
Abstract
Cystic echinococcosis is a socioeconomically important parasitic disease caused by the larval stage of the canid tapeworm Echinococcus granulosus, afflicting millions of humans and animals worldwide. The development of a vaccine (called EG95) has been the most notable translational advance in the fight against this disease in animals. However, almost nothing is known about the genomic organisation/location of the family of genes encoding EG95 and related molecules, the extent of their conservation or their functions. The lack of a complete reference genome for E. granulosus genotype G1 has been a major obstacle to addressing these areas. Here, we assembled a chromosomal-scale genome for this genotype by scaffolding to a high quality genome for the congener E. multilocularis, localised Eg95 gene family members in this genome, and evaluated the conservation of the EG95 vaccine molecule. These results have marked implications for future explorations of aspects such as developmentally-regulated gene transcription/expression (using replicate samples) for all E. granulosus stages; structural and functional roles of non-coding genome regions; molecular ‘cross-talk’ between oncosphere and the immune system; and defining the precise function(s) of EG95. Applied aspects should include developing improved tools for the diagnosis and chemotherapy of cystic echinococcosis of humans.
Similar content being viewed by others
Introduction
Cystic echinococcosis (hydatidosis) of humans is a neglected tropical disease (NTD) caused by the larval (metacestode) stage of the tapeworm (cestode) Echinococcus granulosus (family Taeniidae). This parasite has a complex life cycle, involving definitive hosts (canids) and intermediate hosts (ungulates—such as sheep, goats, cattle, camels—and macropods). Humans become infected when they accidentally ingest eggs released from canids infected with adult tapeworms; motile larvae (oncospheres) hatch from these eggs, penetrate the intestinal wall, enter blood vessels and are then passively transported to key predilection sites, mostly liver and lung. There, oncospheres grow and develop to cysts (over months and years), which internally produce larval stages (protoscoleces) asexually. The growth and propagation of these cysts cause severe disease1.
The prevention and control of echinococcosis rely on breaking transmission from host to host. Although canid definitive hosts can be treated at regular intervals (3–4 weeks) to eliminate adult worms from their small intestines, chemotherapy of people affected by echinococcosis with drugs, such as mebendazole or albendazole, is often ineffective2,3. An effective means of control is to vaccinate intermediate hosts (e.g., sheep) against E. granulosus to prevent them from becoming infected, thus disrupting transmission to definitive hosts4,5,6. Indeed, the development of a recombinant vaccine (called EG95) that protects sheep (with an efficacy of 95–99%) against echinococcosis6,7 has been one of the biggest milestones in the fight against neglected tropical diseases (cestodiases) caused by taeniid cestodes4,8,9.
Despite this breakthrough and the major relevance of this vaccine molecule, there is limited information regarding the genome organisation and fundamental biological role(s) of genes encoding EG95 as well as their conservation/variability and immunobiology. Using classical molecular approaches, key studies revealed that EG95 (encoded by a gene originally called eg95-1) was represented by a family of genes and a pseudogene10,11, and gene products have been localised specifically to the penetration glands (type-1) of the infective larval (oncosphere) stage12. Recent work13 attempted to localise the Eg95 genes in the genome, but did not achieve an outcome because of the fragmentation of draft genomes for E. granulosus (genotype G1) available at the time of investigation, leading to an inability to reliably map the coding genes to these genomes13. Currently available draft genomes for E. granulosus14,16 were assembled from short-read data sets, produced using a ‘second generation’, high throughput sequencing platform (Illumina technology), and one assembly was guided by scaffolding to a well-assembled genome for the congener E. multilocularis14. However, the use of short-read data sets does not allow the accurate assembly of long repeat regions within cestode genomes15, preventing the characterisation of non-coding RNA genes and some gene families within such regions, such as that encoding EG95. This challenge can be overcome by using third-generation, long-read sequencing technologies15. Thus, there has been a major need for a genome of near-chromosomal contiguity to enable (i) the accurate mapping of genes to the genome; (ii) the exploration of the organisation of the Eg95 and other gene families; (iii) fundamental investigations of the molecular biology, biochemistry and genetics of E. granulosus; and (iv) the development of improved diagnostic tools and new chemotherapies against cystic echinococcosis in humans. Here, we employed a combination of sequencing methods to generate complementary data sets to achieve a high-quality (chromosome-scale) genome assembly for genotype G1 of E. granulosus, localised members of the Eg95 gene family in this genome and assessed sequence conservation of the EG95 vaccine molecule. We discuss the implications of this study for future research on Echinococcus/echinococcosis.
Results and discussion
Assembly of the Eg-G1s reference genome
A high-quality reference genome for E. granulosus (genotype G1) was essential to undertake structural and comparative genomic analyses. The long-read data obtained (31.7 Gb, 212-fold coverage; Supplementary Table 1) were combined with available paired-end read data to produce a genome assembly of 173 Mb (designated Eg-G1s; scaffold N50/N90 = 18.7/12.3 Mb; scaffold L50/L90 = 4/8; Table 1). This assembly (Eg-G1s) comprised 9 scaffolds (512 contigs) that were consistent with chromosomes and represented 97% of the genome (Fig. 1), with <4.6 Mb of sequence present in 5 scaffolds (17 contigs). Contiguity of the Eg-G1s assembly (Table 1) was substantially greater than the 371 scaffolds (3736 contigs)14 and 957 scaffolds (8264 contigs)16 achieved for E. granulosus in previous studies. The assembly was ‘polished’ with short-reads (25.1 Gb with 167-fold coverage; Supplementary Table 1) from the same E. granulosus sample to correct indels and single nucleotide alterations, with 97% of these reads mapping back to the “polished” assembly (Supplementary Table 1).

a Circular representation of the Echinococcus granulosus genome (genotype G1; designated Eg-G1s) with nine chromosomes (Ch1 to Ch9); indicated are gene (blue), repeat (orange) and encoded RNA (green; log2) densities ranging from 0 to 100% (bin-size of 100 kb) across the genome and the locations of the four Eg95 genes (Eg95-1, -4, -5 and -6). b Structure of the four Eg95 gene family members—thick and thin bars denote 3 exons and 2 introns, respectively. Black bars indicate 100% identity to Eg95-1; shades of grey to white correspond to sequence identity (%) to Eg95-1 (scale, below). c Structure of the Eg95-1 gene and mRNA. Predicted Goldberg-Hogness box (TATAA), start site (ATG), termination codon (TGA) and polyadenylation signal (AATACG) are indicated; the first and last exons are flanked by non-coding regions; mRNA includes 5ʹ- and 3ʹ-UTRs (white) and coding regions (grey). d Complete amino acid sequence of EG95-1 compared with those predicted for EG95-4, EG95-5 and EG95-6. Dashes indicate gaps inserted for the purpose of the alignment. Pairwise sequence comparisons among these four sequences range from ~77% to 99% identity.
Long genomic repeat regions
Conspicuous in the Eg-G1s genome were five long repeat regions with low densities of genes on chromosomes 1, 2, 4, 7 and 8 (Fig. 1). Repeat regions in chromosome 1 harboured 23 protein-coding genes and ten 5 S ribosomal (r)RNA genes; the beginning of chromosome 2 had 7 genes coding for proteins including histones H2B/H3/H4, and 10 large subunit (LSU), small subunit (SSU), 5.8 S and 5 S rRNA, and U2 spliceosomal small non-coding (sn)RNA genes (Supplementary Data 1; Fig. 1a); the end of chromosome 2 had a long (~ 11.5 Mb) repetitive tract of LSU, SSU, 5.8 S rRNA and U2 spliceosomal snRNA genes (n = 44) and 21 protein-coding genes (Supplementary Data 1; Fig. 1a). Repeat regions were distributed across chromosome 4 and contained LSU, SSU, 5.8 S rRNA and U2 spliceosomal snRNA genes (n = 80) and 104 protein-coding genes including some encoding histones (H2A, H2B, H3 and H4) (Supplementary Data 1; Fig. 1a). Repeat regions at the end of chromosomes 7 and 8 contained 46 and 67 protein-coding genes, respectively, with mainly U3 spliceosomal snRNA genes (n = 8) uniquely present in chromosome 7 (Supplementary Data 1; Fig. 1a). A comparison of the nature and extent of repeat regions (Supplementary Tables 2 and 3; Supplementary Data 2 and 3) revealed that only chromosomes 1 and 2 have similarities in that they share 18 of the most frequent repeat elements with each other, with non-coding RNA gene regions being unique to the latter chromosome; all other chromosomes have distinctly different repeat and gene contents (Supplementary Table 3; Supplementary Data 1, 2, 4 and 5).
Long repetitive genomic regions cannot be assembled using short-read sequencing approaches (e.g., Illumina), which is why previous genome assemblies for Echinococcus species14,16—although likely comprising most protein-coding genes—were one-third smaller than the genome size inferred here (173 Mb; Table 1). In accord with recent studies17,18,19,20, we demonstrate here the advantage of using long-read and complementary sequencing methods to overcome fragmentation in the assembly of complex genomes. The proximity of the Eg95 genes to the long repetitive regions identified here raises a question as to whether these regions play roles in regulating the transcription and/or expression of these genes via non-coding RNAs, warranting future exploration.
Gene content and comparison among species
To support gene predictions and explore transcription in key developmental stages of E. granulosus genotype G1, we produced RNA-seq data for both the adult worm and oncosphere stages and sourced publicly available data for the protoscolex stage14,16. All of these data were mapped and transferred to the assembled genome (Eg-G1s), and 9985 protein-coding genes were identified (Table 1). As this genome assembly exhibited features consistent with ‘reference quality’21, including high contiguity and N50/N90, very few gaps and unplaced sequences, and evidence of high-quality protein-coding genes (cf. Table 1), we defined this version as a chromosome-level reference genome (Eg-G1s; accession no. PRJNA754835 in NCBI) whose proteome-completeness metrics (BUSCO) were similar to those achieved previously for E. granulosus and E. multilocularis14 (Table 1). Having validated assembly-quality, we then assigned functions to 8972 (89.9%) of the 9985 protein-coding genes in the Eg-G1s genome (Supplementary Data 4); 1013 (10.1%) genes could not be annotated, 593 (5.9%) of which were transcribed (‘unknowns’ or orphans) and 208 (2.1%) of which encoded proteins with domains, motifs or signatures consistent with excretory/secretory or membrane-bound molecules (Supplementary Data 4).
In a comparative analyses, we identified more paralogous genes (n = 1642) in Eg-G1s than in previous draft assemblies for E. granulosus (1188 and 1231, respectively)14,16 (Fig. 2d), with ‘novel’ paralogs being discovered in long repeat regions containing gene families encoding proteins including histones H2A, H2B & H4 and variable surface glycoproteins (Supplementary Table 4), and we found fewer unique orphan (unknown) proteins encoded in Eg-G1s than in previous assemblies (Fig. 2d). Although the gene sets predicted for previous E. granulosus draft genomes14,16 contain 1432 protein-coding genes without homologues in Eg-G1s (Supplementary Data 6; Fig. 2d), 486 of them are orphans (Supplementary Data 6). However, before the removal of 652 low-confidence genes from Eg-G1s using stringent criteria (Supplementary Fig. 1; Supplementary Data 7), the differential gene set was 771 genes, including 370 orphans (Supplementary Data 6). Thus, the final Eg-G1s gene set (n = 9985) compares to that of E. multilocularis, with 8998 homologues shared with Taenia multiceps (n = 6611) and Hymenolepis microstoma (n = 7293) (Fig. 2c).

a Synteny of the nine chromosomes (Ch1 to Ch9) of the genome Eg-G1s of Echinococcus granulosus (genotype G1) with scaffolds or chromosomes in the genome assemblies of E. multilocularis14, Taenia multiceps22 and Hymenolepis microstoma23. Each line represents a single copy orthologous (SCO) gene between two species (grey—same orientation; green—reverse orientation). Scale bar (top right) indicates chromosome length (Mb). b Consensus tree showing the genetic relationship of the four cestode species using data for 4040 shared SCOs (nodal support values: 1.0 and 100% for MrBayes and RAxML analyses, respectively; scale bar: substitutions per sequence site). c Venn diagram displaying the numbers of orthogroups between or among the four cestode species obtained using the program OrthoFinder68 (numbers of E. granulosus genes in parentheses). d Venn diagram comparing the numbers of genes (using OrthoFinder) common or distinct between or among the reference genome Eg-G1s (top left) and previously published assemblies14,16. Numbers of paralogous genes (small ovals) and orthologous and/or single copy genes (large ovals and overlaps) are indicated, as are orphan (unknown) genes (in parenthesis). Numbers of gene predicted (n = 1432; 539 + 172 and 539 + 182) from two previous draft genomes of E. granulosus14,16 for which homologous protein-coding genes were not identified in the final gene set of Eg-G1s. White lettering was used only to improve visibility of numbers on dark background.
Marked synteny between E. granulosus and other tapeworms
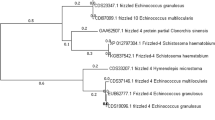
Pairwise comparisons revealed that there was notable synteny between the Eg-G1s and published genome assemblies for E. granulosus14, E. multilocularis14, T. multiceps22 and H. microstoma23 (Fig. 2; Supplementary Fig. 2). As expected, most synteny was observed between Eg-G1s and the E. multilocularis reference genome (Fig. 2; Supplementary Data 8), with 92.8% of nucleotides (n = 98,392,918 bp; 10 scaffolds) of the latter aligning to 72.9% of nucleotides (n = 125,154,915 bp; 11 scaffolds) of the former in 477 syntenic blocks. There was less synteny between Eg-G1s and each T. multiceps and H. microstoma (Fig. 2; Supplementary Data 8), with 42.2% and 61.1% of nucleotides in T. multiceps and H. microstoma aligning to 55.8% to 38.6% of nucleotides in the Eg-G1s genome (9 scaffolds) in 50 to 270 syntenic blocks, respectively (Supplementary Data 8). The observed syntenies (Fig. 2a) were compared with phylogenetic distances (Fig. 2b) between/among species.
Chromosomal localisation of four Eg95 genes and their transcription
The high-quality Eg-G1s reference genome (Table 1; Fig. 1) provided the basis to localise members of the Eg95 gene family in the genome, as previous attempts had failed due to the fragmented nature of draft genomes assembled using short-read data sets alone13. Here, we defined four distinct genes, Eg95-1, Eg95-4, Eg95-5 and Eg95-6. Four of the previously-characterised alleles, eg95-1, eg95-4, eg95-5 and eg95-610, were unequivocally assigned to genes Eg95-1, Eg95-4, Eg95-5 and Eg95-6, and the two other alleles eg95-2 and eg95-310 could be assigned to Eg95-1 and/or Eg95-4 (but not unequivocally to either due to their substantial nucleotide sequence identity: 99.2%). Genes Eg95-1 (encoding protein EG9510) and Eg95-4 were localised to chromosomes 2 and 4, respectively, and genes Eg95-5 and Eg95-6 were at the end of chromosome 2 (Fig. 1). All four Eg95 genes are encoded in repeat-rich regions of the genome; Eg95-5 and Eg95-6 are close to one another and to the end of chromosome 2 (Fig. 1).
Exploring transcription in the distinct developmental stages of E. granulosus gave insight into biological processes and pathways. Genes Eg95-1 (log2 FC = 15) and Eg95-4 (log2 FC = 12) had the highest transcription in the activated oncosphere stage, followed by Eg95-5 (log2 FC = 7) and Eg95-6 (log2 FC = 6), with reference to the protoscolex stage, whereas all four Eg95 genes had low levels of transcription in adult worms of E. granulosus (Supplementary Table 5; Supplementary Data 6; Supplementary Fig. 3). A weighted network analysis defined four distinct clusters (each with sub-clusters ‘+’ and ‘–’) of genes whose transcription was correlated among the protoscolex, adult and oncosphere stages. In the activated oncosphere, the 915 genes that grouped with the four Eg95 family members (all within cluster 1; Fig. 3) were inferred to be linked to key biological pathways, including genetic information processing (ribosome/translation; folding, sorting and degradation/proteasome; protein processing in endoplasmic reticulum; DNA replication and repair); environmental information processing (signal transduction and signalling molecules; Rap1, Ras, PI3K-Akt, Notch and JAK-STAT); cellular processes (focus adhesion and adherens junction); and metabolism (amino acid and energy) (Fig. 3; Supplementary Data 9 and 10). Other clusters of genes were associated with environmental information processing (signalling), organismal systems (endocrine) and cellular processes (e.g., cell motility and cytoskeleton) (cluster 2); metabolism (carbohydrate, vitamin and co-factors), organismal systems (e.g., carbohydrate digestion/absorption, endocrine and excretion/absorption) (cluster 3) or environmental information processing (signal transduction), cellular processes (e.g., regulating pluripotent stem-cells) and organismal systems (nervous/synapse, endocrine and development/regeneration) (cluster 4) (Fig. 3; Supplementary Data 9 and 10). While Eg95 genes do not map to currently-known biological pathways or processes, their high transcription in the oncosphere associates with at least 1102 other cluster 1-genes linked to extensive cellular signalling, metabolism and adhesion, in accord with essential processes required for the parasite to invade/infect the intermediate host animal, including the penetration of the small intestinal wall using oncospheral hooks and excretions/secretions from the penetration glands—in which EG95 is expressed12—to then enter lacteals and/or capillaries for subsequent passive transport to the liver and/or lung, where this stage undergoes post-oncospheral alteration to initiate cyst development (Fig. 3).

a Life cycle of E. granulosus with key developmental stages indicated – adapted from ref. 23 – canid definitive host (DH); intermediate host (IH). b Four distinct clusters (each with sub-clusters + and –; divided according to fold-change (FC) ≥ 4 and FC ≤ −4, respectively) of genes whose transcription correlated among the protoscolex, adult and oncosphere stages, inferred by weighted correlation network analysis (numbers in boxes are gene counts). The four Eg95 genes (within sub-cluster 1 + ) are highly transcribed in the oncospheral stage. Enriched biological (KEGG) pathways representing individual gene clusters/sub-clusters are indicated. White lettering was used only to improve visibility of numbers on dark red background.
Conservation of EG95-1 and related molecules
Studies conducted in a range of countries, including Australia, Argentina and China, have shown that the recombinant EG95 vaccine consistently induces high levels (95–99%) of protection in the intermediate host (sheep) against challenge infection with E. granulosus eggs6,7. However, no study has yet comprehensively assessed sequence variation in Eg95-1 and related genes within E. granulosus in relation to geographical and/or host origin. To explore sequence variation in the gene encoding EG95-1, high-quality short-read genomic data (mean: 31 Gb) for each of 47 E. granulosus samples were mapped (coverage: 100%; depth at each nucleotide position: ≥20; mean depth: >80) to the Eg95-1 gene within the genome (Supplementary Data 11). No fixed nucleotide difference was detected (upon pairwise comparison) in the open reading frame (ORF = 3 exons) of this gene for any of the 47 individual samples when compared with the reference genome sequence (Eg-G1s) (Fig. 4). Although minor polymorphism (allelic variability) was detected at 13 positions in all 3 exonic regions of Eg95-1 (Fig. 4; Supplementary Data 12), the dominant base at each of these positions matched the reference sequence (Supplementary Data 13). This heterozygosity, detected also in PacBio long-read and transcriptomic data sets, was expected because Eg-G1s is presented as a haploid reference representing a diploid organism (i.e., E. granulosus24).

a Genomic DNA samples (n = 47) representing single cysts or adult worms of Echinococcus granulosus (genotype G1 or G3) from 8 distinct host species and 10 different countries were sequenced. b Mapping of sequence data from individual samples to the haploid reference genome (Eg-G1s) sequence detected polymorphism (allelic variability) but no unambiguous (i.e. fixed or homozygous) nucleotide difference in the 3 exons of Eg95-1 for any of the (diploid) sequences from any of the 47 samples with the reference sequence. Black horizontal bars represent the three exons (1 to 3) and black lines denote intervening introns. Polymorphic positions are indicated above each exon: a dominant base (black) matches the Eg-G1s reference sequence; a grey base represents the minor allele (cf. Supplementary Data 13); a fixed nucleotide difference from the reference sequence is indicated at one position; and a dash indicates an indel. c Mapping of allelic variation of EG95-1 to the modelled three-dimensional structure of the vaccine molecule EG95 reveals variable regions (see colour-key for percentage conservation) in the N-terminal α-helix, as well as two β-strands, each of which located in one of the predicted anti-parallel β-sheets. All residue side chains subject to allelic variation are surface exposed, and thus, due to the conservative nature of most mutations (A→T, T→I, G→E, M→R, V→I, R→H, E→D, D→S), overall structural conservation of the vaccine molecule (EG95-1) can be inferred.
The amino acid conservation inferred here for the vaccine molecule (i.e. EG95-1), based on the mapping of short-read data derived from E. granulosus (G1 or G3) from sheep or other host species, including cattle, buffalo, goat, pig, dog/dingo and human from 10 different countries (Fig. 4; Supplementary Data 11) is consistent with the biological evidence of a consistently high degree of protection achieved by the EG95 vaccine in sheep against cystic echinococcosis, irrespective of geographical location6,7, and also in accord with findings from some previous investigations indicating molecular conservation of EG95-1 in E. granulosus genotype G125,26. The evidence of conservation in EG95-1 contrasts the results from one study27, suggesting marked nucleotide variability in Eg95-1, which we interpret might relate to artefacts introduced due to the methodology employed at the time; the PCR-primers employed match both genes Eg95-1 and Eg95-4 and, thus, would have co-amplified these and potentially other genes and/or might have created artefactual ‘chimeras’ in PCR, ultimately being reflected in apparently variable sequences ensuing molecular cloning and sequencing.
Our findings, using both long-read and short-read data, provide clear evidence of heterozygosity in Eg95-1 (Fig. 4) and in the three other Eg95 genes (Supplementary Data 12). At this point, we are not able to conclude whether this allelic variation is present within cells, among distinct cell types within individual developmental stages of E. granulosus12 or among individuals within cysts or worm populations (i.e. samples), but we speculate that this allelic variation for members of the Eg95 gene family is critical for adaptation to distinct host species and survival. Major transcription of Eg95-1 in activated oncospheres, but not in adult worms (containing intact eggs), is consistent with immunohistochemical evidence of pronounced expression of the EG95 protein in penetration glands within activated, infective oncospheres, but substantially less in eggs12. Interestingly, as EG95 appears not to be expressed on the tegument of the activated oncosphere stage, it is proposed that this infective stage is killed by complement-mediated antibody attack in EG95-vaccinated sheep during early post-oncospheral development in tissues in lung or liver12.
Conclusion
In addition to defining the genomic locations, structures and compositions of the Eg95 gene family members and demonstrating the conservation of the EG95-vaccine molecule, the genomic and transcriptomic resources created here pave the way for myriad future molecular explorations of cystic echinococcosis/E. granulosus. Further work will be required to comprehensively explore developmentally regulated gene transcription and expression using at least four replicate samples for individual stages of E. granulosus. Investigating the structural and functional roles of the long stretches of non-coding DNA in the genome would also be interesting. At the host-parasite interface, it would be exciting to explore the molecular ‘cross-talk’ between oncosphere and the immune system, and the function(s) of EG95 as a fibronectin III domain-containing molecule, possibly involving the use of well-defined liver and/or lung organoid systems28,29. On a broader scale, exploring molecular variation within E. granulosus sensu stricto from the host and distributional ranges across the globe, in genome-wide manner, could comprehensively document the population genetic sub-structuring, with implications for understanding transmission patterns of cystic echinococcosis. From an applied perspective, the inference and functional evaluation of essential genes encoded in the Eg-G1s genome could enable the discovery of new intervention targets for the treatment of cystic echinococcosis in people. These are just some of the areas that should be positively impacted by the availability of a chromosome-scale genome and associated data and tools.
Methods
Ethics statement
No ethics permissions were required for this study. Samples of E. granulosus (cf. Supplementary Data 11) were collected from animals by logistical support personnel and professionals, with approval from relevant institutions in individual countries; samples were donated to the investigators of this article.
Genomic sequencing
High molecular weight genomic DNA (1 µg) was isolated from protoscoleces (200 µl packed volume) from an individual cyst of E. granulosus (genotype G1) obtained from a sheep (Ovis aries) from New South Wales, Australia, using an established sodium dodecyl-sulphate–proteinase K digestion protocol and phenol/chloroform extraction10. The DNA amount was determined using a Qubit fluorometer dsDNA HS Kit (Invitrogen), and DNA integrity was verified using a Bioanalyzer 2100 (Agilent). For long-read sequencing, a SMRTbell library was constructed from 8 μg of genomic DNA (≥10 kb) without prior shearing, employing the SMRTbell Template Prep Kit 1.0, following the manufacturer’s protocol, and enriching for templates of >10 kb using the BluePippin system (Sage Scientific, MA, USA). This library was sequenced (chemistry v.2.1) in three SMRT cells using the PacBio Sequel System (Pacific Biosciences, Menlo Park, CA, USA). For short-read sequencing, a paired-end library (insert size: 500 bp) was constructed from 1 μg of genomic DNA using the MGIEasy DNA Library Prep Kit (V1.1, MGI Tech Co., Ltd, Shenzhen, China), employing the recommended protocol, and then sequenced (PE100 chemistry) using the BGISEQ-500RS platform.
RNA-seq and transcription analysis
For the protoscolex stage of E. granulosus (genotype G1), RNA-seq data (8 samples) were obtained from the NCBI Sequence Read Archive (SRA; accession number SRP17251730). For adults and activated oncospheres12, one sample each, of E. granulosus (genotype G1), total RNAs were isolated from hundreds of individuals using the TriPure isolation reagent (Roche Molecular Biochemicals). RNA yield was estimated spectrophotometrically (NanoDrop 1000), and integrity verified using the BioAnalyzer (Agilent). RNA-seq was conducted using an established method31 on a NovaSeq 6000 instrument and relevant data summarized (cf. Supplementary Table 1). The genome-guided assembly of RNA-seq data was performed using a software pipeline, incorporating the program Trimmomatic v0.3632 for read quality filtering, Hisat2 v2.1.033 for read mapping, Trinity v2.8.434 for sequence assembly and CD-HIT-EST v4.8135 for reducing redundancy. EdgeR v3.3236 was used to estimate log2-fold change (FC) in transcription of individual Eg95 genes between each the adult or oncosphere and the protoscolex stage of E. granulosus employing an established protocol37 and using a minimum counts-per-million threshold of 0.35. For EdgeR, expected read-counts were calculated using the program RSEM v1.3.338.
Genome assembly
An established pipeline21 was used to create an initial assembly from PacBio sequence data. In brief, these data were assembled using the program Canu v1.839, polished with both PacBio raw reads using the program Arrow40 and with BGISEQ-500 PE reads employing the software Pilon v1.2241. Redundant sequences were removed from the assembly using the program Purge Haplotigs v1.1.142, and resultant contigs were combined with longer contigs using a customised workflow v0.0.1-publication (https://gitlab.unimelb.edu.au/vetscience/gapmaster), which includes the program RagTag v1.1.0 (https://github.com/malonge/RagTag) for scaffolding and TGS-GapCloser v1.0.343 to close gaps. This workflow was run in an iterative manner, guided by previously published genome sequences for E. granulosus14 and E. multilocularis14, using high-quality, corrected long reads. The quality of gap closure was verified in each iteration; if there was any indication of a break point in the flanking regions of closed gaps, or if they had a repeat content of >50%, scaffolds were broken again into contigs. The process was repeated until no more gaps could be closed. Using Pilon, resultant combined contigs were iteratively polished both with short-read data to remove mismatches and indels, and with RNA-seq data (SRR8281957–SRR8281959, SRR8284434–SRR8284436 and SRR8293717–SRR8293719; for E. granulosus G1) to remove indels of <10 bp in length. Using RagTag, final scaffolding (Supplementary Data 14) was carried out using homologous sequences (without closing gaps) in genomes of E. multilocularis and E. granulosus.
Gene prediction and functional annotation
Gene models were predicted using a custom pipeline (v0.0.1-publication; https://gitlab.unimelb.edu.au/bioscience/annotosis), which employs the programs AUGUSTUS v3.4.044, StringTie v2.1.445, GMAP v2020.10.1446, EMBOSS v6.6.047, TransDecoder v5.5.034 and CD-HIT 4.8.135 using the same RNA-seq data as evidence utilised for gene and genome polishing as well as gene models from a previous E. granulosus assembly14 using the program LiftOver48 and all Swiss-Prot protein sequences within UniProtKB49 (accessed 15 March 2021). The quality of the predicted genes was assessed using a custom pipeline (v0.0.1-publication; https://gitlab.unimelb.edu.au/bioscience/annotosis), which builds on the programs Kmeans in the R language50, fLPS51, table2asn52, InterPro 5.5153 (https://www.ebi.ac.uk/interpro/), bedtools54, OrthoMCL v2.0.455, GeneValidator v2.1.1056 and BUSCO 5.1.257. Genes inferred to be of a low quality, based on the observed ‘steepest curvature before the shoulder point’ in the graph displaying the estimated gene-wise quality scores, were removed. The annotation of each inferred amino acid sequence was achieved using InterPro and sequence homology to proteins in the Swiss-Prot, KEGG58; accessed (30 June 2021) and NCBI NR59; accessed (4 February 2021) databases using BLASTp (threshold E-value: ≤10−8). Genes that were transcribed at ≥ 0.35 counts-per-million but not annotatable were designated as ‘unknowns’ or ‘orphans’. Nuclear LSU, SSU, 5.8 S and 5 S rRNA, spliceosomal snRNA were predicted by applying the program Infernal v1.1.460 with Rfam 14 database61 to the assembly.
Prediction of repeat regions
Genomic repeat elements specific to E. granulosus were first inferred using the programs RECON62 and RepeatScout63. These repeats were processed using the program RepeatModeler64 to obtain custom repeats, which were then combined with known repeats from Repbase v.17.0265 to mask the Eg-G1s assembly employing the program RepeatMasker66.
Assessing genome completeness and synteny
First, the completeness of the E. granulosus genome (Eg-G1s) was assessed using the program BUSCO v5.1.2 (lineage: Metazoa). Second, the synteny of Eg-G1s with the published (repeat-masked) genomes of E. granulosus, E. multilocularis14, T. multiceps and H. microstoma was visually assessed using the program circos v.0.2367 by identifying genomic locations of protein-encoding single-copy orthologs (SCOs) in a pairwise manner and inferred using OrthoMCL v2.0.455. Homologous genes between/among these species and previous E. granulosus assemblies14,16 were inferred using the program OrthoFinder v2.5.468, and the numbers of shared homologous gene groups displayed in Venn diagrams. The numbers of orphan genes in previous E. granulosus assemblies14,16 were established via BioMart in WormBase ParaSite69.
Phylogenetic analysis
Aligned amino acid sequences of SCOs among E. granulosus (genotype G1) genome, E. multilocularis, T. multiceps and H. microstoma were subjected to (unrooted) phylogenetic analyses using Bayesian inference (BI) in MrBayes v.3.2.270,71 and maximum likelihood (ML) in RAxML v.8.0.2472. The evolutionary models were established using the program PartitionFinder v.2.1.173. The number of Markov Chain Monte Carlo (MCMC)74 iterations for BI was 10 million generations, from which the first 25% were discarded as non-converged ‘burn-in’. For ML, nodal support values were assessed by 1000 bootstrap replicates. The resultant trees were then subjected to analysis in the program SumTrees using DendroPy v.3.12.075 to produce a consensus tree, and drawn using the program FigTree v.1.4 (https://www.softpedia.com/get/Science-CAD/FigTree-AR.shtml).
Network analysis, and pathway and functional enrichment
Using the program WGCNA v1.69, we performed a weighted correlation network analysis76 to define correlated clusters (modules) of genes in the protoscolex, adult and oncosphere stages of E. granulosus according to level of transcription (in TMM normalised expected counts) using a minimum cluster size of 500 genes and a scale-free index of 0.6. Qualitative transcription analysis was undertaken, as single adult and oncosphere samples were available (such samples are challenging to source and infective to humans). For the individual developmental stages, the clusters obtained were subdivided according to the log2-fold change (FC): ≥ 2 (high; h) or ≤ −2 (low; l). FCs were compared among the stages using the program egdeR v3.32.036, in accord with best practice for qualitative analysis37. Expected read-counts used in WGCNA and in EdgeR, and TPMs used in a heatmap were calculated using the program RSEM. The enrichment of the Kyoto Encyclopaedia of Genes and Genomes (KEGG) pathways and the BRITE functional hierarchies58 was inferred using a well-established methodology77. For heatmap display, TPM values were averaged for the protoscolex stage.
Short-read sequencing of genomic DNA samples from distinct host species and geographical locations around the world
Genomic DNA samples representing E. granulosus genotypes G1 (n = 41) and G3 (n = 6) (Supplementary Data 11) were available from previous studies78,79,80,81. Total genomic DNA had been extracted from protoscoleces or germinal membrane from single cysts from intermediate hosts species, or single adult worms of E. granulosus from individual canids, using the High Pure PCR Template Preparation Kit (Roche Diagnostics, Mannheim, Germany). DNA amounts were determined using a Qubit fluorometer dsDNA HS kit (Invitrogen). Then, individual DNA samples were whole genome-amplified using the REPLI-g Mini Kit (QIAGEN; cat. no. 150025), and genomic DNA libraries constructed using the MGIEasy FS DNA Library Prep Set (MGI; v2.0) and an established protocol82. The libraries were then sequenced (100 bp paired-end reads) using the DNBSEQ-T1 platform (BGI–Shenzhen, China).
Recording nucleotide variation
For individual samples, raw DNA sequence data in the FASTQ format83 were filtered for quality using SOAPnuke v1.5.684 by removing adapter-contaminated, duplicated and low-quality reads (parameters -l 20, -q 0.3, -n 0.02 and -d). Sequence quality was verified using FastQC v0.11.885 and MultiQC v1.786. Then, high-quality read-pairs were mapped to the nuclear genome sequence of E. granulosus (Eg-G1s) using the Burrows-Wheeler Aligner (BWA) v.0.7.887 and kept in the BAM format. Subsequently, read coverage, depth and mapping quality scores in all four Eg95 genes were assessed for each individual sample using mosdepth v.0.3.188. For each sample, the aligned read data was then used to record single nucleotide polymorphisms (SNPs) at individual positions and insertion/deletion events (indels) in relation to the reference genome sequence using the Genome Analysis Toolkit (GATK) v4.1.3.089. In brief, base quality scores of ‘raw’, aligned read data were re-calibrated twice based on predicted variants; then, SNP sites and indels were identified for each sample using the GATK HaplotypeCaller89 and merged into one ‘variant call format’ (VCF) file – listing all variable sites for all samples – using GATK Combine GVCFs and GenotypeGVCFs. ‘Raw’ SNPs and indels were filtered for quality using GATK VariantFiltration, retaining SNPs if strand bias (FS) < 60, variant confidence (QD) > 1.0, mapping quality (MQ) > 20.0, mapping quality (MQRankSum) > −12.5, read position bias (ReadPosRankSum) > −8.0, and indels if FS < 200, QD > 2.0, ReadPosRankSum > −20.0. Variable sites were verified by eye using the read alignment file and the program Geneious v.11.1.590.
Analyses of nucleotide variation for individual Eg95 genes
For each of the 47 E. granulosus DNA samples, individual FASTA files containing sequence data (including inferred SNPs and indels) for each gene were generated using GATK FastaAlternateReferenceMaker and BCFtools v1.991, with all polymorphic substitutions coded as IUPAC ambiguity characters92. Subsequently, all gene sequences produced here were aligned, as were their amino acid sequences. The open reading frame (ORF) of each gene was verified, conceptually translated, and synonymous or non-synonymous substitutions identified using the program Geneious. All nucleotide sequences were deposited in the GenBank database (accession nos. MZ889937–MZ890124). The three-dimensional structure of EG95-1, conceptually translated from individual nucleotide sequences, was modelled using a deep learning method, employing RoseTTAFold software93 accessed via the protein structure prediction service Robetta (https://robetta.bakerlab.org/) that is continually evaluated through CAMEO (https://www.cameo3d.org/). Visualisation and figure preparation were done with UCSF Chimera (http://preview.cgl.ucsf.edu/chimera/).
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The nucleotide sequence data from this study are publicly available via the NCBI database: BioProject PRJNA754835 (all genomic and transcriptomic data sets relating to genome Eg-G1s); GenBank accession no. JAIKUZ000000000 (Eg-G1s genome sequence); Sequence Read Archive (SRA) accession nos. SRR15522570, SRR15522571 and SRR15522580 (PacBio long read DNA data for the protoscolex stage of E. granulosus genotype G1); SRR15522572 to SRR15522577, SRR15522581 and SRR15522582 (short-read DNA data for the protoscolex stage of E. granulosus genotype G1); SRR15522578 (RNA-seq data for the oncosphere stage of E. granulosus genotype G1); SRR15522579 (RNA-seq data for the adult stage of E. granulosus genotype G1). GenBank accession nos. MZ889937 to MZ890124 (DNA sequences of each of the four Eg95 genes of each of 47 E. granulosus samples (genotype G1 or G3; derived from short read data)).
References
McManus, D. P. Echinococcosis. Lancet 362, 1295–1304 (2003).
Brunetti, E., Kern, P. & Vuitton, D. A. Writing Panel for the WHO-IWGE. Expert consensus for the diagnosis and treatment of cystic and alveolar echinococcosis in humans. Acta Trop. 114, 1–16 (2010).
McManus, D. P., Gray, D. J., Zhang, W. & Yang, Y. Diagnosis, treatment, and management of echinococcosis. BMJ 344, e3866 (2012).
Lightowlers, M. W. Cysticercosis and echinococcosis. Curr. Top. Microbiol. Immunol. 365, 315–335 (2013).
Craig, P. S. et al. Echinococcosis: control and prevention. Adv. Parasitol. 96, 55–158 (2017).
Amarir, F. et al. Control of cystic echinococcosis in the Middle Atlas, Morocco: field evaluation of the EG95 vaccine in sheep and cesticide treatment in dogs. PLoS Negl. Trop. Dis. 15, e0009253 (2021).
Lightowlers, M. W. et al. Vaccination against hydatidosis using a defined recombinant antigen. Parasite Immunol. 18, 457–462 (1996).
Gauci, C., Heath, D., Chow, C. & Lightowlers, M. W. Hydatid disease: vaccinology and development of the EG95 recombinant vaccine. Expert Rev. Vaccines 4, 103–112 (2005).
Wen, H. et al. Echinococcosis: advances in the 21st century. Clin. Microbiol. Rev. 32, e00075–18 (2019).
Chow, C. et al. A gene family expressing a host-protective antigen of Echinococcus granulosus. Mol. Biochem. Parasitol. 118, 83–88 (2001).
Chow, C. et al. Echinococcus granulosus: oncosphere-specific transcription of genes encoding a host-protective antigen. Exp. Parasitol. 106, 183–186 (2004).
Jabbar, A. et al. Oncospheral penetration glands are the source of the EG95 vaccine antigen against cystic hydatid disease. Parasitology 138, 89–99 (2011).
Gauci, C. G., Alvarez Rojas, C. A., Chow, C. & Lightowlers, M. W. Limitations of the Echinococcus granulosus genome sequence assemblies for analysis of the gene family encoding the EG95 vaccine antigen. Parasitology 145, 807–813 (2018).
Tsai, I. J. et al. The genomes of four tapeworm species reveal adaptations to parasitism. Nature 496, 57–63 (2013).
Kamenetzky, L., Maldonado, L. L. & Cucher, M. A. Cestodes in the genomic era. Parasitol. Res. 1-13 https://doi.org/10.1007/s00436-021-07346-x (2021) [ahead of print].
Zheng, H. et al. The genome of the hydatid tapeworm Echinococcus granulosus. Nat. Genet. 45, 1168–1175 (2013).
Kinkar, L. et al. Long-read sequencing reveals a 4.4 kb tandem repeat region in the mitogenome of Echinococcus granulosus (sensu stricto) genotype G1. Parasit. Vectors 12, 238 (2019).
Kinkar, L. et al. Nanopore sequencing resolves elusive long tandem-repeat regions in mitochondrial genomes. Int. J. Mol. Sci. 22, 1811 (2021).
Korhonen, P. K. et al. High-quality nuclear genome for Sarcoptes scabiei—a critical resource for a neglected parasite. PLoS Negl. Trop. Dis. 14, e0008720 (2020).
Young, N. D. et al. High-quality reference genome for Clonorchis sinensis. Genomics 113, 1605–1615 (2021).
Korhonen, P. K., Hall, R. S., Young, N. D. & Gasser, R. B. Common workflow language (CWL)-based software pipeline for de novo genome assembly from long- and short-read data. GigaScience 8, giz014 (2019).
Li, W. et al. The genome of tapeworm Taenia multiceps sheds light on understanding parasitic mechanism and control of coenurosis disease. DNA Res. 25, 499–510 (2018).
Olson, P. D. et al. Complete representation of a tapeworm genome reveals chromosomes capped by centromeres, necessitating a dual role in segregation and protection. BMC Biol. 18, 1–16 (2020).
Špakulová, M. et al. Cytogenetics and chromosomes of tapeworms (Platyhelminthes, Cestoda). Adv. Parasitol. 74, 177–230 (2011).
Zhang, W. et al. Short report: Echinococcus granulosus from Xinjiang, PR China: cDNAs encoding the EG95 vaccine antigen are expressed in different life cycle stages and are conserved in the oncosphere. Am. J. Trop. Med. Hyg. 68, 40–43 (2003).
Pan, W. et al. Genetic diversity and phylogenetic analysis of EG95 sequences of Echinococcus granulosus: implications for EG95 vaccine application. Asian Pac. J. Trop. Med. 10, 524–527 (2017).
Haag, K. L., Gottstein, B. & Ayala, F. J. The EG95 antigen of Echinococcus spp. contains positively selected amino acids, which may influence host specificity and vaccine efficacy. PLoS One 4, e5362 (2009).
Huch, M. et al. Long-term culture of genome-stable bipotent stem cells from adult human liver. Cell 160, 299–312 (2015).
Miller, A. J. et al. Generation of lung organoids from human pluripotent stem cells in vitro. Nat. Protoc. 14, 518–540 (2019).
Fan, J. et al. Transcriptomic features of Echinococcus granulosus protoscolex during the encystation process. Korean J. Parasitol. 58, 287 (2020).
Modi, A., Vai, S., Caramelli, D. & Lari, M. The Illumina sequencing protocol and the NovaSeq 6000 System. Methods Mol. Biol. 2242, 15–42 (2021).
Bolger, A. M. et al. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Kim, D. et al. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 37, 907–915 (2019).
Haas, B. J. et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 8, 1494–1512 (2013).
Fu, L. et al. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 28, 3150–3152 (2012).
Robinson, M. D. et al. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010).
Law, C. W. et al. RNA-seq analysis is easy as 1-2-3 with limma, Glimma and edgeR. F1000Research 5, ISCB Comm J-1408 (2016).
Li, B. & Dewey, C. N. RSEM: accurate transcript quantification from RNA-seq data with or without a reference genome. BMC Bioinform. 12, 323 (2011).
Koren, S. et al. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 27, 722–736 (2017).
Chin, C.-S. et al. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat. Methods 10, 563–569 (2013).
Walker, B. J. et al. Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One 9, e112963 (2014).
Roach, M. J., Schmidt, S. A. & Borneman, A. R. Purge Haplotigs: allelic contig reassignment for third-gen diploid genome assemblies. BMC Bioinform. 19, 460 (2018).
Xu, M. et al. TGS-GapCloser: A fast and accurate gap closer for large genomes with low coverage of error-prone long reads. GigaScience 9, giaa094 (2020).
Stanke, M. et al. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res. 34, W435–W439 (2006).
Pertea, M. et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 33, 290–295 (2015).
Wu, T. D. & Watanabe, C. K. GMAP: a genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics 21, 1859–1875 (2005).
Rice, P., Longden, I. & Bleasby, A. EMBOSS: The European Molecular Biology Open Software Suite. Trends Genet. 16, 276–277 (2000).
Kuhn, R. M., Haussler, D. & Kent, W. J. The UCSC genome browser and associated tools. Brief. Bioinforma. 14, 144–161 (2013).
Magrane, M., the UniProt Consortium. UniProt Knowledgebase: a hub of integrated protein data. Database (Oxf.) 2011, bar009 (2011).
R Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria; http://www.r-project.org/index.html (2020).
Harrison, P. M. fLPS: Fast discovery of compositional biases for the protein universe. BMC Bioinform. 18, 1–9 (2017).
Benson, D. A. et al. GenBank. Nucl. Acids Res. 46, D41–D47 (2018).
Zdobnov, E. M. & Apweiler, R. InterProScan–an integration platform for the signature-recognition methods in InterPro. Bioinformatics 17, 847–848 (2001).
Quinlan, A. R. BEDTools: the Swiss‐army tool for genome feature analysis. Curr. Protoc. Bioinforma. 47, 11.12.11–34 (2014).
Li, L., Stoeckert, C. J. & Roos, D. S. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 13, 2178–2189 (2003).
Drăgan, M. A. et al. GeneValidator: identify problems with protein-coding gene predictions. Bioinformatics 32, 1559–1561 (2016).
Simao, F. A. et al. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015).
Kanehisa, M., Goto, S., Sato, Y., Furumichi, M. & Tanabe, M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 40, D109–D114 (2012).
Pruitt, K. D., Tatusova, T., Brown, G. R. & Maglott, D. R. NCBI Reference Sequences (Refseq): current status, new features and genome annotation policy. Nucleic Acids Res. 40, D130–D135 (2012).
Nawrocki, E. P. & Eddy, S. R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 29, 2933–2935 (2013).
Kalvari, I. et al. Rfam 14: expanded coverage of metagenomic, viral and microRNA families. Nucleic Acids Res. 49(D1), D192–D200 (2021).
Bao, Z. & Eddy, S. R. Automated de novo identification of repeat sequence families in sequenced genomes. Genome Res. 12, 1269–1276 (2002).
Price, A. L., Jones, N. C. & Pevzner, P. A. De novo identification of repeat families in large genomes. Bioinformatics 21(Suppl. 1), i351–i358 (2005).
Smit, A. F. A. & Hubley, R. RepeatModeler Open-1.0. 2008–2015 http://www.repeatmasker.org.
Jurka, J. et al. Repbase Update, a database of eukaryotic repetitive elements. Cytogenet. Genome Res. 110, 462–467 (2005).
Smit, A. F. A., Hubley, R. & Green, P. RepeatMasker Open-4.0. 2013-2015 http://www.repeatmasker.org.
Krzywinski, M. et al. Circos: an information aesthetic for comparative genomics. Genome Res. 19, 1639–1645 (2009).
Emms, D. M. & Kelly, S. OrthoFinder: solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol. 16, 1–14 (2015).
Howe, K. L. et al. WormBase ParaSite—a comprehensive resource for helminth genomics. Mol. Biochem. Parasitol. 215, 2–10 (2017).
Ronquist, F. & Huelsenbeck, J. P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19, 1572–1574 (2003).
Altekar, G. et al. Parallel Metropolis coupled Markov chain Monte Carlo for Bayesian phylogenetic inference. Bioinformatics 20, 407–415 (2004).
Stamatakis, A., Ludwig, T. & Meier, H. RAxML-III: a fast program for maximum likelihoodbased inference of large phylogenetic trees. Bioinformatics 21, 456–463 (2005).
Lanfear, R. et al. PartitionFinder 2: new methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol. Biol. Evol. 34, 772–773 (2017).
Geyer, C. J. Markov-Chain Monte-Carlo Maximum-Likelihood. Computing Science and Statistics. Proceedings of the 23rd Symposium on the Interface, Interface Foundation, Fairfax Station; pp. 156–163 (1991).
Sukumaran, J. & Holder, M. T. DendroPy: A Python library for phylogenetic computing. Bioinformatics 26, 1569–1571 (2010).
Langfelder, P. & Horvath, S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinform. 9, 559 (2008).
Xie, C. et al. KOBAS 2.0: a web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 39, W316–W322 (2011).
Laurimäe, T. et al. Genetic diversity and phylogeography of highly zoonotic Echinococcus granulosus genotype G1 in the Americas (Argentina, Brazil, Chile and Mexico) based on 8279 bp of mtDNA. Infect. Genet. Evol. 45, 290–296 (2016).
Kinkar, L. et al. Global phylogeography and genetic diversity of the zoonotic tapeworm Echinococcus granulosus sensu stricto genotype G1. Int. J. Parasitol. 48, 729–742 (2018a).
Kinkar, L. et al. Genetic diversity and phylogeography of the elusive, but epidemiologically important Echinococcus granulosus sensu stricto genotype G3. Parasitology 145, 1613–1622 (2018b).
Pärn, M. Genetic diversity and phylogenetic relations of Echinococcus granulosus sensu stricto in Armenia and Turkey (The University of Tartu, Tartu, Estonia, 2019).
Huang, J. et al. A reference human genome dataset of the BGISEQ-500 sequencer. GigaScience 6, 1–9 (2017).
Cock, P. J. A. et al. The Sanger FASTQ file format for sequences with quality scores, and the Solexa/Illumina FASTQ variants. Nucleic Acids Res. 38, 1767–1771 (2010).
Chen, Y. et al. SOAPnuke: a MapReduce acceleration-supported software for integrated quality control and preprocessing of high-throughput sequencing data. GigaScience 7, 1–6 (2018).
Andrews, S. FastQC: A quality control tool for high throughput sequence data. http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (2010).
Ewels, P. et al. MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics 32, 3047–3048 (2016).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler Transform. Bioinformatics 25, 1754–1760 (2009).
Pedersen, B. S. & Quinlan, A. R. Mosdepth: quick coverage calculation for genomes and exomes. Bioinformatics 34, 867–868 (2018).
McKenna, A. et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010).
Kearse, M. et al. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649 (2012).
Li, H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 27, 2987–2993 (2011).
Cornish-Bowden, A. Nomenclature for incompletely specified bases in nucleic acid sequences: recommendations 1984. Nucleic Acids Res. 13, 3021–3030 (1985).
Baek, M. et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science 15 Jul 2021: eabj8754; https://doi.org/10.1126/science.abj8754 (2021).
Acknowledgements
Funding from the Australian Research Council (LP180101085 to R.B.G. and B.C.H.C.; LP180101334 to N.D.Y. and P.K.K.), BGI and Yourgene Singapore supported this project. Additional support came from the Horizon 2020 Research and Innovation Programme of the European Union (no. 773830; One Health European Joint Programme—MEME project; https://onehealthejp.eu/jrp-meme/ to A.C.) and from the Estonian Ministry of Education and Research (grant no. PRG1209). Thanks to Gezhen Qiangba and Jiandan Xie for project support.
Author information
Authors and Affiliations
Contributions
M.W.L. and C.G. provided the E. granulosus protoscolex sample for the sequencing and assembly of the Eg-G1s genome, and samples of activated oncospheres and adult worms of E. granulosus for transcriptomic analyses. D.J.J., U.S., T.L., M.R.-N., M.I., H.M., M.S., F.P.-G., S.S., A.C., H.Z., H.A., M.L.R., T.R., M.W., S.A.A. and H.G. provided E. granulosus samples (n = 47) from different countries for genetic analysis. H.C., J.L., J.L., G.Q., H.X., D.W., J.Y. and B.Y. coordinated or organised work at BGI, conducted library preparation, sequencing and/or processed raw sequence data. P.K.K. conducted genomic assembly, annotation as well as synteny, transcriptomic and network analyses. L.K. carried out the analysis of genetic variation, and A.H. conducted the modelling of protein structure. P.K.K., L.K., N.D.Y., M.W.L., C.G., A.J., A.H. and R.B.G. contributed to the interpretation of the results. R.B.G., together with U.S., prepared the research proposal and the project agreement among collaborating parties via the University of Melbourne. P.K.K., L.K. and R.B.G. drafted the manuscript, with editorial inputs from N.D.Y., M.W.L., A.J., and T.W. A.V.K., A.H., U.S., D.J.J., T.L., M.R.-N., M.I., H.M., M.S., F.P.-G., S.S., A.C., H.Z., H.A., M.L.R., T.R., M.W., S.A.A., H.G., H.Y. and B.Y. commented on the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks Laura Kamenetzky and the other, anonymous, reviewers for their contribution to the peer review of this work. Primary Handling Editor: George Inglis.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Korhonen, P.K., Kinkar, L., Young, N.D. et al. Chromosome-scale Echinococcus granulosus (genotype G1) genome reveals the Eg95 gene family and conservation of the EG95-vaccine molecule. Commun Biol 5, 199 (2022). https://doi.org/10.1038/s42003-022-03125-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-022-03125-1
- Springer Nature Limited