TLR1), 6 (TLR6), and 10 (TLR10) genes are located in the same gene cluster on chromosome 4p14, and they play a fundamental role in pathogen recognition and activation of innate immunity15. Previous studies have shown that TLR1 and TLR10 are linked to Graves’ disease16 and Hashimoto’s disease17, which are clinical subtypes of autoimmune thyroid diseases. Furthermore, variants in CD22618, and RASGRP119 were found to be associated with autoimmune thyroid diseases and with thyroid preparations (H03A medication class, which comprises levothyroxine sodium)20.
In addition to the PANTHER gene set enrichment analysis, we identified seven co-clusters comprised of known relationships between genes and phenotypes in relevant tissues (Table 1), providing strong biological relevance. For example, the same gene GGCX appears in two co-clusters in related tissues, co-cluster 161 in heart-left ventricle and co-cluster 11 in Whole Blood (Table 1 and Supplementary Figs. 6 and 7). Gamma-glutamyl carboxylase (GGCX) encodes an integral membrane protein of the rough endoplasmic reticulum that carboxylates glutamate residues of vitamin K-dependent proteins to gamma carboxyl glutamate. Vitamin K-dependent proteins affect a number of physiologic processes including blood coagulation, inflammation, and prevention of vascular calcification21. Furthermore, a meta-analysis including the UKBB data identified an intronic variant in GGCX associated with coronary artery disease (CAD), with inclusion or exclusion of angina22. Taken together, these results suggest that the identified co-clusters contain relevant biological information highlighting functional links between genes and phenotypes.
Comparison between eQTLGen and GTEX
We compared the colocalizated loci found in GTEX with a larger gene expression level dataset. The eQTLGen Consortium identified cis-eQTLs in blood based on a meta-analysis of 37 different cohorts in up to 31,684 individuals23. We reran coloc2 using the same GWAS summary statistics (Neale and SAIGE) as before, but this time using cis-eQTL variants from the eQTLGen consortium (see “Methods”). All colocalization results can be found in Supplementary Data 5. As expected, more unique links between genes and phenotypes (2474) were discovered compared to links found using GTEX Whole Blood tissue (1755), most likely due to increased statistical power. However, we found 430 overlapping loci (a single colocalized locus can contain more than one gene) between GTEX and eQTLGen colocalization results in 251 distinct phenotypes (Supplementary Data 6). After running the biLouvain algorithm, we compared the clustering obtained in the colocalized loci from the eQTLGen dataset with those in the GTEX whole blood tissue. All co-clusters in the eQTLGen colocalization results are listed in Supplementary Data 7. Large co-clusters mostly contained the same phenotypes, although the colocalized genes could differ between eQTLGen and GTEX. One such large co-cluster in eQTLGen (co-cluster 42) includes many of the same phenotypes as in co-cluster 11 in GTEX Whole Blood (Table 1), and many genes are common to both co-clusters (DDAH2, FES, PSRC1, FAM177B, RPS12P26, MCL1).
Colocalization with disease case sample size larger than UKBB
We assessed the effect of using GWAS summary statistics with case sample size in various diseases larger than in UKBB. Increased case sample size as observed in consortia data should in theory generate more colocalized signals than using a population-based cohort such as UKBB. To this end, we evaluated three GWAS meta-analysis case–control datasets as generated by three different consortia: coronary artery disease (CAD) from CARDIoGRAMplusC4D24, schizophrenia from the Psychiatric Genomics Consortium (PGC)25, and type 2 diabetes (T2D) from DIAGRAM26. We reran coloc2 by combining each of these three GWAS datasets with the same GTEX cis-eQTL dataset used before (all 48 tissues). All colocalization results are found in Supplementary Data 8 for CAD, in Supplementary Data 9 for schizophrenia, and in Supplementary Data 10 for T2D.
Regarding CAD and T2D, many colocalized loci discovered using either Neale or SAIGE datasets were replicated using the GWAS meta-analysis consortia data, even though CARDIoGRAMplusC4D24 defined CAD more comprehensively as either myocardial infarction, acute coronary syndrome, chronic stable angina or coronary stenosis >50% (~61,000 CAD cases) while in Neale and SAIGE, such a composite diagnosis is not available, only subphenotypes such as heart attack/myocardial infarction (Neale 20002_1075) or coronary atherosclerosis (SAIGE PheCode 411.4). In DIAGRAM26, the number of T2D cases (~55,000 cases, excluding the UKBB cohort) is about three times the number of cases in Neale (16,183 cases for 2443—Diabetes diagnosed by a doctor) or SAIGE (18,945 cases for PheCode 250.2—Type 2 diabetes), which enhanced the statistical power to detect more colocalized signals. By contrast, using the GWAS summary statistics dataset from the PGC25 largely increased the number of colocalized signals compared to using either Neale or SAIGE datasets. No colocalized signal was found with Neale (337 cases for 20002_1289—Self-reported schizophrenia) and only a few using SAIGE (571 cases for PheCode 295.1—Schizophrenia). The PGC summary statistics (which include ~37,000 schizophrenia cases, about 65 times the number of cases using SAIGE) uncovered many colocalized loci undetected by using either Neale or SAIGE: for example, genes BNIP3L, CNTN4, THOC7, TRPC4, ZNF823, CLCN3, PAX6 were prioritized in the context of synaptic location and function from genome-wide enrichment tests in the latest PGC schizophrenia meta-analysis27.
Discussion
In this study, we have constructed a tissue-level phenome-wide network map, called biPheMap, of colocalized genes and phenotypes using a bipartite network and biLouvain clustering approach on 1411 phenotypes and eQTL data from 48 tissues from the GTEx project. In the phenome-wide map, we observed the following: (1) the majority of colocalized gene and phenotype links are observed in a single tissue, implying that tissue-specific gene regulatory mechanisms drives phenotypic variation; (2) the majority of co-clusters are comprised of a small number of gene and phenotype links; (3) specific co-clusters are enriched with functional gene set annotations; (4) specific co-clusters are identified with biologically relevant gene, phenotype and tissue functions.
While most network analyses have focused on unipartite networks, this study used the less familiar bipartite approach. Many such bipartite networks have been studied in different contexts: actor-movie network in cinema industry, author-scientific paper networks in academia, pollinator-plant in ecological networks, etc., but their topological features and related metrics are unique and different from their more classical unipartite counterpart. A simpler analysis could have been proposed by projecting the bipartite network into two unipartite networks to produce a gene-gene network and a phenotype-phenotype network. However, this projection method entails a loss of information since the original links between genes and phenotypes are no longer available. Such a projection approach was employed in ref. 28 to create a disease-disease network where more than 500 diagnosis codes were linked on the basis of shared variant associations.
An important feature of the phenome-wide map is the exploration and discovery of co-clusters of related genes and phenotypes. So far, few community detection algorithms in bipartite networks could be run in a reasonable amount of time. One fast and precise algorithm is the biLouvain algorithm10 which maximizes bipartite modularity, an extension of the modularity measure found in unipartite network clustering algorithms. The biLouvain creators compared their algorithm against five state-of-the-art bipartite community detection algorithms. In their evaluation, they conclude that biLouvain is always close or equal to the maximum bipartite modularity achieved by any of the five methods while being consistently one of the fastest algorithms for the large real-world datasets tested.
Table 1 displays various examples of gene-phenotype co-clusters confirming known genetic associations and also suggesting unsuspected etiological links between phenotypes. We note that this represents only a small fraction of interesting co-clusters we chose to highlight in our paper. For example, many epidemiological and genetic studies have suggested shared loci between migraine and CAD, and one study identified gene PHACTR1 as the strongest shared locus between the two disorders29. However, some co-clusters might also consist of phenotypes being observed as a consequence of another phenotype. For example, we observed lipid-lowering medications in the same co-cluster as lipid disorders.
There are numerous limitations to our study that deserve mention. First, some phenotypes are highly correlated in UKBB, and therefore, colocalized signals were sometimes redundant in our phenome-wide map. However, this redundancy provided some internal replication of the strongest colocalized signals between Neale and SAIGE association datasets, while sometimes complementing signals found in one dataset but not in the other due to different criteria of case and control definition (ICD-10 codes versus PheCodes)30. The PheCode scheme, as utilized in SAIGE genetic association scans, applies stringent exclusion criteria to prevent contamination by cases in the control group, which could decrease the statistical power of association tests. Second, we applied stringent quality control in our phenotype selection to avoid reporting false colocalized loci. This came at the expense of missing some loci, especially if the leading associated variant in a locus is rare (minor allele frequency <0.1%). Third, the coloc2 method assumes that at most one causal variant affects both the gene expression and the trait association at the locus under consideration. In presence of allelic heterogeneity (more than one causal variant), the false discovery rate is maintained but coloc2 might suffer a loss of power31. In contrast, a method such as the regulatory trait concordance (RTC) score assumes that a GWAS variant and eQTL variant located in the same genomic region delimited by recombination hotspots tag the same functional variant. In ref. 32, the authors proposed to compute a probability of shared functional effect, called P(shared), based on the regulatory trait concordance (RTC) score, and to compare this probability with PPH4 as generated by coloc. Interestingly, their simulation study suggests that, knowing the GWAS and eQTL P values for every variant in the region, coloc is a better choice as it uses all the information in the locus. Fourth, the literature cited to support the links between genes and phenotypes of co-clusters displayed in Table 1 relies heavily on genetic associations found in the GWAS summary statistics of European ancestry participants in UKBB. Incorporation of findings from diverse ancestry populations will be necessary to yield more genetic association phenotypes. Fifth, we used the bipartite network approach over the more common unipartite approach, which limited the set of tools to analyze our results. Fortunately, the bipartite network and its characteristics are gaining more attention in the network literature, and future methodological developments will expand the range of tools and analyses that could be performed in this type of networks. Sixth, the first version of biPheMap currently utilizes Neale v1 GWAS summary statistics and GTEX v7 data. Recently, more GWAS summary statistic data (e.g., Pan-UK Biobank33) and more recent versions of GTEX (e.g., GTEX v834) have been published. Furthermore, additional large-scale phenome-wide association datasets originating from exome sequencing data (e.g., genebass35) have become available. We expect subsequent versions of biPheMap will incorporate these resources as they become publicly available over time. Seventh, while we recognize that the GWAS of many binary (case–control) phenotypes in UKBB are underpowered due to the reduced number of cases for many of them -hence reducing the power to detect colocalization loci- our study was more focused at identifying the strongest shared links between genes and phenotypes in a single population-based cohort. Nonetheless, to increase the number of colocalization loci, an alternative route is to include GWAS summary statistics from other consortia (which usually include more cases than UKBB), in addition to a larger gene expression level dataset, such as the whole blood eQTLGen consortium23. Our colocalization analyses using larger case–control GWAS and/or eQTL datasets independently confirmed many colocalized loci found in the biPheMap while uncovering additional ones that were missed due to the lower number of cases in Neale and SAIGE for CAD, schizophrenia, and T2D. We note that although the eQTLGen whole blood sample size is much larger than the GTEx whole blood sample size, the identified cis-eQTLs are based on a meta-analysis of 37 different cohorts and therefore heterogenous cell type-composition effects might persist. Finally, some tissues in the GTEx project involve an in vitro manipulation such as EBV-transformed lymphocytes. We identified co-clusters and significant pathways within EBV-transformed lymphocytes that warrant cautious interpretation.
In this study, the intention was to report colocalization signals and identify clusters of phenotypes sharing colocalized genes at the tissue level. One possible research avenue could exploit the sharing of eQTLs among biologically related tissues to improve statistical power to detect colocalized genes at the tissue level. The recently published method JTI36 leverages this abundance of shared eQTLs to improve prediction of gene expression levels. This method relies on prediction models of gene expression, and has to be distinguished from colocalization methods. This approach of combining predicted expression data with a colocalization method has been recently proposed37.
In conclusion, we showed that the phenome-wide map can be a useful resource to understand gene, phenotype, and tissue links across a wide spectrum of biological classes and diseases. We expect that further interrogation of the phenome-wide map will yield more biological and clinical discoveries.
Methods
Datasets
This study uses two resources: (a) the UK Biobank (UKBB) project; and (b) the Genotype-Tissue Expression (GTEx) project. The UKBB project is a prospective EHR-linked cohort with deep genetic and rich phenotypic data collected on ~500,000 middle-aged individuals (aged between 40 and 69 years old) recruited from across the United Kingdom1. Ethics approval for the UK Biobank project was obtained from the North West Centre for Research Ethics Committee (11/NW/0382) and all participants provided written informed consent. The GTEx project is a resource database and associated multi-tissue bank aimed at studying the relationship between genetic variation and gene expression in different human tissues5,6,7.
Colocalization method
We integrated multiple association datasets to assess whether two association signals, one from a genome-wide association study (GWAS) on a phenotype, and the other from expression quantitative trait locus (eQTL) analysis in a tissue, overlap in such a matter that they are consistent with a shared causal gene. This approach, referred to as colocalization, was conducted using coloc211, an enhancement of the previously published method coloc38. The coloc method is a Bayesian approach which computes the posterior probability that a genetic variant is both associated with the phenotype and the gene expression level in the tissue. Our coloc2 implementation improves over the original coloc method by (1) aligning eQTL and GWAS summary statistics in each eQTL cis-region; (2) estimating the likelihood of mixture proportions of five hypotheses (H0: no association, H1: GWAS associated only, H2: eQTL associated only, H3: both associated but not colocalized, H4: both associated and colocalized) from genome-wide data. These proportions serve as priors in the empirical Bayesian calculation of the posterior probability of colocalization at each locus. Asymptotic Bayes factors are averaged across three different values of the prior variance term (0.01, 0.1, and 0.5)39. We defined a colocalized signal using a posterior probability for H4 (PPH4) ≥ 0.80, as described previously38.
GWAS and eQTL summary statistics
coloc2 requires both GWAS summary data and eQTL association summary data. For GWAS data, we used two sets from the UKBB project. The first set of results are GWAS association test statistics publicly available from the Neale lab (Round 1 in 2419 phenotypes). We selected variants with minor allele frequency (MAF) > 0.1%. More details on the data quality control and the full list of phenotypes can be found at www.nealelab.is/uk-biobank. We further used a second set of UKBB GWAS association statistics computed by the SAIGE testing method40. In total, 1403 case–control phenotypes (PheCodes) were available. We selected variants with MAF > 0.1%. Full datasets and list of PheCodes can be downloaded at https://www.leelabsg.org/resources. For eQTL association signals, we used data from Analysis V7 of the GTEx project available at https://www.gtexportal.org/home/datasets. We restricted our study to the list of 48 tissues (from 620 donors) having a sample size of at least 80. Cis-eQTLs with MAF > 1% were considered as input for coloc2 (detailed in https://storage.googleapis.com/gtex-public-data/Portal_Analysis_Methods_v7_09052017.pdf available on the GTEx Portal).
Before running coloc2 on the Neale GWAS data, we performed stringent quality control. First, we removed results from phenotypes related to cause of death (since these phenotypes generally had very low number of cases), and we also removed case–control phenotypes with less than 1250 cases (or controls), except phenotypes showing prior gene/locus association as reported in the NHGRI-EBI GWAS catalog (https://www.ebi.ac.uk/gwas/)41. The rationale for excluding phenotypes with less than 1250 cases (or controls) is based on the recommendation by Neale to keep only variants with at least 25 minor alleles in the sample of cases (or controls), in order to avoid inflation in association test statistics due to extreme case–control ratio imbalance and ensuring reliable P value computation as detailed in http://www.nealelab.is/blog/2017/9/11/details-and-considerations-of-the-uk-biobank-gwas. With the Neale GWAS data, we retained coloc2 results for 496 continuous and binary phenotypes. In the same vein, before running coloc2 with SAIGE association data, we excluded case–control phenotypes with less than 200 cases, as recommended by the authors40. With SAIGE data, we generated coloc2 results for 915 case–control (PheCode) phenotypes.
Construction and descriptive statistics of bipartite networks
To construct the phenome-wide map of genes and phenotypes using the colocalized data of 9151 genes and 1411 phenotypes, we created a bipartite network for each tissue. In brief, a bipartite network, also called a two-mode network, is a network in which nodes of one mode (i.e., type) are only connected to nodes of the other mode (for a review, see refs. 8,9). Associations between phenotypes and genes are indicated by links or edges between them.
To characterize and compare colocalization results across tissues, we computed descriptive statistics adapted to bipartite networks. We computed the average degree of colocalized genes and phenotypes in each tissue. The degree of a given gene (respectively, phenotype) is simply the number of unique phenotypes (respectively, genes) connected to it in the network, the average being taken over the total number of genes (respectively, phenotypes)12.
biLouvain clustering algorithm
For each tissue, it is expected that the bipartite network of coloc2 results will tend to cluster in small groups of related phenotypes with their causally associated genes. To uncover clustering within each network, we applied the biLouvain clustering algorithm, an extension of the unipartite Louvain clustering algorithm. This algorithm identifies co-clusters, also called communities, of non-overlapping genes and phenotypes through maximization of a modularity score adapted to bipartite networks (see ref. 10 for details). To decrease the overall computational time of the algorithm, we opted for the Fuse preprocessing step before running the co-clustering step per se. In rare occasions, this fusing step incurred some loss in clustering quality, hence resulting in missing edges in the co-cluster (e.g., Supplementary Fig. 5).
Comparison between eQTLGen and GTEX
The eQTLGen consortium identified cis-eQTLs with MAF > 1% based on a meta-analysis of 37 different cohorts in up to 31,684 individuals23. Every SNP-gene combination within a distance <1 Mb from the center of the gene and tested in at least two cohorts were included. We ran coloc2 using as input the same GWAS summary statistics (Neale and SAIGE) and cis-eQTL variants from the eQTLGen consortium. An overlapping locus between GTEX and eQTLGen was reported if one signal found in one set of colocalization results overlapped within 2 Mb in the other set of results (as a consequence, many genes may appear in the same locus). The biLouvain algorithm was run in eQTLGen colocalization results using the same options as in GTEX results.
Colocalization with disease case sample size larger than UKBB
In order to assess the effect of using GWAS summary statistics with case sample size in various diseases larger than in UKBB, we downloaded three GWAS meta-analysis case–control datasets as generated by three different consortia: coronary artery disease from CARDIoGRAMplusC4D (60,801 cases and 123,504 controls)24, schizophrenia from the Psychiatric Genomics Consortium (36,989 cases and 113,075 controls)25, and type 2 diabetes from DIAGRAM (55,005 cases and 400,308 controls, after excluding UKBB participants)26. We ran coloc2 by combining each of these three GWAS datasets with the same GTEX cis-eQTL dataset used before (all 48 tissues).
PANTHER enrichment analysis
To test if biLouvain co-clusters were enriched with some functional gene classes, we selected 183 co-clusters consisting of 10 genes or more, and input them into the online PANTHER enrichment analysis tools13,14. We applied Fisher overrepresentation tests on four different annotation types: Biological process (2064 Gene Ontology (GO) terms), Cellular component (520 GO terms), Molecular function (532 GO terms), and 164 different Pathways. For each co-cluster and each annotation type, we took the minimal P value of all Fisher tests, and plotted it against the expected minimal P value under the null hypothesis of no enrichment. We assumed that P values within each annotation type are independently distributed uniformly over the interval (0,1), which represents a conservative approach. Note that the minimal P value of n independent P values from a Uniform (0,1) is not uniformly distributed under the null: its cumulative density function is instead given by
In each panel of Fig. 3, we plotted a straight line with slope equal to 1, which crosses the y axis at x = (observed 1st quartile – expected 1st quartile) using the above expected cumulative density function. Gene enrichment was deemed significant if the minimal P value was less than 0.05/(183 × 4) = 6.8 × 10−5.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
All colocalized results analyzed in this study are available through a R Shiny app called biPheMap at https://rstudio-connect.hpc.mssm.edu/biPheMap/. This research has been conducted using the UK Biobank Resource under Application Number “16218”. UK Biobank data is available to researchers upon approval of an application form at https://www.ukbiobank.ac.uk/. The GTEx Analysis V7 dataset can be freely downloaded at https://www.gtexportal.org/home/datasets. At the time of our colocalization analysis, we utilized Round 1 of Benjamin Neale’s lab GWAS summary statistics in the UK Biobank. The Round 1 results are no longer accessible and has since been replaced by a more recent Round 2, which can be freely downloaded at http://www.nealelab.is/uk-biobank. Seunggeun Lee’s lab GWAS summary statistics in the UK Biobank using SAIGE can be freely downloaded at https://www.leelabsg.org/resources. The full cis-eQTL summary statistics from the eQTLGen Consortium are publicly available at https://www.eqtlgen.org/cis-eqtls.html. GWAS summary statistics from CARDIoGRAMplusC4D (http://www.cardiogramplusc4d.org/data-downloads/), the Psychiatric Genomics Consortium (https://pgc.unc.edu/for-researchers/download-results/), and DIAGRAM (https://diagram-consortium.org/downloads.html) are publicly available and can be freely downloaded.
Code availability
R codes to run coloc2 are available at https://github.com/Stahl-Lab-MSSM. The biLouvain algorithm may be downloaded and installed by following instructions at https://github.com/paolapesantez/biLouvain. The PANTHER classification system is available at http://www.pantherdb.org/. R software and packages (https://cran.r-project.org/) were used to analyze the data and generate the figures, except Fig. 1, which contains graphical elements from a free version of Canva (https://www.canva.com/en/).
References
Bycroft, C. et al. The UK Biobank resource with deep phenotyping and genomic data. Nature 562, 203–209 (2018).
Cortes, A. et al. Bayesian analysis of genetic association across tree-structured routine healthcare data in the UK Biobank. Nat. Genet. 49, 1311–1318 (2017).
Cortes, A., Albers, P. K., Dendrou, C. A., Fugger, L. & McVean, G. Identifying cross-disease components of genetic risk across hospital data in the UK Biobank. Nat. Genet. 52, 126–134 (2020).
Bush, W. S., Oetjens, M. T. & Crawford, D. C. Unravelling the human genome–phenome relationship using phenome-wide association studies. Nat. Rev. Genet. 17, 129–145 (2016).
Lonsdale, J. et al. The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 45, 580–585 (2013).
GTEx Consortium. et al. Using an atlas of gene regulation across 44 human tissues to inform complex disease- and trait-associated variation. Nat. Genet. 50, 956–967 (2018).
The GTEx Consortium. The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science 369, 1318–1330 (2020).
Pavlopoulos, G. A. et al. Bipartite graphs in systems biology and medicine: a survey of methods and applications. GigaScience 7, 1–31 (2018).
Pavlopoulos, G. A. et al. Corrigendum to: Bipartite graphs in systems biology and medicine: a survey of methods and applications. GigaScience 9, giz130 (2020).
Pesantez-Cabrera, P. & Kalyanaraman, A. Efficient detection of communities in biological bipartite networks. IEEE/ACM Trans. Comput. Biol. Bioinform. 16, 258–271 (2019).
Dobbyn, A. et al. Landscape of conditional eQTL in dorsolateral prefrontal cortex and co-localization with schizophrenia GWAS. Am. J. Hum. Genet. 102, 1169–1184 (2018).
Latapy, M., Magnien, C. & Vecchio, N. D. Basic notions for the analysis of large two-mode networks. Soc. Netw. 30, 31–48 (2008).
Mi, H., Muruganujan, A., Ebert, D., Huang, X. & Thomas, P. D. PANTHER version 14: more genomes, a new PANTHER GO-slim and improvements in enrichment analysis tools. Nucleic Acids Res. 47, D419–D426 (2019).
Mi, H. et al. Protocol Update for large-scale genome and gene function analysis with the PANTHER classification system (v.14.0). Nat. Protoc. 14, 703–721 (2019).
Kawasaki, T. & Kawai, T. Toll-like receptor signaling pathways. Front. Immunol. 5, 461 (2014).
Xiao, W. et al. Polymorphisms in TLR1, TLR6 and TLR10 genes and the risk of Graves’ disease. Autoimmunity 48, 13–18 (2015).
Li, M. et al. IRAK2 and TLR10 confer risk of Hashimoto’s disease: a genetic association study based on the Han Chinese population. J. Hum. Genet. 64, 617–623 (2019).
Saevarsdottir, S. et al. FLT3 stop mutation increases FLT3 ligand level and risk of autoimmune thyroid disease. Nature 584, 619–623 (2020).
Kichaev, G. et al. Leveraging polygenic functional enrichment to improve GWAS power. Am. J. Hum. Genet. 104, 65–75 (2019).
Wu, Y. et al. Genome-wide association study of medication-use and associated disease in the UK Biobank. Nat. Commun. 10, 1891 (2019).
Lees, J. S., Chapman, F. A., Witham, M. D., Jardine, A. G. & Mark, P. B. Vitamin K status, supplementation and vascular disease: a systematic review and meta-analysis. Heart 105, 938–945 (2019).
Nelson, C. P. et al. Association analyses based on false discovery rate implicate new loci for coronary artery disease. Nat. Genet. 49, 1385–1391 (2017).
Võsa, U. et al. Large-scale cis- and trans-eQTL analyses identify thousands of genetic loci and polygenic scores that regulate blood gene expression. Nat. Genet. 53, 1300–1310 (2021).
Nikpay, M. et al. A comprehensive 1000 Genomes–based genome-wide association meta-analysis of coronary artery disease. Nat. Genet. 47, 1121–1130 (2015).
Ripke, S. et al. Biological insights from 108 schizophrenia-associated genetic loci. Nature 511, 421–427 (2014).
Mahajan, A. et al. Fine-mapping type 2 diabetes loci to single-variant resolution using high-density imputation and islet-specific epigenome maps. Nat. Genet. 50, 1505–1513 (2018).
Trubetskoy, V. et al. Mapping genomic loci implicates genes and synaptic biology in schizophrenia. Nature 604, 502–508 (2022).
Verma, A. et al. Human-disease phenotype map derived from PheWAS across 38,682 individuals. Am. J. Hum. Genet. 104, 55–64 (2019).
Winsvold, B. S. et al. Shared genetic risk between migraine and coronary artery disease: a genome-wide analysis of common variants. PLoS ONE 12, e0185663 (2017).
Wu, P. et al. Mapping ICD-10 and ICD-10-CM codes to phecodes: workflow development and initial evaluation. JMIR Med. Inform. 7, e14325 (2019).
Hukku, A. et al. Probabilistic colocalization of genetic variants from complex and molecular traits: promise and limitations. Am. J. Hum. Genet. 108, 25–35 (2021).
Ongen, H. et al. Estimating the causal tissues for complex traits and diseases. Nat. Genet. 49, 1676–1683 (2017).
Pan-UKB team. https://pan.ukbb.broadinstitute.org. (2020).
GTEx Portal. https://www.gtexportal.org/home/datasets.
Karczewski, K. J. et al. Systematic single-variant and gene-based association testing of thousands of phenotypes in 394,841 UK Biobank exomes. Cell Genomics (2022).
Zhou, D. et al. A unified framework for joint-tissue transcriptome-wide association and Mendelian randomization analysis. Nat. Genet. 52, 1239–1246 (2020).
Pividori, M. et al. PhenomeXcan: mapping the genome to the phenome through the transcriptome. Sci. Adv. 6, eaba2083 (2020).
Giambartolomei, C. et al. Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genet. 10, e1004383 (2014).
Pickrell, J. K. et al. Detection and interpretation of shared genetic influences on 42 human traits. Nat. Genet. 48, 709–717 (2016).
Zhou, W. et al. Efficiently controlling for case-control imbalance and sample relatedness in large-scale genetic association studies. Nat. Genet. 50, 1335–1341 (2018).
Buniello, A. et al. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 47, D1005–D1012 (2019).
Acknowledgements
We thank Dr. Benjamin Neale’s and Dr. Seunggeun Lee’s group for generously sharing GWAS summary statistics in the UK Biobank. We would also like to thank the donors and their families for making organ and tissue donations to the GTEx project. This work was supported in part through the computational resources and staff expertise provided by Scientific Computing at the Icahn School of Medicine at Mount Sinai. R.D. is supported by R35GM124836 from the National Institute of General Medical Sciences of the National Institutes of Health, R01HL139865 and R01HL155915 from the National Heart, Lung, and Blood Institute of the National Institutes of Health. D.M.J. is supported by T32HL00782 from the National Heart, Lung, and Blood Institute of the National Institutes of Health. I.S.F. is supported by T32GM007280 from the National Institute of General Medical Sciences of the National Institutes of Health. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Author information
Authors and Affiliations
Contributions
G.R. and R.D. conceived and planned the study. HHW managed and created the pipeline to generate colocalization results. G.R., D.M.J., H.H.W., I.S.F., Ai.D., Am.D., M.V., and R.D. analyzed and/or interpreted the colocalization results. D.M.J. performed the PANTHER enrichment analysis. S.B. implemented and deployed the biPheMap R shiny app. G.R. created all figures and tables, with assistance from I.S.F., Ai.D., and S.B. G.R. and R.D. drafted and revised the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
R.D. received grants from AstraZeneca, grants and nonfinancial support from Goldfinch Bio, is a scientific co-founder, equity holder and consultant for Pensieve Health (pending), and is a consultant for Variant Bio, all not related to this work. The remaining authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks Adriaan van der Graaf and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editors: Chiea Chuen Khor and George Inglis.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Rocheleau, G., Forrest, I.S., Duffy, Á. et al. A tissue-level phenome-wide network map of colocalized genes and phenotypes in the UK Biobank. Commun Biol 5, 849 (2022). https://doi.org/10.1038/s42003-022-03820-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-022-03820-z
- Springer Nature Limited