
Abstract
Intraocular pressure (IOP) is an important factor in glaucoma development, which involves aqueous humor (AH) dynamics, with inflow from the ciliary body and outflow through the trabecular meshwork (TM). IOP has a circadian rhythm entrained by sympathetic noradrenaline (NE) or adrenal glucocorticoids (GCs). Herein, we investigated the involvement of GC/NE in AH outflow. Pharmacological prevention of inflow/outflow in mice indicated a diurnal outflow increase, which was related to TM phagocytosis. NE showed a non-self-sustained inhibition in phagocytosis of immortalized human TM cells, but not GC. The pharmacological and reverse genetic approaches identified β1-adrenergic receptor (AR)-mediated exchange proteins directly activated by cyclic adenosine monophosphate (EPAC)-SHIP1 signal activation by ablation of phosphatidylinositol triphosphate, regulating phagocytic cup formation. Furthermore, we revealed the phagocytosis involvement in the β1-AR-EPAC-SHIP1-mediated nocturnal IOP rise in mice. These suggest that TM phagocytosis suppression by NE can regulate IOP rhythm through AH outflow. This discovery may aid glaucoma management.
Similar content being viewed by others
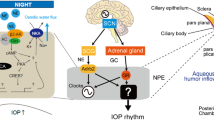


Introduction
In most organisms, multiple physiological processes are controlled by circadian rhythms, which last approximately 24 h. The suprachiasmatic nucleus (SCN) in the anterior hypothalamus acts as a circadian pacemaker in mammals, which senses optical information from the retina, to manage daily body rhythms1. The SCN synchronizes most peripheral tissues and cells through various complex pathways, involving mainly the autonomic nervous system and endocrine signals2. Norepinephrine (noradrenaline, NE) released from the superior cervical ganglion (SCG), a part of the sympathetic nervous system, transmits circadian timing signals to the ciliary body of the eye to regulate pupil size, among other functions3. Furthermore, for most peripheral tissues, glucocorticoids (GCs) secreted from the adrenal glands via the hypothalamus-pituitary-adrenal axis-mediated SCN act as strong endocrine timing signals because GC receptors (GRs) are expressed in most peripheral cell types4.
Glaucoma is a leading cause of blindness in elderly people; however, no effective cure exists. Abnormal intraocular pressure (IOP) inside the eye, for example, high IOP, contributes to glaucoma development and progression characterized by vision loss5. IOP is balanced by the aqueous humor (AH) and has a circadian rhythm. In humans, nocturnal IOP increases irrespective of posture6. IOP is also elevated at night in nocturnal or diurnal animals7, which is controlled by the SCN in mice8. Nocturnal IOP is also elevated in patients with glaucoma6,9, and the IOP rhythm undergoes phase shifts in patients with primary open-angle glaucoma (POAG) and normal-tension glaucoma (NTG)9. NTG was also reported to involve an abnormal IOP rhythm10. Aging desynchronizes the IOP rhythm in older healthy subjects11. Interestingly, IOP rhythm is disrupted in night-shift workers12. A recent study reported that a disrupted circadian IOP rhythm causes optic nerve damage and increases the risk of glaucoma11. Thus, regulation of IOP rhythm, especially during the night, is central to glaucoma management, and the circadian mechanism in AH dynamics is important for glaucoma therapy. We have previously demonstrated the dual pathway by which both NE and GCs transmit timing information to the eye to form the IOP rhythm in mice13. However, the molecular mechanisms of this axis remain unknown.
In AH dynamics, the non-pigmented epithelial cell (NPE) of the ciliary body participates in AH production14. In contrast, the trabecular outflow pathway is responsible for homeostatic regulation of IOP and is regulated by the coordinated generation of AH outflow resistance mediated by the constituent cells of the trabecular meshwork (TM) and Schlemm’s canal (SC)15,16, and partially, uveoscleral outflow is known to be involved in human IOP rhythm17. Most of this resistance is believed to be generated in the inner wall region comprising the juxtacanalicular tissue and the inner wall endothelium of the SC and its pores (small openings in the giant vacuoles)18. The mechanisms that regulate aqueous outflow resistance in normal and glaucomatous eyes remain unclear, and a few newly approved medications target this site of resistance. TM phagocytosis can decrease particulate material and debris from AH, attenuating outflow resistance, and contribute to IOP reduction15. Phagocytosis is thought to play an important role in the normal functioning of the outflow pathway by keeping the drainage channels free of debris. Conversely, actin remodeling, such as fragmentation or polymerization, also regulates AH outflow, and the actin cytoskeleton of TM cells is a therapeutic target in glaucoma patients. Small GTPase Rho-associated coiled-coil-containing protein kinase (ROCK) inhibitors lower IOP, not only by relaxation of the TM by disruption of actin stress fibers, but also by the activation of phagocytosis19,20,21. However, since all phagocytic processes are driven by a finely controlled rearrangement of the actin cytoskeleton22,23, it has been difficult to verify the effects of these two factors separately.
Long-term dexamethasone (Dex) treatment is known to decrease TM phagocytosis and increase actin fiber stress in the human eye, as well as primary TM cells24,25, leading to increased AH outflow resistance, and ultimately, glaucoma development. In addition, NE suppresses wound macrophage phagocytic efficiency through α- and β-adrenergic receptor (AR)-dependent pathways26, and can promote actin polymerization in the retina27. However, the detailed effect of NE on TM phagocytosis has not yet been elucidated. Hence, this study aimed to uncover the effects of GC/NE on trabecular phagocytosis and its molecular mechanisms. We addressed the effect of AH outflow on diurnal IOP changes in mice and found the involvement of TM phagocytosis in the AH outflow. In addition, the effect of NE/GC exposure on phagocytic activity was monitored in real time using immortalized human TM cells. Pharmacological approaches and RNA interference have identified AR-related pathways involved in phagocytosis regulation. Furthermore, the role of the identified regulatory pathway in IOP rhythm regulation was assessed using pharmacological instillation in mice.
Results
AH outflow increased in the daytime in mice
To address the effects of ciliary AH-production on nocturnal increase in IOP, we injected the Na+/K+ATPase antagonist, ouabain, into the anterior chamber of the mouse eye at zeitgeber time (ZT) 10 (Fig. 1a). Ouabain allowed a nocturnal IOP increase (Fig. 1b), but prevented IOP increase in individual data (p < 0.01) (Fig. 1c), indicating the effect of AH outflow. Since AH drainage suppression from the TM by microbeads intraocularly (i.o.) injection elevates IOP in rats28, to identify the role of AH outflow on mouse IOP rhythm, microbeads were injected into the anterior chamber of the eye to mildly suppress the AH outflow (Fig. 1d), as previously reported28. After 2 weeks, IOP in bead-injected mice at ZT6 increased up to the nocturnal level (Fig. 1e), consistent with previous reports28,29. Day-night differences in individual IOP were arrested by bead injection (Fig. 1f), indicating a daytime decrease in IOP due to AH outflow. To confirm diurnal changes in AH drainage in the TM, we administered small fluorescent particles to the anterior chamber at ZT0 and ZT12, and subsequently observed the anterior eye extracted at ZT6 and ZT18, respectively, and measured the fluorescence intensity (Fig. 1g), where we assumed that the particles were taken into the SC or passed through the TM (Fig. 1h). The fluorescence intensity at ZT6 was significantly stronger than that at ZT18 (Fig. 1h, i), consistent with a previous rabbit AH outflow study30. Thus, it seems plausible that AH outflow through TM/SC may be involved in the daytime decrease in IOP in mice.

a–c Effect of Na+/K+ATPase antagonist, ouabain, treatment on nocturnal intraocular pressure (IOP) increase. a Ouabain was injected to anterior chamber of mouse eye at ZT10, and IOP were measured before injection and 5 h after injection (ZT15). b Ouabain allowed a nocturnal IOP increase (paired t-test, *p < 0.05, ***p < 0.001). Data are presented as box-and-whisker plots with individual data (n = 11). Variability is shown in the box from the 25th to 75th percentiles, and min to max (whiskers). c Ouabain individually prevents a nocturnal IOP increase (t-test, **p < 0.01). Data are presented as scatter plots with mean ± SEM (n = 11). d–f Diurnal changes of IOP (ZT6 and ZT15) were measured 2 weeks after intraocular injection of beads. e IOP at ZT6 was increased up to nocturnal level (paired t-test, **p < 0.01). Data are presented as box-and-whisker plots with individual data (n = 7). f Day-night differences in individual IOP were arrested by bead injection. Data are presented as scatter dot plots with mean ± SEM (n = 7). g–i Fluorescent particles were injected into the anterior chamber of the eye at ZT0 or ZT12, and after 6 h; the fluorescence of extracted cornea was observed with a microscope (Ex: 480 nm, Em: >510 nm) and quantified by microplate reader (Em: 525 nm). h Diurnal change of the particles at the edge of cornea (speculating the TM) was detected (arrowhead). i Fluorescent intensity at ZT6 was significantly higher than that at ZT18 (t-test, *p < 0.05). Data are presented as bar graphs (mean ± SEM) with scatter dot plots (n = 8). ZT Zeitgeber time.
TM phagocytosis is involved in diurnal IOP reduction
Phagocytosis and remodeling of the actin cytoskeleton are related to AH outflow15,19,20,21. Dynasore, an inhibitor of dynamin GTPase activity, but not other small GTPases, has been widely studied in clathrin-mediated endocytosis and phagocytosis, also in HTMC31 (Fig. 2a). Dynamin is thought to interact with actin filaments when the edges of the phagocytic cup close32. Latrunculin A and cytochalasin D, drugs that interfere with actin–myosin contraction, also lower IOP33,34 and suppress TM phagocytosis33,34,35 (Fig. 2a). To examine their involvement in daytime IOP reduction, we first administered mice dynasore, cytochalasin D, latrunculin A, and RKI1447 (ROCK inhibitor; phagocytosis enhancer and cytoskeleton disruptor [Fig. 2a]) at ZT4 and measured IOP at ZT9 (Fig. 2b). Instillation with dynasore significantly increased IOP, whereas latrunculin A decreased IOP (Fig. 2c). Individual data also showed that dynasore enhanced diurnal IOP (Fig. 2d), indicating that the daytime activation of TM phagocytosis may suppress IOP, which can be canceled by depolymerization of actin fibers. In contrast, instillation with cytochalasin D, latrunculin A, and RKI1447 at ZT10 arrested the nocturnal IOP increase, especially latrunculin A significantly suppressed IOP, but not dynasore (Fig. 2e, f). At the individual level, these drugs, regardless of their effects on phagocytosis, also suppressed the nocturnal IOP rise (Fig. 2g), indicating the role of TM actin polymerization in nocturnal IOP rise. This IOP suppression limited to nighttime by ROCK inhibitor was similar to a previous report demonstrating that RhoA blocking by AAV prevents nocturnal IOP elevation in rats36. Taken together, these results suggest that AH outflow regulation by the cytoskeleton seems to be night-limited or time-independent, while TM phagocytosis may mediate the daytime increase in AH outflow.

a–d Effect of phagocytosis regulatory drugs (a) on diurnal IOP increase in mice. b Mice were administered an instillation of Dynamin-mediated phagocytosis inhibitor (Dynasore; 1 mM), inhibitors of actin polymerization and phagocytosis (cytochalasin D; 100 µM, latrunculin A; 100 µM), and cytoskeleton disruptor/phagocytosis activator (ROCK inhibitor RKI1447; 1 mM) at ZT4 and IOP were measured at ZT9. c Instillation with Dynasore significantly increased IOP, whereas latrunculin A decreased IOP (paired t-test, *p < 0.05, ***p < 0.001). Data are presented as box-and-whisker plots with individual data (n = 6–18). d Individual data also showed that Dynasore enhanced diurnal IOP (**p < 0.01, one-way ANOVA [p < 0.001], Dunnett’s multiple comparison test). Data are presented as scatter plots with mean ± SEM (n = 6–18). e–g Effect of phagocytosis-regulatory drugs on nocturnal IOP increase in mice. Mice were administered an instillation of drugs at ZT10, and IOP was measured at ZT15. f Instillation of cytochalasin D, latrunculin A, and RKI1447 arrested the nocturnal IOP increase, especially latrunculin A significantly suppressed IOP (paired t-test, *p < 0.05, ***p < 0.001). Data are presented as box-and-whisker plots with individual data (n = 5–16). g All inhibitors except Dynasore prevented an increase in nocturnal IOP (***p < 0.001 vs. DMSO, one-way ANOVA [p < 0.001], Dunnett’s multiple comparison test). Data are presented as scatter plots with mean ± SEM (n = 5–16).
Driven-output stimuli of NE may suppress phagocytosis in the TM
To investigate the effect of GC and NE on phagocytosis in the TM, we used pH-sensitive pHrodo particles to evaluate phagocytosis in real time in immortalized human TM cells (iHTMCs) (Fig. 3a). We first validated these iHTMCs by analyzing gene expression (direct GC-induced clock gene PER1, trabecular meshwork-inducible GC response protein [MYOC]37, and GC-suppressed matrix metalloproteinase MMP338,39), 6 h (early) and 48 h (late) after Dex stimulation. Early induction of PER1, late induction of MYOC, and late suppression of MMP3 were detected (Supplementary Fig. 1), indicating that this iHTMC has TM cell properties40.

a Immortalized human TM cells (iHTMC) were placed on type I collagen-coated plates, and after 24 h, pH-sensitive pHrodo Zymosan was added with norepinephrine (NE) and dexamethasone (Dex) to evaluate phagocytosis in real time over 3 days. b This ability was confirmed by the phagocytosis inhibitor, Dynasore (*p < 0.05, ***p < 0.001 vs. DMSO, one-way ANOVA [p < 0.001], Dunnett’s multiple comparison). Data are presented as scatter plots with mean ± SEM (n = 3 independent experiments). Representative images 24 h after medium changes including NE or Dex (0.1, 1, and 10 µM) (c, e), and quantified phagocytic activity (d, f). Scale bar = 300 μm. Continuous NE exposure dose-dependently prevented phagocytic activity in iHTMC during 2 days of culture (*p < 0.05 vs. DMSO, one-way ANOVA [p < 0.01], Dunnett’s multiple comparison) (d), but not Dex (f). Data b, d, f are presented as scatter plots with mean ± SEM (n = 3 independent experiments). g driven output regulation, or self-sustainable regulation after a single stimulation in diurnal phagocytosis changes. h, i Exposure to NE (1, 10, and 100 µM) or Dex (0.1, 1, and 10 µM) stimulation for 30 min to iHTMC and real-time phagocytic activity was observed in iHTMC for 3 days. Although we did not observe any circadian phagocytosis rhythm in the normalized fluorescence signal, a high NE pulse stimulation (yellow bar; 10 µM) suppressed phagocytosis for about 1 day (***p < 0.001 vs. DMSO, one-way ANOVA [p < 0.001], Dunnett’s multiple comparison) (h, i), which was later restored to normal level (i), but Dex pulse stimulation (yellow bar; 10 µM), did not modulate phagocytosis rhythm (h, i). Data are presented as bar graphs (mean ± SEM) with scatter dot plots (h) and mean ± SEM (i) of independent experiments (n = 3–4).
Next, the validity of this phagocytosis assay was confirmed using a control with Dynasore (Fig. 3b). After medium changes, including NE or Dex, phagocytosis was significantly suppressed by NE (Fig. 3c, d). When we performed an in vitro NE clearance assay, the half-life of medium NE (~18.6 h, y = 335.9 e−0.000621 x, r2 = 0.979) (Supplementary Fig. 2) was far greater than that of blood NE (a few minutes), suggesting that in vitro NE treatment continuously act on cells. Indeed, NE treatment dose-dependently prevented phagocytic activity in iHTMCs during 2 days of culture (Fig. 3d). Although Dex alone had no effect (Fig. 3e, f and Supplementary Fig. 3a), Dex with NE-suppressed phagocytosis in a dose-dependent manner (Supplementary Fig. 3b), consistent with previous in situ studies24,25. Long-term Dex exposure did not alter cell viability (Supplementary Fig. 3c) or senescence (Supplementary Fig. 3d), indicating no effect of apoptosis or senescence on phagocytosis inhibition by Dex with NE. GC also binds to TM in humans41, and its receptor localizes in mouse TM13. Thus, the interaction between GCs and NE can generate an appropriate AH drainage rhythm. Taken together, these results suggest the importance of NE in TM phagocytosis.
Circadian time signals can generate rhythmicity through self-sustainable autonomous rhythm by single stimulation or a driven-output system by stimulation of circadian factors such as NE and GCs once a day (Fig. 3g). Since NE from the SCG appears to show a nocturnal peak in rodents42, the circadian rhythm of SCG-NE can regulate diurnal changes in TM phagocytosis. To investigate the short-term effect of NE and Dex on phagocytic circadian rhythm and activity, we exposed iHTMC to NE and Dex for 30 min, and subsequently observed the phagocytic activity in iHTMCs in real-time for 3 days (Fig. 3h). Although we could not observe the circadian phagocytosis rhythm in the normalized fluorescence signal by control, high NE pulse stimulation immediately began to suppress phagocytosis, reaching a minimum after 9 h and showing an inhibitory effect over 24 h, while Dex stimulation did not modulate phagocytosis rhythm (Fig. 3i). These findings suggest the possibility that diurnal changes in phagocytosis in TM may occur as a result of a driven output, but are not self-sustainable after a single NE stimulation (Fig. 3g).
β1-AR mainly attenuates phagocytosis in iHTMCs
As NE suppresses wound macrophage phagocytic efficiency through α- and β-AR dependent pathways26, we first confirmed the gene expression of nine AR subtypes in iHTMC. Strong expression of ADRA2A, ADRA2B, ADRA2C, ADRB1, and ADRB2 was observed, while very weak expression of ADRA1s and ADRB3 (Supplementary Fig. 4a) was observed, consistent with the gene expression patterns analyzed from previous single-cell RNA-seq data in human TM macrophages (Supplementary Fig. 4b)43. To identify the AR regulating SC phagocytosis, we subsequently treated several agonists of AR (L-adrenaline, β1-AR agonists [L-NE and dobutamine], β1β2-AR agonist [isoproterenol], selective α1-AR agonist [phenylephrine], direct-acting α2-AR agonist [clonidine], and β2-AR agonists [formoterol and salbutamol]) to iHTMC for single screening. As a result, we observed a dose-dependent robust suppression of phagocytosis by L-NE, dobutamine, and isoproterenol for 3 days (Fig. 4a), even though some showed slight changes in cell viability after 72 h of treatment (Supplementary Fig. 5). Further detailed analysis revealed the significant suppression of phagocytosis by L-NE, dobutamine, and isoproterenol, as well as by NE, but not salbutamol (Fig. 4b), indicating the involvement of β1-AR in phagocytosis suppression. Furthermore, the AR antagonists timolol (β1β2), betaxolol (β1), and ICI-118.551 (β2) tended to rescue NE-suppressed phagocytosis but not phentolamine (α-AR antagonist) in iHTMC, whereas antagonist alone showed no effect (Fig. 4c), indicating the involvement of β1β2-AR in phagocytosis suppression. To clarify this, we analyzed the effect of RNA interference on β1β2-AR and iHTMC phagocytosis, using siRNA against ADRB1 and ADRB2 (Fig. 4d), after verifying the inhibitory effect on gene expression (Supplementary Fig. 6a). ADRB1 siRNA significantly suppressed ADRB1 protein levels (Supplementary Fig. 6b) and increased NE-reduced phagocytic activity, but not ADRB2 siRNA (Fig. 4e). This difference may be due to the conversion of Gαs to Gαi by PKA activation in β2-AR in HEK293 cells44. Taken together, these results present clear evidence that β1-AR mainly mediates NE effects in phagocytosis.

a Effects of adrenergic receptor (AR) agonists on immortalized human TM cell (iHTMC) phagocytosis for 3 days. Data are presented as the mean (n = 1; quadruple experiment). Phagocytosis activities were normalized to the control DMSO. Dose-dependent robust suppression of phagocytosis by AR-agonist L-adrenaline, β1-AR agonists (L-NE and dobutamine), and β1β2-AR agonist (isoproterenol) for 3 days, and no effect of selective α1-AR agonist (phenylephrine), direct-acting α2-AR agonist (clonidine), and β2-AR agonists (formoterol and salbutamol). b Detailed analysis revealed the significant suppression of phagocytosis by L-NE, dobutamine, and isoproterenol as well as NE, but not by the β2-AR agonist salbutamol during day 1 (***p < 0.001 vs. DMSO, one-way ANOVA [p < 0.001], Dunnett’s multiple comparison). Data are presented as bar graphs (mean ± SEM) with scatter dot plots of independent experiments (n = 4). c AR antagonists timolol (β1β2), betaxolol (β1), and ICI-118.551 (β2) simultaneously with NE (5 µM) tended to rescue NE-suppressed normalized phagocytic activity but not phentolamine (α-AR antagonist) in iHTMC during day 1 (*p < 0.05, ***p < 0.001, one-way ANOVA [p < 0.001], Tukey’s multiple comparison). Data are presented as bar graphs (mean ± SEM) with scatter dot plots of independent experiments (n = 3−5). d, e Effect of RNA interference on β1β2-AR by siRNA against ADRB1 and ADRB2 on iHTMC phagocytosis. d After 24 h siRNA (finally 0.04 µM) exposure with accell siRNA delivery medium, media were changed to those containing NE (1 µM). e ADRB1 siRNA significantly increased phagocytic activity, but not ADRB2 siRNA (***p < 0.001, one-way ANOVA [p < 0.01], Tukey’s multiple comparison). Data are presented as bar graphs (mean ± SEM) with scatter dot plots of independent experiments (n = 3−4).
β1-AR attenuates phagocytosis via the cAMP-EPAC pathway
β1-AR is a typical G protein-coupled receptor (GPCR) that preferentially binds to stimulatory G protein Gs to induce cyclic adenosine monophosphate (cAMP) production. After dobutamine stimulation in iHTMC, we detected the downstream phosphorylation of cAMP response element binding protein (CREB), but not other Gq downstream Ca2+/calmodulin-dependent protein kinase II (CaMKII), and Gi/Gq downstream protein kinase C (Fig. 5a–c and Supplementary Fig. 7), indicating the activation of the Gs-coupled GPCR. In fact, prostaglandin E2 (PGE2), which activates Gs-coupled GPCR (EP4 receptor), prevented iHTMC phagocytosis in a dose-dependent manner (Fig. 5d). In addition, dose-dependent intracellular cAMP accumulation by cAMP inducers forskolin (FSK; Fig. 5e) and β1-AR agonist (L-NE and dobutamine; Fig. 5f) were observed, while betaxolol suppressed dobutamine-induced cAMP accumulation (Fig. 5g). These data demonstrate the importance of functional Gs-coupled β1-AR in HTMC phagocytosis.

a–c Effect of 6-h dobutamine (1 µM) stimulation on GPCR-related signaling pathways. b, c Western blot analysis revealed that phosphorylated CREB (pCREB) (Gαi/Gαs-related marker) was increased by dobutamine (t-test, **p < 0.01), but not phospho-CaMKII alpha (pCaMKII) (Gαq-related maker) and phospho-PKCα/βII [pPKCα/β] (Gαi/Gαq). Data are normalized by unphosphorylated protein or total protein and are presented as bar graphs (mean ± SEM) with scatter dot plots of independent experiments (n = 4). d Prostaglandin E2α (PGE2α), activating Gs-coupled GPCR, dose-dependently prevented iHTMC phagocytosis (*p < 0.05, **p < 0.01 vs. DMSO, one-way ANOVA [p < 0.001], Dunnett’s multiple comparison). Data are presented as bar graphs (mean ± SEM) with scatter dot plots of independent experiments (n = 3). Phagocytosis activity was normalized to that of control DMSO. Dose-dependent intracellular cAMP accumulation by cAMP inducers forskolin (FSK) (e), and by L-NE and dobutamine (f) (*p < 0.05, **p < 0.01, ***p < 0.001 vs. DMSO, one-way ANOVA [p < 0.001], Dunnett’s multiple comparison). g betaxolol dose-dependently suppressed L-NE (10 µM)- or dobutamine (1 µM)-induced cAMP accumulation (*p < 0.05, **p < 0.01 vs. DMSO, one-way ANOVA [p < 0.001], Dunnett’s multiple comparison). Data e–g are presented as bar graphs (mean + SEM) with scatter dot plots of independent experiments (n = 4). h iHTMC phagocytic activity was slightly but significantly reduced by cAMP inducers (FSK and IBMX) and cAMP analogs, such as PKA activator Sp-cAMP and EPAC/PKA activator 8-CPT-cAMP (*p < 0.05, **p < 0.01, ***p < 0.001 vs. DMSO, one-way ANOVA [p < 0.001], Dunnett’s multiple comparison). Phagocytosis activity was normalized to that of control DMSO. i siRNA (PRKACA, RAPGEF3, and RAPGEF4) administration significantly rescued β1-AR-mediated suppression of iHTMC phagocytosis (**p < 0.01, one-way ANOVA [p < 0.001], Tukey’s multiple comparison). j The recovery effect of PKA and EPAC1/2 antagonists (KT5720 and ESI09, respectively) simultaneously added with dobutamine (1 µM) dose-dependently rescued dobutamine-suppressed iHTMC phagocytosis (*p < 0.05, **p < 0.01, ***p < 0.001 vs. DMSO, one-way ANOVA [p < 0.001], Tukey’s multiple comparison). The antagonists were added at the same time as dobutamine. Data h–j are presented as bar graphs (mean ± SEM) with scatter dot plots of independent experiments (n = 3–4).
cAMP binds to activate protein kinase A (PKA) or exchange proteins directly activated by cAMP (EPACs)45. In microglia and peritoneal macrophages, myelin phagocytosis occurs with the involvement of both EPAC1 and PKA46. iHTMC phagocytic activity was slightly but significantly reduced by cAMP inducers (FSK and 3-Isobutyl-1-methylxanthine [IBMX]) and cAMP analogs, such as the PKA activator Sp-cAMP and the EPAC/PKA activator 8-CPT-cAMP (Fig. 5h). To clarify the importance of PKA and EPAC in phagocytosis, RNA interference against PRKACA, RAPGEF3, and RAPGEF4, encoding PKA, EPAC1, and EPAC2, respectively, rescued β1-AR-suppressed iHTMC phagocytosis to a control level (p > 0.05, vs. control; Fig. 5i). Furthermore, blocking of PKA and EPAC1/2 by antagonists (KT5720 and ESI09, respectively) prevents β2-AR- and PGE2-suppressed neutrophil phagocytosis47, which dose-dependently rescued dobutamine-mediated phagocytosis inhibition (Fig. 5j), indicating the involvement of both pathways. NE and isoproterenol suppress the phagocytosis of microglia cells via EPAC activation48, while these findings clearly suggest that Gs-coupled β1-AR modulates iHTMC phagocytosis via PKA and EPAC.
NE decrease PIP3 through SHIP activation via EPAC and PKA
PI(3,4,5)P3 (PIP3) is necessary for phagocytosis49 (Fig. 6a). To verify the role of PIP3 in iHTMC phagocytosis, the PIP3 antagonist, PITenin-7, dose-dependently and significantly suppressed the phagocytic activity of iHTMC (Fig. 6b). PI3K produces PIP3 from PI(4,5)P250(Fig. 6a). Class I PI3K has four isoforms: α, β, γ, and δ. PI3Kγ, but not α, β, and δ, modulates the phagocytosis of microglia, which is suppressed by cAMP-mediated EPAC activation51. When we exposed LY294002 (broad-spectrum inhibitor of PI3Kαβδ) and CAY10505 (selective PI3Kγ inhibitor) to iHTMC, only CAY10505 slightly but significantly restrained phagocytosis (Fig. 6c). Furthermore, we detected a decrease in PIP3 levels following dobutamine treatment (Fig. 6d). These results clarified that β1-AR-mediated PIP3 modulates phagocytosis in iHTMCs.

a The hypothesized phagocytosis suppression mechanism of β1-AR-cAMP. PIP3 is a hub molecule that induces phagocytic cup formation through phosphorylation of AKT and ERK, and PI3K, PTEN, and SHIP1 regulate PIP3 levels. However, the involvement of EPAC and PKA in iHTMCs in this system is unknown. b Administration of the PIP3 antagonist PITenin-7 dose-dependently and significantly suppressed the phagocytic activity of iHTMC (**p < 0.01, ***p < 0.001 vs. DMSO, one-way ANOVA [p < 0.001], Dunnett’s multiple comparison). c Effect of LY294002 (inhibitor of PI3Kαβδ) and CAY10505 (selective PI3Kγ inhibitor) administration on phagocytosis (**p < 0.01, DMSO, one-way ANOVA [p < 0.01], Dunnett’s multiple comparison). d PIP3 levels after 6 h exposure to dobutamine with several antagonists in iHTMC. Dobutamine (1 µM) decreased PIP3, which was improved by KT5720 and ESI09. In addition, bpV (pic) (PTEN inhibitor) and 3AC (SHIP1 inhibitor) also improved it (**p < 0.01, vs. control, one-way ANOVA [p < 0.001], Dunnett’s multiple comparison). Data b–d are presented as bar graphs (mean ± SEM) with scatter dot plots of independent experiments (n = 3). e Western blot analysis of SHIP1 phosphorylation in iHTMCs. Dobutamine induced phosphorylation of SHIP1, which was prevented by ESI09 but not by KT5720 (**p < 0.01, ***p < 0.001 vs. DMSO, one-way ANOVA [p < 0.001], Dunnett’s multiple comparison). Data are normalized to unphosphorylated SHIP1 and are presented as bar graphs (mean ± SEM) with scatter dot plots of independent experiments (n = 4). f Involvement of the β1-AR signaling pathway in AKT or ERK1/2 phosphorylation. Dobutamine inhibited this phosphorylation, which was recovered significantly by ESI09, bpV (pic), and 3AC, but not by KT5720 (*p < 0.05, **p < 0.01, vs. control, one-way ANOVA [p < 0.001], Dunnett’s multiple comparison). Data are normalized by unphosphorylated proteins and are presented as bar graphs (mean ± SEM) with scatter dot plots of independent experiments (n = 4). g recovery efficacy of bpV(pic), K118 (SHIP1 inhibitor), or 3AC on dobutamine (1 µM)-mediated suppression of phagocytosis (**p < 0.01, ***p < 0.001, one-way ANOVA [p < 0.001], Tukey’s multiple comparison). Phagocytosis activity was normalized to that in the control. Data are presented as bar graphs (mean ± SEM) with scatter dot plots of independent experiments (n = 3).
In macrophages, PKA does not inhibit phagocytosis, whereas Epac1 exerts an inhibitory effect mainly through the activation of the tyrosine phosphatase SHIP152 (Fig. 6a). SHIP1 converts PIP3 to PI(3, 5)P250 (Fig. 6a). In addition, activated SHIP1 has been reported to dephosphorylate PTEN, catalyzing PIP3 in macrophages52, and single-cell RNA-seq analysis has suggested that both SHIP1 and PTEN are expressed in human TM macrophages43 (Supplementary Fig. 8). In fact, exposure of iHTMC with PKA and EPAC inhibitors improved the PIP3 suppression efficacy of dobutamine (Fig. 6d). In addition, the PTEN inhibitor bisperoxovanadium (pyridine-2-carboxyl) [bpV(pic)]53, SHIP1 inhibitor 3-a-aminocholestane (3AC)54 and K11855 also improved this efficiency (Fig. 6d), indicating the involvement of the β1-AR-signaling pathway in PIP3 reduction. Based on the results presented above, we next performed western blot analysis of SHIP1 phosphorylation to determine the effects of β1-AR-mediated PKA and EPAC signaling in iHTMCs. Dobutamine induced the phosphorylation of SHIP1, which was prevented by EPAC inhibition, but not by PKA inhibition (Fig. 5e), indicating the importance of β1-AR-mediated SHIP1 activation through EPAC.
As PIP3 stimulates AKT and ERK1/2 signaling to modulate phagocytosis in macrophages52, we next verified the effect of the signaling pathway of NE on AKT/ERK1/2 phosphorylation (Fig. 6a, f). Dobutamine cleanly inhibited their phosphorylation, which recovered significantly with EPAC inhibitors, but not with PKA inhibitors (Fig. 6f). AKT/ERK1/2 phosphorylation suppressed by dobutamine was upregulated by PTEN and SHIP1 inhibitors (Fig. 6f). These results indicate the involvement of SHIP/PTEN-reduced PIP3 in the EPAC-mediated suppression of AKT/ERK activation. Furthermore, when we monitored the effect of PTEN and SHIP1 inhibitors on the phagocytosis suppression effect of dobutamine, only SHIP1 inhibitors dose-dependently rescued it up to the near control level, but no effect was seen for the PTEN inhibitor or the antagonist alone (Fig. 6g). These results suggest that, at least in vitro, β1-AR-mediated SHIP1 activation through EPAC reduces PIP3 to suppress phagocytic cup formation.
Nocturnal activation of the β1-AR-EPAC-SHIP1 pathway enhanced IOP in mice
Based on the results presented above, we next performed immunohistochemical analysis to determine the localization of Adrb1 and Ship1 in the mouse eye (Supplementary Fig. 9). This demonstrated the colocalization of Adrb1 and Ship1 within the endothelial cells in the SC and the NPE cells in the ciliary body (Supplementary Fig. 9), providing evidence of the existence of the β1-AR-EPAC-Ship1 system in mice.
Since NE in AH released from the SCG has a circadian rhythm with nocturnal increase in rodents56, which seems to also be true in humans, a nocturnal increase of NE in TM may cause IOP increase by inhibiting phagocytosis in the TM. To validate this hypothesis, we administered mice β1-AR-SHIP1 pathway inhibitors by instillation at ZT10 and measured IOP at night (ZT15) (Fig. 7a, b). First, the β1-AR antagonist betaxolol is well used for glaucoma therapy57, lowers topically mouse IOP58, it blocked the nocturnal IOP increase (Fig. 7a). Furthermore, the effects of KT5720, ESI09, and bpV(pic) on retinal photoreceptor death in rodents59,60 have been reported but not on IOP, while that of the 3AC, promoting phagocytosis54, on IOP in animals remains unknown. Interestingly, instillation of ESI09 and 3AC among these reagents significantly suppressed the nocturnal IOP increase (Fig. 7a) and also at the individual level (p < 0.01); however, the PKA inhibitor did not show this effect (Fig. 7b), suggesting a role for the β1-AR-EPAC-SHIP1 pathway in nocturnal IOP increase. However, since EPAC inhibitor ESI09 suppressed nocturnal IOP more than betaxolol (Fig. 7b), and RAPGEF3 and RAPGEF4 are expressed in human SC endothelial cells43, it may also affect mechanisms such as cytoskeleton and cell adhesion in the TM, the giant vacuoles in the endothelial cells of the SC, or AH inflow in the NPE of the ciliary body other than TM phagocytosis because Epac1 mediates retinal neurodegeneration in mouse models of ocular hypertension60.

a, b Effect of β1-AR-EPAC-SHIP1 pathway-related inhibitors on nocturnal IOP increase in mice. Mice were administered betaxolol (100 µM/0.1% DMSO PBS), KT5720 (100 µM), ESI09 (100 µM), 3AC (1 mM), K118 (1 mM), and bpV(pic) (100 µM) at ZT10, and IOP was measured at ZT15. a Instillation of KT5720 and ESI09 significantly suppressed the nocturnal IOP increase (paired t-test, **p < 0.01, ***p < 0.001). Data are presented as box-and-whisker plots with individual data (n = 7–16). b All inhibitors except bpV (pic) prevented an IOP increase (*p < 0.05, **p < 0.01, ***p < 0.001 vs. DMSO, one-way ANOVA [p < 0.001], Dunnett’s multiple comparison test). Data are presented as scatter plots with mean ± SEM (n = 7–16). c Day-night changes in IOP one day before and after 3AC instillation. Data are presented as mean ± SEM (n = 10) with individual data. One day after single instillation, IOP showed a normal nocturnal increase (**p < 0.01, ***p < 0.001, one-way ANOVA [p < 0.001], Bonferroni’s multiple comparison test), providing evidence of driven-output IOP enhancement by nocturnal NE (d). e, f Validation of IOP inhibitory effects of drugs on β1-AR-mediated IOP regulation using instillation of dobutamine (100 µM) at ZT4. After 5 h, the IOP was measured. Dobutamine significantly enhanced IOP, while pretreatment with betaxolol, ESI09, and 3AC prevented this effect (paired t-test, **p < 0.01, ***p < 0.001). f The same was true for individual data (**p < 0.01, ***p < 0.001 vs. Dobutamine+DMSO, one-way ANOVA [p < 0.001], Dunnett’s multiple comparison test). Data are presented as e box-and-whisker plots with individual data and f as scatter plots with mean ± SEM (n = 6–16). g Regulatory model of time-dependent systems in which nocturnal NE suppresses phagocytosis to induce AH outflow resistance through β1-AR-EPAC-SHIP1 activation, leading to an IOP increase at night. The AH produced from the ciliary body is drained by two pathways: trabecular outflow and uveoscleral outflow.
As we demonstrated the possibility of a driven-output phagocytic activity (Fig. 3), we next measured day-night changes in IOP 1 day before and after instillation to verify whether SHIP1 inhibition suppresses IOP rhythm in a self-sustainable manner (Fig. 7c). Interestingly, SHIP1 inhibition by single instillation did not show any inhibitory effect on nocturnal IOP increase the day after instillation (Fig. 7c), providing evidence of driven-output IOP enhancement by nocturnal NE (Fig. 7d). This regulatory pathway is consistent with β1-AR-mediated circadian regulation of the pineal gland61. Since NE from the SCG appears to show a nocturnal peak in rodents42, the circadian rhythm of SCG-NE can regulate diurnal changes in TM phagocytosis (Fig. 7d).
To verify the IOP inhibitory effects of drugs on β1-AR-mediated IOP regulation in vivo, we administered dobutamine instillation at ZT4 to increase IOP (Fig. 7e). Five hours after administration, IOP was significantly and clearly enhanced (Fig. 7e, f). To clarify the involvement of cytoskeletal rearrangement and phagocytosis in this enhancement, we next evaluated cytochalasin D, latrunculin A, and RKI1447. Interestingly, only RKI1447, a phagocytosis promoter, prevented this increase (Fig. 7e, f), indicating the contribution of TM phagocytosis in β1-AR-mediated IOP increase rather than actin rearrangement. In other words, the decrease in AH outflow by phagocytosis suppression outweighed that of actin rearrangement in the other TM cells. Conversely, since these inhibitors suppressed nocturnal IOP increase (Fig. 2), the increase in AH outflow by actin depolymerization may increase at night. In addition, importantly, preinstillation with a β1-AR antagonist prevented this increase (Fig. 7e, f), confirming the effect of β1-AR. To determine the role of EPAC and SHIP1 in this effect, we blocked dobutamine-activated PKA, EPAC, or SHIP1 (Fig. 7e, f). Preinstillation of EPAC and SHIP1 inhibitors significantly suppressed IOP increase (Fig. 7e, f) and prevented IOP increase in individual data (p < 0.01); however, the PKA inhibitor did not show this effect (Fig. 7f), indicating the importance of the β1-AR-EPAC-SHIP1 pathway in IOP regulation. Taken together, these findings revealed that nocturnal NE suppresses phagocytosis-mediated AH outflow through β1-AR-EPAC-SHIP1 activation, leading to an increase in IOP at night (Fig. 6g).
Discussion
Previous studies have indicated that sympathetic NE and adrenal GC transmit circadian timing signals to the eye to generate IOP13. However, the involvement of NE and GC in TM phagocytosis contributing to IOP regulation remains unknown62,63. In this study, we found that the suppression of AH drainage in mice may partially contribute to the increased nocturnal IOP. In addition, we demonstrated that driven-out suppressed TM phagocytosis by NE in vitro, which is regulated by β1-AR-cAMP-EPAC-SHIP1 activation. Although NE is known to suppress macrophage phagocytosis26, and PIP3 stimulates AKT and ERK1/2 signaling to modulate phagocytosis49,52, the connection between NE and PIP3-triggered phagocytosis remains unclear. In this study, we identified β1-AR -suppressed TM phagocytosis by silencing PIP3-AKT or -ERK signaling through the cAMP-EPAC-SHIP1 pathway. In the TM of the POAG donors, the cAMP signaling pathway and CREB were activated64, while ERK phosphatase activity was downregulated65. These results support those of the present study. Further understanding of this pathway is necessary to fully explain the complex cellular mechanisms by which occupancy of specific ARs regulates AH dynamics, which may contribute to the establishment of chronotherapy.
In the present study, the time-dependent mechanism of the inhibitory effect of NE on TM phagocytosis was elucidated, but the circadian regulation of NE/GC in other important key factors such as cytoskeleton, cell adhesion, and IOP-independent uveoscleral outflow to the ciliary muscle (Fig. 6i) in the AH outflow remain unknown. Long-term stimulation of GC increases the actin polymerization of TM and induces fibrosis66. In the TM, cAMP/PKA activation and downstream RhoA inactivation lead to a loss of actin stress fibers and focal adhesions and disassembly of the matrix network67. Ship1−/− in neutrophils upregulates basal actin polymerization68. However, the time-dependent efficacy may explain their contribution to some extent. The effects of actin polymerization inhibitors on IOP reduction seem to be limited to the dark period in mice (Figs. 2, 7). In fact, the decrease in AH outflow by phagocytosis suppression on day appears to outweigh that by actin rearrangement in the TM. The cell-intrinsic circadian clock regulates numerous cytoskeletal regulators in fibroblast69. In contrast, in mice, 30–42% of AH passes through the uveoscleral pathway70. To understand the AH dynamic rhythm completely, these determinants need to be elucidated. The discovery of a phagocytosis activator independent of the cytoskeleton or single-cell analysis separated by the function of TM will be able to elucidate them completely in the future.
In general, β-AR blockers, including betaxolol, effectively reduce IOP by decreasing AH inflow in patients71, while several studies have demonstrated the opposite effect of the sympathetic role in AH outflow72,73. β-AR may be involved in the increase of AH outflow by reducing the size of cells in the TM72. Continuous electrical stimulation of the cervical sympathetic nerve decreased IOP only during 1 h73. However, these studies demonstrated the role of β2-AR. In fact, POAG involves a greater increase in IOP at night when SCG-NE release is thought to be higher than during day10. Furthermore, a recent study revealed that betaxolol is the top compound most opposed to POAG signatures calculated by microarray database analysis74. β1-AR may contribute to nocturnal AH outflow resistance.
Since, in a previous study demonstrating that the double knockout mouse model of β1 and β2 ARs maintains IOP rhythm7, these mice had different genetic backgrounds compared with the control mice, we think it is impossible to assess the IOP rhythmicity such as its amplitude. However, it is certain that β1/2-ARs are not essential for IOP rhythm formation7. In the present study, betaxolol did not completely prevent nocturnal IOP rise (Fig. 7). The removal of SCG in mice attenuated the IOP rhythm13, and betaxolol instillation resulted in IOP rhythms with nocturnal peak flattening in both POAG and NTG patients57. The contribution of the β1-AR-mediated circadian rhythm of AH resistance for IOP rhythm formation may not be so high, and Dex or the other AR can regulate AH-outflow/inflow. β2-AR- or GC-mediated AH production in the NPE of the ciliary body is related to the IOP rhythm13. This may explain why β-AR1/2 double knockout mice maintain this IOP rhythm.
In the present study, we found that NE was a necessary condition for GC-mediated phagocytosis in the TM. Both β-adrenergic signaling and GCs are mediators of SCN timing signals in osteoblasts75. Interactions between the sympathetic nervous system and GCs have also been previously reported. In particular, GC transcriptionally modulates β2-AR expression by modulating GC-response elements on the promoter76. Interestingly, GCs rapidly activate cAMP production via Gαs to initiate non-genomic signaling, which contributes to one-third of their canonical genomic effects77. In fact, betaxolol prevented steroid-induced IOP increase78. Thus, in TM, Gs-bound GR may enhance the β1-AR-Gs signal to suppress phagocytosis and generate an appropriate AH drainage rhythm.
Our study has several inherent limitations. First, although we provide a model of IOP induction in nocturnal NE, the circadian rhythm of NE released from SCG in humans remains unclear. Although GC secretion peaks at the light offset79, and NE is released with a nocturnal peak from the SCG in rodents42, GC rhythms are anti-phasic but not SCG-NE in diurnal animals42,80. Nocturnal NE release from SCG generally stimulates melatonin synthesis in humans, as well as in other mammals81. In humans, β-blockers inhibit nocturnal melatonin levels82, and suppress IOP increase during late night to morning83. Furthermore, in nocturnal rabbits, β-blockers suppress IOP increase only at night84. These results indicate the involvement of nocturnal NE release from the SCG in regulating the IOP rhythm. Second, we cannot fully explain the differences in IOP rhythms in diurnal and nocturnal animals using the present model. The IOP rhythm peaks early at night in nocturnal animals7, while in healthy humans, it appears to be elevated during the night and peaks from late night to early morning6,9. In phasic SCG-NE and anti-phasic GC, the action of both factors in IOP increase may explain such differences. Third, protein downregulation by RNA interference in the present study might not be sufficient for the contribution of target proteins, so knock-out analysis in vitro or tissue-specific gene modification techniques using virus in vivo can help the understanding.
Taken together, these results suggest a potential circadian role for NE in the modulation of phagocytosis and AH outflow resistance by TM, contributing to the IOP rhythm. Although the TM accounts for most outflow and is the major site of AH outflow resistance70,85, until the approval of the ROCK inhibitor, no clinically administered drug had a direct effect on the TM. In the present study, we suggest the possibility of therapeutic drug development targeting TM. In addition, the synergistic regulatory mechanism of GC in the presence or absence of NE in AH dynamics remains unknown. Since genomic signaling may also contribute to this mechanism77 (Fig. 7g), further understanding of the time-dependent efficacy of GC and NE on AH inflow/outflow will lead to a complete elucidation of the regulatory mechanisms of IOP rhythm. Although new therapeutics with new mechanisms, such as chronotherapy, are urgently needed for glaucoma treatment, the development of multiple types of drugs using this interaction could be especially useful for glaucoma treatment in the future.
Methods
Animals
Five-week-old male C57BL/6JJmsSlc mice (N = 130; Japan SLC Inc., Shizuoka, Japan) were purchased and housed in plastic cages (170 W × 240 D × 125 H mm; Clea, Tokyo, Japan) under a 12 h light (200 lx of fluorescent light)/dark cycle (12L12D, 0800 light ON, 2000 light OFF), maintained at a constant temperature (23 ± 1 °C). Food (CE-2; CLEA) and water were provided ad libitum13. All animal experiments were approved by the Committee of Animal Care and Use of the Aichi Medical University. All experimental procedures were conducted in accordance with the institutional guidelines for the use of experimental animals13.
IOP measurement
IOP measurements were performed using a tonometer (Icare TonoLab, TV02; Icare Finland Oy, Espmoo, Finland), as previously reported8,13. All mice were kept under 12L12D conditions for more than 2 weeks before IOP measurements. Unanesthetized mice were gently held using a sponge. IOPs were measured during the light phase under light (200 lx) conditions and during the dark phase under dim red-light conditions. For the analysis of phagocytosis-related drugs and dobutamine-mediated IOP induction, IOP was measured before drug instillation at zeitgeber time (ZT) 4 and measured at ZT9. ZT0 (0800) was defined as the time of light ON. To analyze the nocturnal IOP increase, IOP was obtained by measuring the IOP at ZT10 and ZT15. We calculated back from ZT15 (the peak of IOP) for 5 h and set it to ZT10 when IOP was low. For diurnal changes in IOP, IOP was measured at ZT6 (IOP trough) and ZT15 (IOP peak)13, 2 weeks after bead injection.
Intraocular injection and detection of fluorescence particles
To investigate the effect of AH inflow on nocturnal IOP increase, mice were anesthetized by isoflurane inhalation (2%; WAKO, Saitama, Japan), supplemented with topical proparacaine HCl (0.5%; P2156, Tokyo Chemical Industry [TCI], Tokyo, Japan), and were treated with an intraocular injection of an Na+/K+ATPase inhibitor ouabain (100 µM/0.1% dimethyl sulfoxide [DMSO], phosphate-buffered saline [PBS], 3 µL) into anterior chamber of the right eye with a 34-gauge needle (0.18 × 8 mm, Pasny; Unisis) connected to a Hamilton syringe at ZT10. To precisely control the small volume (3 μL) of anterior chamber injection, 3 μL PBS (0.1% DMSO) was injected into the left eye with a 34-gauge needle connected to a Hamilton syringe.
For microbead injection to prevent AH outflow, mice were anesthetized by isoflurane inhalation (2%; WAKO, Japan), supplemented with topical proparacaine HCl. IOP elevation was induced unilaterally in adult C57BL/6J mice by injection of 3 µL of 1/10 diluted fluosphere polystyrene microspheres (15 µm, yellow-green fluorescent, F8844, Invitrogen, Carlsbad, CA) into the anterior chamber of the right eye with a 34-gauge needle connected to a Hamilton syringe. Microbeads were then resuspended in PBS at 5.0 × 106 beads/mL. To precisely control the small volume (3 μL) of anterior chamber injection, 3 μL PBS was injected into the left anterior chamber with a 34-gauge needle, connected to a Hamilton syringe.
To visualize the AH outflow in the SC, mice were anesthetized by isoflurane inhalation (2%; WAKO, Japan), supplemented with topical proparacaine HCl, and were treated with an intraocular injection of 3 µL of carboxylate-modified microspheres (0.5 µm, yellow-green fluorescent, 2% solids, F8813, Invitrogen) at ZT0 and ZT12. After 6 h, the injected mice were anesthetized, and their anterior eyes were extracted and fixed in 4% paraformaldehyde (26126-25, Nakarai Tesk)/PBS for 5 min. After washing with PBS, anterior eye cups were placed in Hanks‘ Balanced Salt Solution (HBSS; 082-08961 WAKO) in a 96-well microplate to observe the fluorescence from the bottom using a digital fluorescent microscope (Dino-Lite Edge M Fluorescence TGFBW; Opto Science Inc., Tokyo, Japan), and the fluorescence intensity (525 nm) was measured using a microplate reader, SpectraMax M5 (Molecular Devices).
Drug instillation
Drug instillation was performed as described in our previous report13. Unanesthetized 8-week old male mice were used in this study. For the analysis of phagocytosis and/or cytoskeleton-related drugs, mice were instilled with a single drop (30 μL) of Dynamin-related phagocytosis inhibitor Dynadore (1 mM/0.1% DMSO PBS; D5461, TCI), actin polymerization and phagocytosis inhibitor cytochalasin D (100 µM; 11330, Cayman), latrunculin A (100 µM; 125-04363, WAKO), and ROCK inhibitor RKI1447 (1 mM; 16278, Cayman) at ZT4 using a micropipette into bilateral eyes, and IOP was measured at ZT10. For nocturnal IOP increase analysis, mice were instilled with a single drop of Dynadore, cytochalasin D, latrunculin A, RKI1447, β1-AR antagonist betaxolol hydrochloride (100 µM/0.1% DMSO PBS; B4474, TCI), PKA inhibitor KT5720 (100 µM; 10011011, Cayman), EAPC1/2 inhibitor ESI09 (100 µM; 19130, Cayman), 3α-aminocholestane (1 mM, 3AC; HY-19776, MCE), pan-SHIP1 inhibitor K118 (1 mM; B0344, Echelon Biosciences, Salt Lake City, USA), and PTEN inhibitor bpV (pic) (100 µM; SML0885, Sigma-Aldrich) at ZT10 using a micropipette in both eyes, and IOP was measured at ZT15. To analyze the inhibitory efficacy of dobutamine-induced IOP increase, the above antagonists were preinstillated at ZT4, after 10 min, a single drop of dobutamine (100 µM/0.1% DMSO PBS) was added. During instillation, the mice were gently restrained with necks held back.
iHTMC culture
The immortalized human TM-SV40 cell line (iHTMC) derived from primary human SC and TM regions was purchased from Applied Biological Materials Inc. (T-371-C, ABM Inc., Richmond, BC, Canada) and cultured in TM cell medium (6591, Sciencell, Carlsbad, CA, USA) supplemented with 2% fetal bovine serum (0010, ScienCell), 1% growth supplement (TMCGS, 6592, Sciencell) and 1% penicillin/streptomycin (0503, Sciencell) in type I collagen-coated 100-mm dish (3020-100, IWAKI, Japan). Experiments were performed on type I collagen-coated plates. Upon reaching confluence, iHTMCs were split 1:4 using 0.05% trypsin/PBS. Cell viability was determined using trypan blue (0.4%) exclusion.
Phagocytosis assay
For phagocytosis assay, iHTMCs (T0371, abm) were plated in collagen I-coated 96-well microplates (4860-010; IWAKI) at a density of 5.0 × 103 cells/well in trabecular meshwork cell medium (TMCM) supplemented with 1% penicillin/streptomycin and growth factors (6591; Sciencell). To measure phagocytosis, pHrodo Green Zymosan Bioparticles (P35365; ThermoFisher) were suspended in TMCM and vortexed to disperse. After 90% confluence, the medium was removed by aspiration, and 100 μL of serum-free TMCM was immediately added. After 24 h, the medium was replaced with serum-free TMCM containing pHrodo Zymosan (2.5 µg/well) in the presence of several kinds of drugs, and the plate was placed in the IncuCyte ZOOM instrument (Essen Bioscience, Ann Arbor, MI, USA), installed in a 5% CO2 incubator at 37 °C. Each well was imaged at 3 points, every 0.5 h or 1 h for more than 72 h using the channels of green fluorescence and phase and the 10× objective (Nikon). No pHrodo Zymosan was used for fluorescent control using background fluorescent intensity because of autofluorescence in TMCM, and vehicle control included 0.1% DMSO. The green fluorescence intensity at each time point in each well was measured using the IncuCyte ZOOM 2015A software (Essen Bioscience). To perform a detailed analysis of the pulse stimulation, we calculated the difference from the control. We calculated the average daily fluorescence intensity (0–23 h [Day 1], 24–47 h [Day 2], and 48–71 h [Day 3]) normalized to that of the control DMSO-treated group to show statistical changes. For pulse stimulation of drug, phagocytosis index is calculated by the difference with the control. The phagocytosis assay was independently repeated thrice using three or four biological replications.
Drug treatment
For the phagocytosis assay, iHTMCs were simultaneously treated with (−)-NE (0.1, 1, and 10 µM; S9507, Selleck) and/or dexamethasone (Dex; 0.1, 1, and 10 µM; 11107-51, Nakarai Tesk, Kyoto, Japan) and pHrodo Zymosan (2.5 µg/well), and were exposed over 3 days without washout. To confirm the phagocytic activity of iHTMCs, we treated iHTMCs with the phagocytosis inhibitor Dynasore (0.1, 1, and 10 µM; D5461, TCI). For a detailed analysis of pulse stimulation, serum-free TMCM containing NE (1, 10, and 100 µM) or Dex (0.1, 1, and 10 µM) was added to iHTMC for 30 min. After washing with TMCM, pHrodo zymosan-containing TMCM was added to the iHTMC. For analysis of effect of agonists on phagocytosis, we treated several kinds of agonists for AR agonist L-adrenaline (A0173, TCI), β1-AR agonists [L-Noradrenaline Bitartrate Monohydrate (L-NE, A0906, TCI) and dobutamine hydrochloride (15582, Cayman)], β1β2-AR agonist [Isoproterenol Hydrochloride (I0260, TCI)], selective α1-AR agonist [L-Phenylephrine (P0395, TCI)], direct-acting α2-AR agonist [Clonidine HCl (038-14291, WAKO)], β2-AR agonist [Formoterol fumarate hydrate (F0881, TCI)], short-acting β2-AR agonist (Salbutamol Hemisulfate [S0531, TCI]), PGE2 (0.1, 1, and 10 µM; P1884, TCI), cAMP inducers [Forskolin (10 µM FSK; F0855, TCI) and 3-Isobutyl-1-methylxanthine (10 µM IBMX; 095-03413, WAKO)], and cAMP analogs as PKA activator Sp-cAMP (10 µM; 14983, Cayman) and as EPAC/PKA activator 8-CPT-cAMP (10 µM; 12011, Cayman) to iHTMC. To analyze the antagonists for ARs, NE (5 µM) was simultaneously added with antagonists: β1β2-AR antagonist timolol maleate (1, 10, and 100 µM; T2905, TCI), β1-AR antagonist betaxolol hydrochloride (1, 10, and 100 nM; B4474, TCI), β1-AR antagonist ICI-118.551 hydrochloride (0.01, 0.1, and 1 µM; HY-13951, MCE), and α2-AR antagonist phentolamine mesylate (100 µM; P1985, TCI). For pathway antagonists, dobutamine (1 µM) was simultaneously added with antagonists: PKA inhibitor KT5720 (0.001, 0.01, 0.1, and 1 µM; 10011011, Cayman), EAPC1/2 inhibitor ESI09 (0.1, 1, 10, and 100 µM; 19130, Cayman), PTEN inhibitor bpV (pic) (0.001, 0.01, 0.1, and 1 µM; SML0885, Sigma-Aldrich), and 3α-aminocholestane (0.01, 0.1, 1, and 10 µM; 3AC; HY-19776, MCE). To regulate PIP3 content, we treated iHTMC with the PIP3 antagonist PITenin-7 (0.01, 0.1, 1, and 10 µM; 524618, Calbiochem), PI3Kαβδ inhibitor LY294002 (70920, Cayman), and selective PI3Kγ inhibitor CAY10505 (HY-13530, MCE).
Western blot analysis
Western blot analysis was performed as described previously in our report86. Protein extraction was performed using cell lysis buffer (9803, Cell Signaling Technology, Tokyo, Japan) containing a protease inhibitor cocktail (P8340, Sigma) and phosphatase inhibitor cocktail 1 (P2850, Sigma) according to the manufacturer’s instructions. Total protein transferred to the PVDF membrane was detected using EzStainAQua MEM (WSE-7160, ATTO) and used for normalization. After destaining, membranes were incubated with the following primary antibodies: goat polyclonal antibody against ADRB1 (1:2000; NB600-978, Novus Biologicals), rabbit monoclonal antibodies against phospho-CREB (Ser133) (1:1000; 9198, Cell Signaling Technology), CREB (1:1000; 9192, Cell Signaling Technology), Akt (pan) (C67E7) (1:1000; 4691, Cell Signaling Technology), rabbit polyclonal antibody against phospho-PKCα/βII (Thr638/641) (1:1000; 9375, Cell Signaling Technology), phospho-Akt (Thr308) (1:1000; 9275, Cell Signaling Technology), phosphor-CaMKII alpha (Thr286) (1:1000; ab5683,abcam), ERK1/ERK2 (1:1000; A16686, ABclonal), phospho-ERK1(T202/Y204)/ERK2(T185/Y187) (1:1000; AP0472, ABclonal), INPP5D (SHIP1) (1:1000; A0122, ABclonal), phosphor-INPP5D (Tyr1021) (1:1000; PA903060, CSB), and mouse monoclonal antibody against CaMKIIα/β/γ/δ (G-1) (1:500; sc-5306, Santa Cruz Biotechnology). Membranes were washed and then incubated with HRP-conjugated goat polyclonal antibody against mouse and rabbit IgG (1:10,000; 7074, Cell Signaling Technology). Chemiluminescent images were detected using an Amersham Imager 600 (Cytiva Lifescience).
cAMP measurement
cAMP measurements were performed with a homogeneous TR-FRET immunoassay using the LANCE cAMP Detection Kit (AD0262, PerkinElmer, USA), according to the manufacturer’s instructions (PerkinElmer). After confluence, iHTMCs in collagen I-coated 96-well microplates (4860-010; IWAKI) were washed with PBS (0.2 g/L EDTA), and then washed with stimulation buffer (HBSS, 5 mM HEPES, 0.5 mM IBMX, and 0.01% BSA at pH 7.4). After aspiration, iHTMC was added to 10 µL of tested compounds with FSK (0.001, 0.01, 0.1, and 1 µM), L-NE (0.01, 0.1, 1, and 10 µM), and dobutamine (0.001, 0.01, 0.1, 1, and 10 µM), along with 10 µL of Alexa Fluor 647 anti-cAMP antibody diluted with stimulation buffer. The cells were stimulated for 60 min at room temperature. The antagonist response analysis was performed using L-NE or dobutamine as the reference agonist. To analyze the antagonistic effect on β1-AR, the β1-AR agonist L-NE or dobutamine was used at submaximal concentrations (10 and 1 µM, respectively) to stimulate cAMP accumulation. These agonists and the β1-AR antagonist betaxolol (0.05, 0.5, 5, and 50 µM) were simultaneously added. After incubation, the reaction was stopped, and cells were lysed by the addition of 20 µL working solution (10 µL Eu-cAMP and 10 µL ULight-anti-cAMP), and incubated for 1 h at room temperature. The TR-FRET signal was read using a microplate reader SpectraMax M5 (Molecular Devices). cAMP concentrations were determined using GraphPad Prism software (version 6.0; GraphPad Software Inc., San Diego, CA, USA).
Small interfering RNA knockdown
iHTMCs were seeded in quadruplicate into collagen I-coated 96-well microplates (4860-010; IWAKI) at a density of 5.0 × 103 cells/well with serum-free TMCM (1% penicillin/streptomycin; #6591; Sciencell). Twenty-four hours later, cells were transfected with 0.04 µM Accell SMARTpool siRNA against ADRB1, ADRB2, RAPGEF3, RAPGEF4, and PRKACA (Supplementary Table 1, Dharmacon), Accell GAPDH pool-human siRNA (D-001930) was used as a positive control, and Accell Non-targeting pool siRNA (D-001910) as a negative control using 100 µL Accell Delivery Media (B-005000-100; Dharmacon), according to the manufacturer’s instructions. Twenty-four hours later, NE (1 µM; 20 µL/well) or 0.1% DMSO with PBS containing pHrodo Green Zymosan Bioparticles (2.5 µg/well; P35365) were added. After 24 h of exposure to siRNA, total RNA was extracted and purified as described above for knockdown confirmation by qPCR.
PIP3 extraction and quantification
PIP3 measurements were performed using PIP3 Mass ELISA kit (K-2500s, Echelon Biosciences) according to the manufacturer’s instructions. iHTMCs were seeded at a density of 1.2 × 106 cells/collagen I coated six well dish (IWAKI). After 14 h of treatment with dobutamine (1 μM) with or without agonists, KT5720 (10 μM), ESI09 (1 μM), bpV (pic) (1 μM), and 3AC (10 μM); the mediums were removed, and ice-cold 0.5 M tricarboxylic acid (1 mL) was immediately added. Scraped iHTMCs were transferred into a 1.5 mL tube, and centrifuged at 3000 rpm for 7 min at 4 °C. The pellet was resuspended in 5% TCA/1 mM EDTA (0.5 mL), vortexed for 30 s, and centrifuged at 3000 rpm for 5 min at room temperature. The washing step was repeated. Next, we added 0.5 mL of MeOH: CHCl3 (2:1) to extract neutral lipids, vortexed for 10 min at room temperature, and centrifuged at 3000 rpm for 5 min. We next added 0.5 mL MeOH: CHCl3: 12 M HCl (80:40:1) to extract the acidic lipids, vortexed for 25 min at room temperature, and centrifuged them at 3000 rpm for 5 min. After transferring the supernatant to a new 1.5 mL tube, 0.15 mL of CHCl3 and 0.27 mL of 0.1 M HCl were added, vortexed, and centrifuged at 3000 rpm for 5 min to separate organic and aqueous phases. The lower organic phase 0.3 mL was transferred into a new vial, and dried for 4 h at 4 °C. PIP3 samples were resuspended in 125 μL of PBS-Tween including 3% protein stabilizer (provided by the Echelon kit). Samples were sonicated in an ice-water bath for 5 min, vortexed, and spun down before being added to the ELISA. All experiments were performed three times, each performed in triplicate. The lipid amount (60 μL) was used and was run twice for each sample. After color reaction for 30 min in the dark, the 96-well plate was read by measuring the absorption at 450 nm with a SpectraMax M5 (Molecular Device). PIP3 concentrations were determined using GraphPad Prism software (version 6.0; GraphPad Software Inc., San Diego, CA, USA).
Statistics and reproducibility
Results are shown as the mean ± standard error of the mean (SEM) from at least three independent experiments and five mice. Statistical comparisons were made using GraphPad Prism 6 (GraphPad Software Inc., San Diego, CA) or Excel-Toukei 2012 software (Social Survey Research Information Co. Ltd., Osaka, Japan). Paired or Student’s t-tests were used to compare two groups, and one-way analysis of variance (ANOVA) with Tukey’s multiple comparison test or Dennett’s multiple comparison test for more than three groups. Differences were considered statistically significant at p < 0.05.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
Data of gene expression in human AH outflow-related cells have been deposited in the Gene Expression Omnibus accession number GSE14618843. All data is available from the corresponding author on reasonable request. Source values for each of the following figures are available in the corresponding Supplementary Data files: Fig. 1 - Supplementary Data 1; Fig. 2 - Supplementary Data 2; Fig. 3 - Supplementary Data 3; Supplementary Fig. 4 - Supplementary Data 4; Fig. 5 - Supplementary Data 5; Fig. 6 - Supplementary Data 6; Fig. 7 - Supplementary Data 7; Supplementary Fig. 1 - Supplementary Data 8; Supplementary Fig. 2 - Supplementary Data 9; Supplementary Fig. 3 - Supplementary Data 10; Supplementary Fig. 5 - Supplementary Data 11; Supplementary Fig. 6 - Supplementary Data 12.
References
Ralph, M. R., Foster, R. G., Davis, F. C. & Menaker, M. Transplanted suprachiasmatic nucleus determines circadian period. Science 247, 975–978 (1990).
Mohawk, J. A., Green, C. B. & Takahashi, J. S. Central and peripheral circadian clocks in mammals. Annu. Rev. Neurosci. 35, 445–462 (2012).
Szabadi, E. Functional organization of the sympathetic pathways controlling the pupil: Light-inhibited and light-stimulated pathways. Front. Neurol. 9, 1069 (2018).
Kalsbeek, A. & Fliers, E. Daily regulation of hormone profiles. in Circadian Clocks (eds. Kramer, A. & Merrow, M.) 185–226 (Springer, 2013).
Bergeå, B., Bodin, L. & Svedbergh, B. Impact of intraocular pressure regulation on visual fields in open-angle glaucoma. Ophthalmology 106, 997–1004 (1999).
Liu, J. H. K. et al. Nocturnal elevation of intraocular pressure in young adults. Investigative Ophthalmol. Vis. Sci. 39, 2707–2712 (1998).
Tsuchiya, S., Higashide, T., Toida, K. & Sugiyama, K. The Role of beta-adrenergic receptors in the regulation of circadian intraocular pressure rhythm in mice. Curr. Eye Res. 42, 1013–1017 (2017).
Tsuchiya, S., Buhr, E. D., Higashide, T., Sugiyama, K. & Van Gelder, R. N. Light entrainment of the murine intraocular pressure circadian rhythm utilizes non-local mechanisms. PLoS One 12, 1–12 (2017).
Agnifili, L. et al. Circadian intraocular pressure patterns in healthy subjects, primary open angle and normal tension glaucoma patients with a contact lens sensor. Acta Ophthalmol. 93, e14–e21 (2015).
Renard, E. et al. Twenty-four hour (nyctohemeral) rhythm of intraocular pressure and ocular perfusion pressure in normal-tension glaucoma. Investigative Ophthalmol. Vis. Sci. 51, 882–889 (2010).
Mansouri, K., Weinreb, R. N. & Liu, J. H. K. Effects of aging on 24 h intraocular pressure measurements in sitting and supine body positions. Investigative Ophthalmol. Vis. Sci. 53, 112–116 (2012).
Grippo, T. M. et al. Twenty-four-hour pattern of intraocular pressure in untreated patients with ocular hypertension. Investigative Ophthalmol. Vis. Sci. 54, 512–517 (2013).
Ikegami, K., Shigeyoshi, Y. & Masubuchi, S. Circadian regulation of IOP rhythm by dual pathways of glucocorticoids and sympathetic nervous system. Invest Ophthalmol. Vis. Sci. 61, 26 (2020).
Shigekuni, O., Toichiro, K. & I, R. S. Selective destruction of the pigmented epithelium in the ciliary body of the eye. Science 184, 1298–1299 (1974).
Stamer, W. D. & Clark, A. F. The many faces of the trabecular meshwork cell. Exp. Eye Res. 158, 112–123 (2017).
Tamm, E. R. The trabecular meshwork outflow pathways: Structural and functional aspects. Exp. Eye Res. 88, 648–655 (2009).
Sit, A. J., Nau, C. B., McLaren, J. W., Johnson, D. H. & Hodge, D. Circadian variation of aqueous dynamics in young healthy adults. Investigative Ophthalmol. Vis. Sci. 49, 1473–1479 (2008).
Johnson, M. ‘What controls aqueous humour outflow resistance?’. Exp. Eye Res. 82, 545–557 (2006).
Fujimoto, T., Sato-Ohira, S., Tanihara, H. & Inoue, T. RhoA activation decreases phagocytosis of trabecular meshwork cells. Curr. Eye Res. 46, 496–503 (2021).
Dang, Y. et al. RKI-1447, a Rho kinase inhibitor, causes ocular hypotension, actin stress fiber disruption, and increased phagocytosis. Graefe’s Arch. Clin. Exp. Ophthalmol. 257, 101–109 (2019).
Chen, W. et al. Rho-associated protein kinase inhibitor treatment promotes proliferation and phagocytosis in trabecular meshwork cells. Front. Pharmacol. 11, 1–10 (2020).
Mylvaganam, S., Freeman, S. A. & Grinstein, S. The cytoskeleton in phagocytosis and macropinocytosis. Curr. Biol. 31, R619–R632 (2021).
May, R. C. & Machesky, L. M. Phagocytosis and the actin cytoskeleton. J. Cell Sci. 114, 1061–1077 (2001).
Zhang, X., Ognibene, C. M., Clark, A. F. & Yorio, T. Dexamethasone inhibition of trabecular meshwork cell phagocytosis and its modulation by glucocorticoid receptor β. Exp. Eye Res. 84, 275–284 (2007).
Matsumoto, Y., Bahler, C. K., Hann, C. R. & Johnson, D. H. Dexamethasone decreases phagocytosis in human trabecular meshwork. Investigative Ophthalmol. Vis. Sci. 37, 1902–1907 (1996).
Gosain, A., Muthu, K., Gamelli, R. L. & DiPietro, L. A. Norepinephrine suppresses wound macrophage phagocytic efficiency through alpha- and beta-adrenoreceptor dependent pathways. Surgery 142, 170–179 (2007).
Kureli, G. et al. F-actin polymerization contributes to pericyte contractility in retinal capillaries. Exp. Neurol. 332, 113392 (2020).
Fu, L., Lai, J. S. M., Lo, A. C. Y. & Shih, K. C. Induction of significant intraocular pressure diurnal fluctuation in rats using a modified technique of microbead occlusion. Int. J. Ophthalmol. 11, 1114–1119 (2018).
Bunker, S. et al. Experimental glaucoma induced by ocular injection of magnetic microspheres. J. Visualized Exp. https://doi.org/10.3791/52400 (2015).
Zhao, M., Hejkal, J. J., Camras, C. B. & Toris, C. B. Aqueous humor dynamics during the day and night in juvenile and adult rabbits. Investigative Ophthalmol. Vis. Sci. 51, 3145–3151 (2010).
Bradley, J. M., Aga, M. & Acott, T. S. Dynasore—a dynamin inhibitor alters outflow facility and endosomal uptake in trabecular meshwork cells. Investigative Ophthalmol. Vis. Sci. 53, 3238 (2012).
Marie-Anaïs, F., Mazzolini, J., Herit, F. & Niedergang, F. Dynamin-actin cross talk contributes to phagosome formation and closure. Traffic 17, 487–499 (2016).
Peterson, J. A. et al. Latrunculins’ effects on intraocular pressure, aqueous humor flow, and corneal endothelium. Investigative Ophthalmol. Vis. Sci. 41, 1749–1758 (2000).
Liu, X. et al. Low dose latrunculin-A inhibits dexamethasone-induced changes in the actin cytoskeleton and alters extracellular matrix protein expression in cultured human trabecular meshwork cells. Exp. Eye Res. 77, 181–188 (2003).
Johnson, D. H. The effect of cytochalasin D on outflow facility and the trabecular meshwork of the human eye in perfusion organ culture. Investigative Ophthalmol. Vis. Sci. 38, 2790–2799 (1997).
Borrás, T., Buie, L. K., Spiga, M. G. & Carabana, J. Prevention of nocturnal elevation of intraocular pressure by gene transfer of dominant-negative RhoA in rats. JAMA Ophthalmol. 133, 182–190 (2015).
Joe, M. K. et al. Analysis of glucocorticoid-induced MYOC expression in human trabecular meshwork cells. Vis. Res. 51, 1033–1038 (2011).
El-Shabrawi, Y. et al. Synthesis pattern of matrix metalloproteinases (MMPs) and inhibitors (TIMPs) in human explant organ cultures after treatment with latanoprost and dexamethasone. Eye 14, 375–383 (2000).
Bermudez, J. Y. et al. A comparison of gene expression profiles between glucocorticoid responder and non-responder bovine trabecular meshwork cells using RNA sequencing. PLoS One 12, 1–20 (2017).
Shepard, A. R. et al. Delayed secondary glucocorticoid responsiveness of MYOC in human trabecular meshwork cells. Investigative Ophthalmol. Vis. Sci. 42, 3173–3181 (2001).
Hernandez, M. R. et al. Glucocorticoid target cells in human outflow pathway: Autopsy and surgical specimens. Investigative Ophthalmol. Vis. Sci. 24, 1612–1616 (1983).
Drijfhout, W. J., Van Der Linde, A. G., Kooi, S. E., Grol, C. J. & Westerink, B. H. C. Norepinephrine release in the rat pineal gland: The input from the biological clock measured by in vivo microdialysis. J. Neurochem. 66, 748–755 (2010).
van Zyl, T. et al. Cell atlas of aqueous humor outflow pathways in eyes of humans and four model species provides insight into glaucoma pathogenesis. Proc. Natl Acad. Sci. USA 117, 10339–10349 (2020).
Daaka, Y., Luttrell, L. M. & Lefkowitz, R. J. Switching of the coupling of the β2-adrenergic receptor to different g proteins by protein kinase A. Nature 390, 88–91 (1997).
Schmidt, M., Dekker, F. J. & Maarsingh, H. Exchange protein directly activated by cAMP (epac): A multidomain cAMP mediator in the regulation of diverse biological functions. Pharmacol. Rev. 65, 670–709 (2013).
Makranz, C., Cohen, G., Reichert, F., Kodama, T. & Rotshenker, S. cAMP cascade (PKA, Epac, adenylyl cyclase, Gi, and phosphodiesterases) regulates myelin phagocytosis mediated by complement receptor-3 and scavenger receptor-AI/II in microglia and macrophages. Glia 53, 441–448 (2006).
Scott, J. et al. Exchange protein directly activated by cyclic AMP (EPAC) activation reverses neutrophil dysfunction induced by β2-agonists, corticosteroids, and critical illness. J. Allergy Clin. Immunol. 137, 535–544 (2016).
Steininger, T. S., Stutz, H. & Kerschbaum, H. H. Beta-adrenergic stimulation suppresses phagocytosis via Epac activation in murine microglial cells. Brain Res. 1407, 1–12 (2011).
Swanson, J. A. Phosphoinositides and engulfment. Cell. Microbiol. 16, 1473–1483 (2014).
Nizami, S., Hall-Roberts, H., Warrier, S., Cowley, S. A. & Di Daniel, E. Microglial inflammation and phagocytosis in Alzheimer’s disease: Potential therapeutic targets. Br. J. Pharmacol. 176, 3515–3532 (2019).
Schmidt, C. et al. Phosphoinositide 3-kinase γ mediates microglial phagocytosis via lipid kinase-independent control of cAMP. Neuroscience 233, 44–53 (2013).
Canetti, C. et al. Activation of phosphatase and tensin homolog on chromosome 10 mediates the inhibition of FcγR phagocytosis by prostaglandin E 2 in alveolar macrophages. J. Immunol. 179, 8350–8356 (2007).
Zhang, Y., Hoppe, A. D. & Swanson, J. A. Coordination of Fc receptor signaling regulates cellular commitment to phagocytosis. Proc. Natl Acad. Sci. USA 107, 19332–19337 (2010).
Ramakrishnan, G. S. & Humphrey, M. B. SHIP-1 inhibitors improve human microglial-like cell function. J. Immunol. 206, 111.22 LP-111.22 (2021).
Pedicone, C. et al. Pan-SHIP1/2 inhibitors promote microglia effector functions essential for CNS homeostasis. J. Cell Sci. 133, jcs238030 (2020).
Yoshitomi, T., Horio, B. & Gregory, D. S. Changes in aqueous norepinephrine and cyclic adenosine monophosphate during the circadian cycle in rabbits. Investigative Ophthalmol. Vis. Sci. 32, 1609–1613 (1991).
Saccà, S. C., Macrì, A., Rolando, M. & Ciurlo, G. Effect of betaxolol on primary open-angle glaucoma and normal-tension glaucoma patients. J. Ocul. Pharmacol. Therapeutics 14, 191–201 (1998).
Millar, J. C., Clark, A. F. & Pang, I. Assessment of aqueous humor dynamics in the mouse by a novel method of constant-flow infusion. Investigative Ophthalmol. Vis. Sci. 52, 685–694 (2011).
Mao, D. & Sun, X. Reactivation of the PI3K/Akt signaling pathway by the bisperoxovanadium compound bpV(Pic) attenuates photoreceptor apoptosis in experimental retinal detachment. Investigative Ophthalmol. Vis. Sci. 56, 5519–5532 (2015).
Liu, W. et al. Neuronal Epac1 mediates retinal neurodegeneration in mouse models of ocular hypertension. J. Exp. Med. 217, e20190930 (2020).
Perreau-Lenz, S. et al. Suprachiasmatic control of melatonin synthesis in rats: Inhibitory and stimulatory mechanisms. Eur. J. Neurosci. 17, 221–228 (2003).
Jain, A. et al. Effects of thailanstatins on glucocorticoid response in trabecular meshwork and steroid-induced glaucoma. Investigative Ophthalmol. Vis. Sci. 54, 3137–3142 (2013).
Yarangümeli, A. & Kural, G. Are there any benefits of Betoptic S (betaxolol HCl ophthalmic suspension) over other beta-blockers in the treatment of glaucoma? Expert Opin. Pharmacother. 5, 1071–1081 (2004).
Zhavoronkov, A. et al. Pro-fibrotic pathway activation in trabecular meshwork and lamina cribrosa is the main driving force of glaucoma. Cell Cycle 15, 1643–1652 (2016).
Qiu, H., Zhu, B. & Ni, S. Identification of genes associated with primary open-angle glaucoma by bioinformatics approach. Int. Ophthalmol. 38, 19–28 (2018).
Wordinger, R. J. & Clark, A. F. Effects of glucocorticoids on the trabecular meshwork: Towards a better understanding of glaucoma. Prog. Retinal Eye Res. 18, 629–667 (1999).
Shen, X., Koga, T., Park, B., Sundarraj, N. & Yue, B. Y. J. T. Rho GTPase and cAMP/protein kinase A signaling mediates myocilin-induced alterations in cultured human trabecular meshwork cells. J. Biol. Chem. 283, 603–612 (2008).
Nishio, M. et al. Control of cell polarity and motility by the PtdIns(3,4,5)P3 phosphatase SHIP1. Nat. Cell Biol. 9, 36–44 (2007).
Hoyle, N. P. et al. Circadian actin dynamics drive rhythmic fibroblast mobilization during wound healing. Sci. Transl. Med. 9, 1–11 (2017).
Cameron Millar, J., Phan, T. N., Pang, I. H. & Clark, A. F. Strain and age effects on aqueous humor dynamics in the mouse. Investigative Ophthalmol. Vis. Sci. 56, 5764–5776 (2015).
Shim, M. S., Kim, K. Y. & Ju, W. K. Role of cyclic AMP in the eye with glaucoma. BMB Rep. 50, 60–70 (2017).
Alvarado, J. A., Murphy, C. G., Franse-carman, L., Chen, J. & Underwood, J. L. Effect of β-adrenergic agonists on paracellular width and fluid flow across outflow pathway cells. Investigative Ophthalmol. Vis. Sci. 39, 1813–1822 (1998).
Belmonte, C., Barrels, S. P., Liu, J. H. K. & Neufeld, A. H. Effects of stimulation of the ocular sympathetic nerves on IOP and aqueous humor flow. Investigative Ophthalmol. 28, 1649–1654 (1987).
Zhu, J., Wang, Y., Hu, Q., Yuan, R. & Ye, J. Rottlerin acts as a therapeutic in primary open-angle glaucoma by targeting the trabecular meshwork via activation of Rap1 signaling. Pharmacol. Res. 159, 104780 (2020).
Komoto, S., Kondo, H., Fukuta, O. & Togari, A. Comparison of β-adrenergic and glucocorticoid signaling on clock gene and osteoblast-related gene expressions in human osteoblast. Chronobiol. Int. 29, 66–74 (2012).
Emorine, L. J. et al. Structure of the gene for human beta 2-adrenergic receptor: Expression and promoter characterization. Proc. Natl Acad. Sci. USA 84, 6995–6999 (2006).
Nuñez, F. J. et al. Glucocorticoids rapidly activate cAMP production via Gαs to initiate non-genomic signaling that contributes to one-third of their canonical genomic effects. FASEB J. 34, 2882–2895 (2020).
Kałuzny, B. J. Betaxolol for prevention of steroid induced intraocular pressure elevations in patients after radial keratotomy. Klinika Ocz. 105, 171–174 (2003).
Ishida, A. et al. Light activates the adrenal gland: Timing of gene expression and glucocorticoid release. Cell Metab. 2, 297–307 (2005).
Copinschi, G. & Challet, E. Endocrinology: Adult and Pediatric (eds. Jameson, J. L. & Groot, L. De) 147–173 (Elsevier, 2016).
Stehle, J. H. et al. A survey of molecular details in the human pineal gland in the light of phylogeny, structure, function and chronobiological diseases. J. Pineal Res. 51, 17–43 (2011).
Rommel, T. & Demisch, L. Influence of chronic β-adrenoreceptor blocker treatment on melatonin secretion and sleep quality in patients with essential hypertension. J. Neural Transm. 95, 39–48 (1994).
Orzalesi, N., Rossetti, L., Invernizzi, T., Bottoli, A. & Autelitano, A. Effect of timolol, latanoprost, and dorzolamide on circadian IOP in glaucoma or ocular hypertension. Investigative Ophthalmol. Vis. Sci. 41, 2566–2573 (2000).
Gregory, D. S. Timolol reduces IOP in normal NZW rabbits during the dark only. Investigative Ophthalmol. Vis. Sci. 31, 715–721 (1990).
Johnson, M., McLaren, J. W. & Overby, D. R. Unconventional aqueous humor outflow: A review. Exp. Cell Res. 158, 94–111 (2017).
Ikegami, K. et al. Effect of expression alteration in flanking genes on phenotypes of St8sia2-deficient mice. Sci. Rep. 9, 1–11 (2019).
Acknowledgements
We are grateful to the Institute of Comprehensive Medical Research, Division of Animal Research Promotion Division (Aichi Medical University), for maintaining the mice, providing advanced research promotion for the expression analysis of genes and proteins, and performing measurements of cAMP, fluorescence, and phagocytosis. We also thank Dr. Sachiko Takamura (Department of Microbiology and Immunology, Aichi Medical University) for their valuable advice. This study was supported by the JSPS KAKENHI (Grant Number 19K09962), the 24th General Assembly of the Japanese Association of Medical Sciences, Terumo Life Science Foundation, Takeda Science Foundation, Suzuken Memorial Foundation, Kato Memorial Bioscience Foundation, the UBE Foundation, and Toyoaki Scholarship Foundation.
Author information
Authors and Affiliations
Contributions
K.I. contributed conceptualization, methodology, investigation, writing original draft, funding acquisition. K.I. and S.M. contributed writing review & editing, resources, supervision.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks Ponugoti Vasantha Rao and Diego Sbardella for their contribution to the peer review of this work. Primary Handling Editors: Karli Montague-Cardoso and Christina Karlsson Rosenthal. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ikegami, K., Masubuchi, S. Suppression of trabecular meshwork phagocytosis by norepinephrine is associated with nocturnal increase in intraocular pressure in mice. Commun Biol 5, 339 (2022). https://doi.org/10.1038/s42003-022-03295-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-022-03295-y
- Springer Nature Limited
This article is cited by
-
Circadian rhythm of intraocular pressure
The Journal of Physiological Sciences (2024)