
Abstract
Changes in a few key transcriptional regulators can lead to different biological states. Extracting the key gene regulators governing a biological state allows us to gain mechanistic insights. Most current tools perform pathway/GO enrichment analysis to identify key genes and regulators but tend to overlook the gene/protein regulatory interactions. Here we present RegEnrich, an open-source Bioconductor R package, which combines differential expression analysis, data-driven gene regulatory network inference, enrichment analysis, and gene regulator ranking to identify key regulators using gene/protein expression profiling data. By benchmarking using multiple gene expression datasets of gene silencing studies, we found that RegEnrich using the GSEA method to rank the regulators performed the best. Further, RegEnrich was applied to 21 publicly available datasets on in vitro interferon-stimulation of different cell types. Collectively, RegEnrich can accurately identify key gene regulators from the cells under different biological states, which can be valuable in mechanistically studying cell differentiation, cell response to drug stimulation, disease development, and ultimately drug development.
Similar content being viewed by others


Introduction
The advances in high throughput technologies such as genomics, transcriptomics, and proteomics have provided unprecedented opportunities to mechanistically understand the genetic and epigenetic alterations in diseases, cellular development, cell stimulation, and immune activation1,2. Typically, alterations in the expression of key genes and proteins play a central role in many of these biological states. Thus, to understand the differences between these states, one of the fundamental steps is to identify a set of genes or proteins that are differentially expressed3. To understand the underlying biological process and functions of these molecules, annotation enrichment methods, such as pathway and gene ontology (GO) term enrichment, have been used widely4. Although the enrichment analysis provides crucial clues about the underlying biological processes and pathways, the lack of information about the underlying regulation hinders us to mechanistically understand how these biological states can be achieved.
To study the function, regulation, and dynamics of individual genes (or proteins) in a complex biological system, network biology is emerging as an important tool5. Several studies have demonstrated that constructing gene/protein interaction networks allows us to gain important insights into the regulatory mechanisms that govern different biological states, including disease, cellular activation, and differentiation6,7. In a gene/protein interaction network, densely connected genes (or hub genes) are crucial for the network’s integrity and the corresponding biological state6,7. However, considering only topological parameters (such as hubness or degree) of a network may overlook key regulators5. So, to gain regulatory insights, we should consider both network topology and the corresponding alterations in gene or protein expression.
Transcription factors and co-factors (TFs) can directly (and/or indirectly) regulate the expression of multiple target (and/or downstream) genes and proteins8,9,10. Some studies took advantage of curated TF–target networks11,12,13, or predicted network based on ChIP-seq data14,15 and used Fisher’s exact test and other enrichment algorithms to identify key/master regulators of the genes of interest16,17,18. However, current curated networks are incomplete, and increasing studies have shown that regulatory interactions may differ over time, upon different conditions and cellular states in the same organism8,19,20. So, analyses based on these incomplete static networks might not be sufficient to unveil functional regulatory patterns in complex biological processes.
State/cell/condition-specific gene regulatory network can directly be inferred from the gene or protein expression data (data-driven network)8,21. Using these data-driven networks and results from differential expression analyses, one can deduce key regulators. For example, ARACNE and ARACNe-AP, two packages based on ARACNe algorithm, have been used in the reverse engineering field, reconstruct a gene regulatory network from gene expression profile datasets based on mutual information22,23. NeTFactor algorithm uses this type of network and applies topological, statistical, and optimization methods to identify key regulators24. An R package called VIPER takes advantage of this network and uses t-statistics (or other measurements) by comparing gene expression of different conditions to compute the final enrichment p values for TFs based on analytic rank-based enrichment analysis (aREA) algorithm and a null model generated by sample bootstrapping25. VIPER has been successful in identifying master regulators in many studies26,27,28,29,30. However, it currently utilizes the network reverse-engineered by ARACNe that typically requires a large number of samples, which may not always be fulfilled, to successfully build a robust network, thus hampering VIPER’s broader application.
Here, we developed “RegEnrich”, an open-source R package for gene regulator enrichment analysis (Fig. 1). The RegEnrich pipeline aims to identify the key regulators based on their differential expression and enrichment of their potential downstream targets from a given gene set. Because the gene regulators do not act alone but function as part of a complex network, by using RegEnrich, one can refine a key gene regulatory network to study the biological process and visualize the derived network.

RegEnrich consists of four major steps: differential expression analysis, regulator-target network construction, enrichment analysis, and regulator ranking and visualization.
Results
Time consumption and memory usage by RegEnrich
The most time- and memory-consuming procedure of the RegEnrich pipeline is inferencing a regulator-target network from the gene expression data. Here, we benchmarked the time consumption and memory usage of different methods in the RegEnrich package using Intel® Xeon® Processors with 1, 4, 8, and 16 cores on CentOS Linux 7 operating system on high-performance computing facility at University Medical Center Utrecht (Fig. 2 and Supplementary Fig. 1). And the gene expression data were simulated with different numbers of samples (10, 20, 50, 100 for the COEN method and 50, 100, 200 for the random forest method (GRN)) and different numbers of genes (from 2000 to 40,000). Overall, the speed of both methods decreased with the increase of the number of genes, and the speed was also dependent on the sample size for only the GRN method (Fig. 2a). More specifically, the consumed time of the COEN method increased quadratically with the number of genes, while independent of the sample size. The COEN method was around 1 ~ 100 times faster, compared to the GRN method, when the number of genes was below 20,000, and the number of samples was over 50. However, since the GRN method is linearly, rather than quadratically, dependent on the number of genes, The COEN method spent more time when the number of genes was above 25,000 and the sample size was below 100. Network construction using the GRN method running on 4, 8, and 16 CPU cores was on average about 2, 4, and 8 times faster than the single-threading implementation, respectively (Supplementary Data 1 and Supplementary Fig. 1).

With one CPU core, (a) the time consumed and (b) maximum memory used by RegEnrich when analyzing a gene expression dataset with different numbers of genes (ranging from 2000 to 40,000) and different numbers of samples (ranging from 10 to 100 and from 50 to 200 for COEN and GRN network, respectively).
The maximum memory usage of the COEN method again increased quadratically with the increase of the number of genes and independent with the number of samples (Fig. 2b). Given the same size of the simulated expression data, the memory used by the COEN method was more than that by the GRN method in almost all circumstances. In general, using a dataset of 50 samples based on 1 CPU core, when constructing a protein-coding network, which comprises about 2 × 104 genes, it costs ~3.4 h (38.5 Gb) and ~4.1 h (8.2 Gb) by the COEN and by the GRN method, respectively. And it costs 1.6 h (7.8 Gb) by the GRN method using 4 CPU cores. The figure may be different depending on the computing power of the processors, but the order of magnitude will less likely change. So, the users can roughly expect the time and memory usage when performing the RegEnrich analysis according to Fig. 2 and Supplementary Fig. 1 and Supplementary Data 1.
Comparisons of key regulators obtained by different methods
Increasing studies predict key gene regulators by the hubs in a network, which is defined by topological features, such as degree and closeness centrality31,32. The degree of a node is the total number of nodes connected to this node in a network. The out-degree of a node is the number of nodes pointed by this node in a directed network. The closeness of a node is defined as the reciprocal of the sum of the shortest path length between this node and all other nodes in the network33. And the out-closeness of a node is defined as the reciprocal of the sum of the shortest path length from this node to all other nodes in a directed network.
We first checked how many key/master regulators identified by the RegEnrich package are hubs defined by the network topological properties. We downloaded a publicly available RNA-sequencing transcriptomic dataset, a total of 97 samples, from the Gene Expression Omnibus (GEO) database34. In that study, peripheral blood mononuclear cell (PBMC) samples from 29 patients with acute Lyme disease and 13 healthy controls were used to investigate longitudinal changes of the transcriptomes from the time of diagnosis (V1) to immediately after the completion of a 3-week course of doxycycline treatment (V2), and to 6 months after the completion of treatment (V5)34. Here, our interest is to investigate which regulators played the important role in regulating the transcriptomic changes of patients between V1 and V2. Thus, we retained 26 paired RNA-seq samples (three are removed because of quality control according to34) on V1 and V2 for RegEnrich analysis. And the network hubs are identified by ranking out-degree and out-closeness of the network generated by RegEnrich. As a result, we obtained three sets of hubs defined by 50 regulators with the highest out-degree (degree hubs), out-closeness (closeness hubs), or RegEnrich ranking score (RegEnrich key regulators). We found that 20% (10) of regulators were overlapping between degree hubs and closeness hubs, while RegEnrich key regulators were barely overlapping with degree hubs or closeness hubs (Fig. 3a).

Venn diagram shows the overlap between the top 50 hubs/regulators (a) defined by out-degree, out-closeness and the RegEnrich score (using COEN network and FET enrichment method), (b) defined by out-degree, out-closeness and the VIPER package (using ARACNe network). (c) Venn diagram shows the overlap between the top 50 hubs/regulators defined by RegEnrich (using different parameter combinations) and those by the VIPER package. (d) Ladder plots compare the rank of regulators identified by RegEnrich using different network inferencing methods, and those by RegEnrich and by VIPER. Lines connect the same regulators. The expression pattern of top three key regulators and their targets identified by (e) RegEnrich using COEN network and FET method and by (f) VIPER. Orange lines are the normalized expression of regulators and grey lines are the normalized expression of the targets of the regulators. The brown bars below the x-axis indicate samples at week 1, and purple bars the samples at week 3. The analyses were performed using the data obtained from34.
Given the successful applications of the VIPER package in identifying master regulators in studying many biological questions26,27,28,29,30, we subsequently checked how many master regulators identified by the VIPER package are network hubs. A gene regulatory network is built based on those 26 paired RNAseq samples by the ARACNE-AP package and we applied out-degree, out-closeness, and the VIPER package to identify hubs (and master regulators) in this network. Similarly, a little more overlapping regulators were observed between degree hubs and closeness hubs in the network constructed using the ARACNE-AP package, and these hubs are also barely the key regulators identified by the VIPER package (Fig. 3b). Different network-inferencing methods and enrichment methods within RegEnrich showed consistent top-ranked regulators (Fig. 3c, d), in which ~40% were also identified by VIPER (Fig. 3c). Altogether, both RegEnrich and VIPER tend to rank the nonhub regulators to be the key regulators. Using the network reverse-engineered from the full dataset of all 97 samples in34 did not show the exactly the same results but still ranked the nonhub regulators to be the key regulators (Supplementary Fig. 2). These results suggest that looking for a gene/regulator with a high degree or other centrality parameters may not be ideal for identifying key gene regulators in every biological process.
Although ~40% of regulators were commonly ranked in the top 50 regulators by both RegEnrich and VIPER, most of the top 10 regulators identified by these two packages are unique (Fig. 3d). Clearly, the expression of the three topmost-ranked regulators (CREG1, DEK, and E2F2) identified by RegEnrich was differential between V1 and V2 and showed a very similar pattern to that of corresponding targets (Fig. 3e and Supplementary Fig. 3). Although the expression of three topmost-ranked regulators identified by VIPER was also differential, only the expression of the targets of ZFP36L1 and CNOT7 was modestly correlated with that of the regulators themselves (Fig. 3f). One may argue that it is inevitable that the expression of a regulator and its targets are highly correlated in COEN networks and this correlation may not hold for the network that was built by ARACNE-AP using mutual information. However, such correlations between the regulator and its targets were still observed when the network is built using random forest, which is also a nonlinear method (Supplementary Figs. 3 and 4). Thus, RegEnrich can successfully, as intended, identify the key regulators that both their expression and their targets’ expression associate with the biological process of interest.
RegEnrich can identify the key regulators in gene-silencing studies
RNA interference (RNAi)-mediated gene-silencing has been widely used to study the biological function of the silenced gene. In a gene regulator silencing experiment, the successfully silenced gene is typically the key regulator that has been malfunctioned. The performance metrics of RegEnrich and VIPER were compared by the ability to rank the silenced gene as one of the top key regulators. Here nine gene-silencing experiments from four independent datasets16,17,35,36 were used to benchmark RegEnrich using COEN network with either FET or GSEA enrichment methods, and VIPER using the network built by the ARACNE-AP package. Multiple cell lines/types, different silenced genes, and varied numbers of samples were deliberately included in these datasets to evaluate the bias induced by these variables (Table 1). RegEnrich with either FET or GSEA enrichment method outperformed VIPER in all these datasets, and within RegEnrich, GSEA outperformed FET in most cases. For example, using the GSEA method, STAT3 and FOXM1 were ranked as the top key regulators when STAT3 and FOXM1 were silenced in the BTIC and ST486 cell lines, respectively (Table 1). STAT3 and FOXM1 were also ranked high (the second key regulators) in these experiments when we applied RegEnrich with the FET method. Interestingly, in the GSE17172 dataset, although FOXM1 was not ranked as the first regulator using RegEnrich FET method, other two genes (FOXN3 and FOXG1) from the same FOX transcription factor family were ranked as the first and fourth regulator, respectively (Supplementary Table 1). This implies that at least several members of the FOX family were perturbed by FOXM1 silencing due to either off-targeting or downstream transcriptional signaling and can be inferred by RegEnrich. It may also attribute to the “pleiotropy of regulators”25 or “shadow effect”17, meaning that some of the transcriptional targets of FOXM1 are also part of the regulons of other FOX family members. By applying VIPER to this dataset, STAT3 was ranked as the 7th regulator. However, FOXM1 was failed to be identified by using VIPER maybe because the sample size is small (Table 1).
Similarly, STAT3 and CEBPB in SNB19 cell line, CHAF1A in IMR32 cell line, and BCL6 in Burkitt lymphoma cell line were identified by RegEnrich, with the GSEA method, as one of the top 20 key regulators in each corresponding dataset. The rankings of these regulators were considered high because these were regulators popping up from a total list of 1712 regulators in these RegEnrich analyses. Meanwhile, we also assessed the RegEnrich using two datasets, where STAT3 and CEBPB were tried to be simultaneously silenced in either BTIC or SNB19 cell lines. Even though these two genes were intended to be silenced, only one gene was successfully silenced. More specifically, STAT3 but not CEBPB was successfully silenced in the BTIC cell line, and CEBPB but not STAT3 was successfully silenced in the SNB19 cell line. Thus, only STAT3 and CEBPB were expected to be top-ranked as key regulators in the BTIC cell line and SNB19 cell line, respectively, which were the results returned by RegEnrich (Table 1). To evaluate RegEnrich’s ability to filter the false-positive results, we included a dataset where CEBPB was not successfully silenced in the BTIC cell line. All three approaches did not rank CEBPB as one of the top regulators (Table 1).
Since the ARACNe algorithm needs a large sample size to reverse-engineer a robust gene regulation network, and the sample sizes of the datasets here are small, we then used the publicly available network to perform master regulator inference with the viper package (see supplementary methods). RegEnrich (GSEA) out-performed the VIPER algorithm except in one of the experiments (Table 1 and Supplementary Table 2). Altogether, RegEnrich with COEN network and GSEA method is robust to identify the key regulators in well-controlled in vitro experiments even when the sample size is small.
RegEnrich retrieves interferon related regulators
In human, there are three types of interferons (IFN): type I IFNs (IFNα, β, ε, κ, and ω); type II IFN (only IFNγ); and type III IFNs (IFNλ1, λ2, λ3, and λ4)37,38. Due to the great therapeutic value of IFNs against virus infection and cancer, multiple studies have been performed to study the regulatory mechanisms of IFNs and interferon-stimulated genes (ISGs). For example, it has been revealed that extracellular IFNs activate cells by a signal transduction cascade, including activating transcription factors STATs and/or IRFs, leading to the induction of hundreds of ISGs, and forming a frontline of defense against virus infections37,38. However, the mechanisms underlying the regulation of most of these ISGs may vary between different cell types and tissues and remain incompletely understood.
Given the potential of identification of key regulators by RegEnrich in a biological process, we sought to identify the key regulators by which IFNs stimulated cells to express ISGs. We retrieved and analyzed 11 microarray or RNAseq datasets from the GEO database, comprising 21 in vitro experiments, in which different cells were stimulated by either type I or type II IFN (Table 2). We found that RegEnrich identified STAT transcription factor family members, including STAT1 and STAT2, in most IFN stimulation experiments, which is supported by the well-known IFN signaling pathway38. In addition, IRF (interferon regulatory factors) transcription factor family members, such as IRF9 and/or IRF7, were also identified as key regulators in a majority of the type I IFN stimulation experiments (Table 2). These IRFs have been reported to play important roles in producing type I IFN downstream receptors that detect viral RNA and DNA, and in regulating interferon-driven gene expression39.
Recent work has shown that ELF1 (a member of the ETS transcription factor family) is induced by IFN, but does not feed-forward to induce interferons, and transcriptionally programs cells with potent antiviral activity40. Interestingly, ELF1 was identified by RegEnrich as one of the key regulators in most of the type I IFN stimulation experiments (Table 2). We further investigated whether any other members of the ETS transcription factor family were also identified by RegEnrich. Interestingly, we found another ETS transcription factor family member, ETV7, in the lists of top regulators from more than half of type I IFN stimulation experiments and from almost all type II IFN stimulation experiments. A more recent study showed that ETV7 preferentially targeted a subset of antiviral ISGs crucial for IFN-mediated control of viruses, such as influenza and SARS-CoV-241.
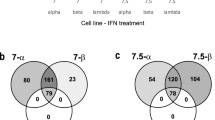
Different cells may respond differently to IFN stimulation with different durations. We further investigated the common regulators involved in IFN stimulation among different cells. Thus, we summarized the most common regulators within type I and type II IFN stimulation experiments. It showed that the ISGs of type I IFNs were strongly regulated by STAT family, TRIM family, IRF family, ETS family, SP100/SP140 family (transcriptional coactivator of ETS family TFs). Similarly, type II IFN ISGs were largely regulated by STAT family, IRF family, ETS family, MCM family, SP100/SP140 family (Table 2 and Fig. 4). One of the most commonly identified regulators of type II IFN ISGs was the MHC class II transactivator (CIITA), which has been very recently shown with the potential to induce cell resistance to the Ebola virus and SARS-CoV-242. Altogether, these results suggest that RegEnrich successfully identified key regulators related to IFN signaling in IFN stimulation experiments.

Key regulators in (a) type I interferon stimulation and (b) type II interferon stimulation datasets. The full list has been shown in Table 2. The top 35 regulators in each dataset were included as key regulators, and only the regulators identified in more than 25% of datasets were shown.
Discussion
High throughput technologies like microarray, RNA-seq, and protein mass spectrometry offer easy, fast, and affordable profiling of the gene/protein expression. These technologies generate massive data facilitating us to study the alterations in gene/protein expression, thereby helping us identify the biomarkers for diseases and biological states. However, it is still challenging to predict which genes play major roles in these biological contexts. To address this problem, we developed RegEnrich, an open-source R/Bioconductor package integrating differential gene expression analysis, network inference, enrichment analysis, and regulator ranking. RegEnrich is able to identify the key regulators by providing gene/protein expression data from multiple high throughput technologies. We benchmarked the speed and maximum memory usage of network inference methods in the RegEnrich, which shows that the COEN method runs much faster than the random forest method does when the number of genes is below 20,000, and the speed of the multithreaded random forest version is significantly improved. Traditionally, COEN is considered a method for depicting linear relations between genes, while the random forest for nonlinear relations. Strikingly, in the Lyme disease transcriptomics dataset, the results from the COEN and GRN methods were consistent. This might be because the COEN methods re-evaluate the edge weights by considering the information of neighbor nodes; as a result, such a network was constructed not only based on a linear relationship.
Since hub nodes have been found to be important in many networks, hub genes that are defined by gene regulatory network properties are also expected to be crucial in biology and have drawn much attention over the last decades43. We compared the hubs identified by the network properties, i.e., degree and closeness, with the key regulators by RegEnrich. Interestingly, only a very small number of the key regulators from either RegEnrich or VIPER were hubs (Fig. 3a, b and Supplementary Fig. 2). One possible reason is that these hub genes are so important in maintaining the major functions of cells that too strong perturbations of these hubs could be fatal for cells44. Therefore, these hub genes are not necessarily the key gene expression regulators in a specific context. For example, Gaiteri et al. showed that differentially expressed genes primarily reside on the periphery of co-expression networks for neuropsychiatric disorders such as depression, schizophrenia, and bipolar disorder45.
As one easy-to-use software/package whose functionality was similar to RegEnrich’s, the VIPER R package was used to compare with RegEnrich to find the final key regulators. According to the VIPER package instruction on Bioconductor, VIPER needs an ARACNE network to perform the analysis. Such network is generated by an independent package, such as minet46, GPU-ARACNE47, and ARACNe-AP23. These packages either are not R packages or fail to construct the network when the sample size is small. In contrast, RegEnrich is an all-in-one package, and a detailed tutorial document is provided along with RegEnrich, which facilitates users to use it more easily. More importantly, RegEnrich can identify the key regulators whose expression and targets’ expression correlate with the experimental phenotypes. In addition, RegEnrich is able to find key regulators not only between two conditions but also in a time series experimental setting. Since RegEnrich is modular and is intended to be a flexible pipeline, we allow users to provide custom regulator lists and to have options for multiple methods at different steps in case needed.
Since the datasets in the benchmarking analyses were of a small sample size. We asked whether the sample size influences the performance of the RegEnrich pipeline and the VIPER algorithm. To answer this question, we generated a dataset comprising of N samples (N = 100), simulating the gene expression of two cell states. These samples were evenly assigned into two groups. We first assumed that all samples had m common gene regulators (m = 500), of which k were bona fide key regulators (k = 20). The expression of each bona fide key regulator followed a Gaussian distribution in each group and was on purpose simulated to be differential between two groups. While the expression of the other regulators followed a Gaussian distribution with the same mean and standard deviation in each group, thus not necessarily differential between the two groups. Then we assumed that each regulator had t targets (t is a random number ranging from 3 to 50). The expression of the targets of a certain regulator was dependent on the expression of this regulator (the expression of this regulator plus Gaussian-distributed values). This resulted in an expression dataset with M genes (M = 13,789) and N samples (N = 100). Subsequently, we reduced the sample size by a down-sampling strategy, resulting in datasets of 50, 20, 10, and 6 samples. Based on these datasets, we checked whether those bona fide key regulators that we predefined were top-ranked by the RegEnrich pipeline. The results showed that with the decrease of the sample size, less bona fide key regulators were top-ranked (Supplementary Fig. 5), meaning that the performance of the RegEnrich pipeline could be improved by increasing the sample size. As expected, the random forest (GRN) method in RegEnrich and ARACNe failed to build a network when the sample size was smaller than 20. Next, we checked whether the performance would be increased when we used the network that was built based on the complete dataset (all 100 samples) (Supplementary Fig. 6). Indeed, all methods showed a higher performance. Although we observed that the ARACNe + VIPER packages showed a slightly better performance when the sample size was smaller than 20, the network was built based on the full dataset of 100 samples, which is an ideal scenario and hard to achieve in reality. Thus, we believe that RegEnrich is important especially in the area where not so much gene expression data are available.
The regulation of genes is a very complex process, in which many aspects affect the expression of a gene, such as the accessibility of chromatin, single nucleotide polymorphisms, DNA modifications, histone modifications, the expression of its upstream regulators, RNA degradation, and post-translational modifications. Thus, as one of the major steps of the RegEnrich pipeline, assuming the key regulators to be differentially expressed between different biological states may not hold in all circumstances. RegEnrich would be failed to predict those regulators whose post-translational modifications but not gene expression changes regulate their downstream gene expression. In addition, an accurate gene regulatory network is very important in identifying the key regulators25. RegEnrich inferred the data-driven network by only gene/protein expression data by default. Glass et al. have shown that integration of protein-protein interaction, protein-gene interaction, and gene expression can increase the accuracy of regulatory network inference48. Currently, we provide an option for the users to provide their gene regulatory network, which can be derived from other epigenetic datasets such as ChIP-seq, ATAC-seq data, protein-protein interactions, etc., thus, granting RegEnrich an ability to integrate multi-omic data.
By analyzing the datasets of dozens of IFN-stimulation experiments, RegEnrich identified STAT and IRF transcription factor family members, including STAT1, STAT2, IRF9, and IRF7, which have been extensively shown to play important roles in IFN signaling pathways38,39. Meanwhile, RegEnrich also identified several ETS transcription factor family members, such as ELF1 and ETV7, as key regulators in IFN signaling. Interestingly, ELF1 transcriptionally program cells with potent antiviral activity and ETV7 targeted antiviral ISGs crucial for IFN-mediated control of viruses, including influenza and SARS-CoV-240,41. These antiviral activities are typically the fundamental role of IFN in innate immunity. By analyzing the most commonly top-ranked regulators, RegEnrich predicted a list of candidate key regulators, such as CIITA and SP100/SP140 family members. Given that CIITA has been recently reported with antivirus ability42, further study may be carried out to investigate the antivirus potential of SP100/SP140 family members, such as SP100 and SP110, which might facilitate the mechanistic studies of IFN-ISG signaling and ultimately drug development.
Recently, using the RegEnrich pipeline, we predicted a network of key regulators that leads monocyte-derived dendritic cells (moDCs) to differentiate into a different trajectory upon CXCL4 stimulation compared to the moDCs without CXCL4 stimulation. We also experimentally validated the RegEnrich pipeline’s prediction by silencing one of the top-ranked regulators in the predicted network, i.e., CIITA49. More recently, we studied the mechanism of human T regulatory (Treg) cell programming under inflammatory conditions. Using RegEnrich, we predicted a network of key regulators important for effector Treg differentiation, including the vitamin D receptor (VDR), which is further validated by H3K27ac and H3K4me1 ChIP-seq experiments50. These two independent experimental studies support that RegEnrich is able to accurately rank the key gene regulators that are mechanistically involved in immune cell development and functions.
Understanding the key regulators between different biological states is essential for gaining mechanistic insights, designing functional experiments, and rational drug development. To this end, here, we presented RegEnrich, a Bioconductor R package for inference of key regulators in biological conditions. There are four major steps to obtain the list of key regulators in RegEnrich, i.e., differential expression analysis, regulator-target network inference, enrichment analysis, and regulator ranking. For differential expression analysis, the methods in DESeq2 and limma packages are provided, which grants RegEnrich the ability to predict the key regulators not only for gene expression data of two conditions but also for time series data. Meanwhile, two regulator-target network inference methods (WGCNA and random forest) are provided, which allows the network to not only contain the linear information but also include a nonlinear relationship between genes. FET and GSEA algorithms are optional for users to perform enrichment analysis. RegEnrich can identify the key regulators whose expression and their targets’ expression correlate with the experimental phenotypes. Using datasets from gene-silencing studies, RegEnrich using the GSEA method performed the best to retrieve the key regulators and outperformed the VIPER package. Further, by analyzing dozens of in vitro interferon-stimulation gene expression datasets, RegEnrich revealed that not only IRF and STAT transcription factor families played an important role in cells responding to IFN but also several ETS transcription factor family members, such as ELF1 and ETV7, were highly associated with IFN stimulations. Above all, RegEnrich can accurately identify, in a data-driven manner, key gene regulators from the cells under different biological states, which can be valuable in mechanistic studies of cell differentiation, cell response on drug stimulation, and disease development, ultimately in drug development.
Methods
The RegEnrich is a modular pipeline and consists of four major steps: (a) differential expression analysis; (b) regulator-target network construction; (c) enrichment analysis; and (d) regulator ranking and visualization.
Differential expression analysis
RegEnrich pipeline can be applied to multiple gene expression datasets, including RNA sequencing (RNAseq), microarray, and proteomic data. The first step of finding the key regulators is to obtain differentially expressed genes or proteins (DEs), corresponding differential significance p values (\({P}_{D}\)), and fold changes between conditions. Concerning two-group comparison, here, RegEnrich incorporates the Wald significance test from the DESeq2 package and the empirical Bayes method-based linear modeling from limma package to perform the differential expression analysis on RNAseq data and microarray/proteomic data, respectively51,52. Regarding the comparisons in experiments with multiple groups or more complex scenarios such as time-series study, the negative binomial generalized linear model-based likelihood ratio test from DESeq2 package51 and linear model-based likelihood ratio test are implemented for RNAseq data and microarray/proteomic data, respectively.
Regulator-target network inference
There are two major types of gene regulatory networks (or regulator-target networks) proposed: static network and dynamic network53,54,55. In a static network, genes are expressed in a steady state thus cannot describe the dynamics of an evolving process, while genes are dynamical in a dynamic network56. These networks can be constructed by many different computational approaches21,22,57,58,59,60. Here, the regulator-target network inference is based on four assumptions: (1) the gene regulatory network is a snapshot of a dynamic network within the users’ experiments; (2) It is a directed network, where the edges are from a regulator to its targets, or from a regulator to its targeted regulators; (3) the potential regulators are transcription factors and co-factors (this can be changed in RegEnrich by users); and (4) the expression change of a regulator can lead to the expression change of its downstream targets. Here, the targets are not only direct targets that the regulator binds to but also the downstream genes whose expression can be perturbed by the regulator. Presently, RegEnrich provides users two basic options to infer regulator-target network, i.e., COEN (co-expression network) and GRN (based on the random forest algorithm).
For COEN, here, the co-expression network is constructed using WGCNA (weighted gene co-expression network analysis) algorithm58. And it can be summarized as three major procedures. First, a similarity matrix is calculated using correlations in expression data to measure the relationship strength between each pair of genes (nodes). Second, by applying the approximate scale-free topology criterion, raising the co-expression similarity to a power to define the weighted network adjacency matrix. Third, this adjacency matrix is then used to calculate the topological overlap measure (TOM), which reflects not only the similarity of each pair of nodes but also their neighbors’ similarity6. The TOM defines the final co-expression network58.
For GRN, this ensemble regression tree-based method was initially described in GENIE3, which was the best performer in the DREAM4 In Silico Multifactorial challenge21. The basic idea of GENIE3 is that each gene is regressed in turn against all other genes to obtain network weights (edge weights), which quantify the strength of the dependence of each pair of genes. The edge weight (Wij) is the importance of gene i in the tree model predicting gene j, which can be interpreted as the fraction of variance of the expression of gene j that can be explained by gene i60. However, the GENIE3 package is slow especially when it is deployed on genome-wide studies with a large number of experiments. So, to facilitate usage and improve speed, we implemented this algorithm by allowing users to define their regulators and by supporting parallel computing (Supplementary Fig. 7). In addition, in this random forest-based method we found the expressions of some genes were hardly predicted by other genes. So, we modified this algorithm by adding a filtering procedure to remove the poor random forest models (Supplementary Fig. 7). In other words, this procedure removed the genes and corresponding edges from the final network, whose expression was hardly predicted by the expression of the predefined regulators.
We provide users with an option to either supply a list of regulators of their interest or use the default list of regulators provided in RegEnrich, which were retrieved from three studies61,62,63. Using either COEN or GRN network, we then extract the regulator-target network by retaining the top-ranked edges (default = top 5% edges) between the regulators and their targets and subsequently filtering out nonconnected nodes. Apart from the data-driven network, RegEnrich also allows users to provide their regulator-target network, which can be derived from the literature, databases, or defined by the user using their data of other types.
Enrichment analysis
The regulators are considered key regulators if they are differentially expressed along with their targets in a differentially expressed gene set. In other words, not only these regulator genes but also their target genes are differentially expressed upon different conditions. Finding these key regulators is an enrichment task, which is similar to retrieving the most overrepresented (enriched) biological annotations, such as gene ontology and pathways terms, of a list of interesting genes. Presently, RegEnrich provides users two options: Fisher’s exact test (FET) and gene set enrichment analysis (GSEA).
Fisher’s exact test (FET), also known as the hypergeometric test, calculates probability using the hypergeometric distribution (Eq. 1). This distribution describes the probability of the number of draws being successful (k) within a sequence of draws (M), without replacement, from a finite population (N) consisting of two types of elements (the total number of successful types is s).
Then the p value, depicting the probability of observing K (and more) differential targets by chance, of regulator i being overrepresented, is calculated by
where N is the total number of genes in the previously constructed regulator-target network; M is the number of genes in the list of users’ interests (the genes not in the network are excluded), which is typically the differential genes between conditions; \({s}_{i}\) is the number of target genes of regulator i in the network; K is the number of target genes that are also in the list of users’ interests. This process repeats for all regulators that are predefined by users.
Gene set enrichment analysis (GSEA) is one of the most widely used methods to study the biological function of groups of genes and to interpret gene expression data64. GSEA takes into account all of the genes in an experiment, unlike FET that takes into account only those genes above a fold-change or significance cutoff. Here, RegEnrich takes two basic inputs, the TF-target network and a named vector of decreasingly sorted ranking metrics (r, z-score scaled negative logarithm of differential significance p-values) of all genes. Briefly, there are three major steps in this analysis:
-
1.
Calculation of an Enrichment Score (ES) by:
$${{{{{\rm{ES}}}}}}=\left\{\begin{array}{c}{{\max }}\left(\Delta P\right),{{\max }}\left(\Delta P\right)\ge {{\max }}\left(-\Delta P\right)\hfill\\ {{\min }}\left(\Delta P\right), {{\max }}\left(\Delta P\right) \, < \, {{\max }}\left(-\Delta P\right)\end{array}\right.,$$(3)where
$$\Delta {P}_{i}={P}_{{hit}}(S,i)-{P}_{{miss}}(S,i),$$(4)where
$${P}_{{hit}}\left(S,i\right)=\mathop{\sum}\limits _{{g}_{j}\in S,j\le i}\frac{\left|{r}_{j}\right|}{\mathop{\sum }\limits_{{g}_{j}\in S}\left|{r}_{j}\right|},{P}_{{miss}}(S,i)=\mathop{\sum}\limits_{{g}_{j}\notin S,j\le i}\frac{1}{(N-{N}_{H})}$$(5)Here, i is the index of decreasingly sorted ranking metrics r, S is the set of target genes of one particular regulator, NH is the number of genes in S, and N is the total number of valid genes in the regulator-target network.
-
2.
Randomly shuffle the ranking metrics of genes and re-compute ES. And repeat this process for 1000 permutations to generate ESNULL that establishes an empirical distribution. Estimate empirical p value for S from ESNULL by only the positive portion of the distribution corresponding to the sign of the observed ES.
-
3.
Perform steps 1 and 2 for each regulator, generating a numeric vector (\({P}_{E}\)) in which each value is an enrichment p value for each regulator.
Regulator ranking and visualization
After the enrichment analysis by either FET or GSEA, the overall ranking scores of regulators were calculated by:
where \(f(x)=\frac{x-{\min }\left(x\right)}{{\max }\left(x\right)-{\min }\left(x\right)}\), and \({P}_{D}\) is the vector of p values of regulators obtained from differential expression analysis, \({P}_{E}\) is the vector of p-values of regulators obtained from the enrichment analysis.
In the RegEnrich package, we have implemented several functions for visualizing the information of a regulator and its targets (Fig. 1). For example, “plotRegTarExpr” function is to plot the expression pattern of a regulator and its targets.
Master regulator inference analysis based on ARACNe + viper algorithm
According to the official tutorial on GitHub (https://github.com/califano-lab/ARACNe-AP), we reverse-engineered the regulatory networks using the gene expression datasets obtained from either GEO database or the simulated dataset, based on the ARACNe-AP package with default parameters except the TF list. To make the results comparable between viper and RegEnrich, here, the TF list is set as the same as the default regulators in RegEnirch. The regulon object is generated from the ARACNe network file and the corresponding expression dataset using the aracne2regulon function from the viper package with default parameters. Either the paired t-test or t-test was applied to compare the gene expression change between groups, depending on whether the experiment is a paired study. Meanwhile the sample permutation and paired t-test or t-test were used to generate a null model. The t-statistics and corresponding p-values, and the null model were used to perform master regulator inference analysis with msviper function from viper package. To confirm the results by using the network that we built, we reanalyzed the gene-silencing datasets using the public network of corresponding cancer type from the aracne.networks package (version 1.16.0).
Statistics and reproducibility
T-test was used to compare the gene expression changes between groups to obtain master regulon by VIPER when the samples between groups are not paired, while paired T-test was used when the samples between groups are paired. To perform the enrichment analysis in the third step of RegEnrich pipeline, either GSEA method or Fisher’s exact test was used. To compare the performance of RegEnrich and the VIPER package, the ranks of key/master regulators identified by both methods using the simulated gene expression data were compared using the GSEA method. To evaluate the influence of the sample size on the performance of RegEnrich and VIPER package, the maximum sample size is set to 100 and the sample size is reduced to 50, 20, 10, and 6 by down-sampling strategy. The datasets of gene knock-down experiments and IFN stimulation experiments with sample sizes in each condition ≥ 3 were used for evaluation. All of the statistical analyses were performed using the R software (version 4.01).
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
All of the gene expression data were downloaded from GEO database according to the GEO accession ID listed in the Tables 1 and 2. The source data underlying Fig. 2 are provided in Supplementary Data 1. The source data underlying Figs. 3 and 4 are provided as Supplementary Data 2. Any other relevant data are available upon reasonable request.
Code availability
The source codes of RegEnrich package are publicly available on Bioconductor (https://www.bioconductor.org/packages/release/bioc/html/RegEnrich.html). The source codes used for analyzing the public data and the simulated data are available on GitHub (https://github.com/paodan/RegEnrich_CustomCode).
References
Linnarsson, S. & Teichmann, S. A. Single-cell genomics: Coming of age. Genome Biol. 17, 97 (2016).
Wagner, A., Regev, A. & Yosef, N. Revealing the vectors of cellular identity with single-cell genomics. Nat. Biotechnol. 34, 1145–1160 (2016).
Clark, N. R. et al. The characteristic direction: a geometrical approach to identify differentially expressed genes. BMC Bioinforma. 15, 1–16 (2014).
Nguyen, N. T., Lindsey, M. L. & Jin, Y.-F. Systems analysis of gene ontology and biological pathways involved in post-myocardial infarction responses. BMC Genomics 16, S18 (2015).
Walley, J. W. et al. Integration of omic networks in a developmental atlas of maize. Sci. (80-.) 353, 814–818 (2016).
Barabási, A.-L., Gulbahce, N. & Loscalzo, J. Network medicine: a network-based approach to human disease. Nat. Rev. Genet 12, 56–68 (2011).
Bhattacharyya, M. & Chakrabarti, S. Identification of important interacting proteins (IIPs) in Plasmodium falciparum using large-scale interaction network analysis and in-silico knock-out studies. Malar. J. 14, 70 (2015).
Aibar, S. et al. SCENIC: single-cell regulatory network inference and clustering. Nat. Methods 14, 1083–1086 (2017).
Almalki, S. G. & Agrawal, D. K. Key transcription factors in the differentiation of mesenchymal stem cells. Differentiation 92, 41–51 (2016).
Lesage, K. M. et al. Cooperative binding of ApiAP2 transcription factors is crucial for the expression of virulence genes in Toxoplasma gondii. Nucleic Acids Res. 46, 6057–6068 (2018).
Lachmann, A. et al. ChEA: transcription factor regulation inferred from integrating genome-wide ChIP-X experiments. Bioinformatics 26, 2438–2444 (2010).
Garcia-Alonso, L. et al. Transcription factor activities enhance markers of drug sensitivity in cancer. Cancer Res. 78, 769–780 (2018).
Keenan, A. B. et al. ChEA3: transcription factor enrichment analysis by orthogonal omics integration. Nucleic Acids Res. 47, W212–W224 (2019).
Wang, Z. et al. BART: a transcription factor prediction tool with query gene sets or epigenomic profiles. Bioinformatics 34, 2867–2869 (2018).
Puente-Santamaria, L., Wasserman, W. W. & Del Peso, L. TFEA. ChIP: a tool kit for transcription factor binding site enrichment analysis capitalizing on ChIP-seq datasets. Bioinformatics 35, 5339–5340 (2019).
Carro, M. S. et al. The transcriptional network for mesenchymal transformation of brain tumours. Nature 463, 318–325 (2010).
Lefebvre, C. et al. A human B-cell interactome identifies MYB and FOXM1 as master regulators of proliferation in germinal centers. Mol. Syst. Biol. 6, 377 (2010).
Qi, D., Wu, B., Tong, D., Pan, Y. & Chen, W. Identification of key transcription factors in caerulein-induced pancreatitis through expression profiling data. Mol. Med. Rep. 12, 2570–2576 (2015).
Peter, I. S. & Davidson, E. H. Evolution of gene regulatory networks controlling body plan development. Cell 144, 970–985 (2011).
Voordeckers, K., Pougach, K. & Verstrepen, K. J. How do regulatory networks evolve and expand throughout evolution? Curr. Opin. Biotechnol. 34, 180–188 (2015).
Huynh-Thu, V. A., Irrthum, A., Wehenkel, L. & Geurts, P. Inferring Regulatory Networks from Expression Data Using Tree-Based Methods. PLoS One 5, e12776 (2010).
Margolin, A. A. et al. ARACNE: An Algorithm for the Reconstruction of Gene Regulatory Networks in a Mammalian Cellular Context. BMC Bioinforma. 7, S7 (2006).
Lachmann, A., Giorgi, F. M., Lopez, G. & Califano, A. ARACNe-AP: Gene network reverse engineering through adaptive partitioning inference of mutual information. Bioinformatics 32, 2233–2235 (2016).
Ahsen, M. E. et al. NeTFactor, a framework for identifying transcriptional regulators of gene expression-based biomarkers. in Proceedings of the 11th ACM International Conference on Bioinformatics, Computational Biology and Health Informatics 1–13 (2020).
Alvarez, M. J. et al. Functional characterization of somatic mutations in cancer using network-based inference of protein activity. Nat. Genet. 48, 838–847 (2016).
Thorsson, V. et al. The immune landscape of cancer. Immunity 48, 812–830 (2018).
Elyada, E. et al. Cross-species single-cell analysis of pancreatic ductal adenocarcinoma reveals antigen-presenting cancer-associated fibroblasts. Cancer Disco. 9, 1102–1123 (2019).
Chari, A. et al. Oral selinexor–dexamethasone for triple-class refractory multiple myeloma. N. Engl. J. Med 381, 727–738 (2019).
Califano, A. & Alvarez, M. J. The recurrent architecture of tumour initiation, progression and drug sensitivity. Nat. Rev. Cancer 17, 116–130 (2017).
Guzzi, P. H., Mercatelli, D., Ceraolo, C. & Giorgi, F. M. Master regulator analysis of the SARS-CoV-2/human interactome. J. Clin. Med. 9, 982 (2020).
Gargouri, M. et al. Identification of regulatory network hubs that control lipid metabolism in Chlamydomonas reinhardtii. J. Exp. Bot. 66, 4551–4566 (2015).
Zhuang, D. Y., Jiang, L. I., He, Q. Q., Zhou, P. & Yue, T. Identification of hub subnetwork based on topological features of genes in breast cancer. Int. J. Mol. Med 35, 664–674 (2015).
Wasserman, S., Faust, K. & others. Social network analysis: Methods and applications. vol. 8 (Cambridge university press, 1994).
Bouquet, J. et al. Longitudinal transcriptome analysis reveals a sustained differential gene expression signature in patients treated for acute Lyme disease. MBio 7, (2016).
Barbieri, E. et al. Histone chaperone CHAF1A inhibits differentiation and promotes aggressive neuroblastoma. Cancer Res. 74, 765–774 (2014).
Basso, K. et al. Integrated biochemical and computational approach identifies BCL6 direct target genes controlling multiple pathways in normal germinal center B cells. Blood, J. Am. Soc. Hematol. 115, 975–984 (2010).
Schneider, W. M., Chevillotte, M. D. & Rice, C. M. Interferon-Stimulated Genes: A Complex Web of Host Defenses. Annu. Rev. Immunol. 32, 513–545 (2014).
Schoggins, J. W. Interferon-Stimulated Genes: What Do They All Do? Annu. Rev. Virol. 6, 567–584 (2019).
Jefferies, C. A. Regulating IRFs in IFN driven disease. Front. Immunol. 10, 325 (2019).
Seifert, L. L. et al. The ETS transcription factor ELF1 regulates a broadly antiviral program distinct from the type I interferon response. PLoS Pathog. 15, e1007634 (2019).
Froggatt, H. M., Harding, A. T., Heaton, B. E. & Heaton, N. S. ETV7 limits antiviral gene expression and control of SARS-CoV-2 and influenza viruses. bioRxiv 851543 (2020).
Bruchez, A. et al. MHC class II transactivator CIITA induces cell resistance to Ebola virus and SARS-like coronaviruses. Sci. (80-.) 370, 241–247 (2020).
Langfelder, P., Mischel, P. S. & Horvath, S. When is hub gene selection better than standard meta-analysis? PLoS One 8, e61505 (2013).
He, X. & Zhang, J. Why do hubs tend to be essential in protein networks? PLoS Genet 2, e88 (2006).
Gaiteri, C., Ding, Y., French, B., Tseng, G. C. & Sibille, E. Beyond modules and hubs: the potential of gene coexpression networks for investigating molecular mechanisms of complex brain disorders. Genes, Brain Behav. 13, 13–24 (2014).
Meyer, P. E., Lafitte, F. & Bontempi, G. Minet: A r/bioconductor package for inferring large transcriptional networks using mutual information. BMC Bioinforma. 9, 461 (2008).
He, J., Zhou, Z., Reed, M. & Califano, A. Accelerated parallel algorithm for gene network reverse engineering. BMC Syst. Biol. 11, 83 (2017).
Glass, K., Huttenhower, C., Quackenbush, J. & Yuan, G.-C. Passing messages between biological networks to refine predicted interactions. PLoS ONE 8, e64832 (2013).
Silva-Cardoso, S. C. et al. CXCL4 links inflammation and fibrosis by reprogramming monocyte-derived dendritic cells in vitro. Front. Immunol. 11, 2149 (2020).
Mijnheer, G. et al. Conserved human effector Treg signature is reflected in transcriptomic and epigenetic landscape. bioRxiv (2020).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Ritchie, M. E. et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47 (2015).
Wang, J. et al. Reconstructing regulatory networks from the dynamic plasticity of gene expression by mutual information. Nucleic Acids Res. 41, e97 (2013).
Greenham, K. & Robertson McClung, C. Time to build on good design: Resolving the temporal dynamics of gene regulatory networks. Proc. Natl. Acad. Sci. USA. 115, 6325–6327 (2018).
Chen, X., Li, M., Zheng, R., Wu, F. X. & Wang, J. D3GRN: A data driven dynamic network construction method to infer gene regulatory networks. BMC Genomics 20, 1–8 (2019).
Zhu, H., Shyama Prasad Rao, R., Zeng, T. & Chen, L. Reconstructing dynamic gene regulatory networks from sample-based transcriptional data. Nucleic Acids Res. 40, 10657–10667 (2012).
Chai, L. E. et al. A review on the computational approaches for gene regulatory network construction. Computers Biol. Med. 48, 55–65 (2014).
Langfelder, P. & Horvath, S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinforma. 9, 559 (2008).
Marbach, D. et al. Wisdom of crowds for robust gene network inference. Nat. Methods 9, 796–804 (2012).
Huynh-Thu, V. A. & Sanguinetti, G. Gene Regulatory Network Inference: An Introductory Survey. in Methods in Molecular Biology vol. 1883 1–23 (Humana Press Inc., 2019).
Han, H. et al. TRRUST: a reference database of human transcriptional regulatory interactions. Sci. Rep. 5, 11432 (2015).
Liu, Z.-P., Wu, C., Miao, H. & Wu, H. RegNetwork: an integrated database of transcriptional and post-transcriptional regulatory networks in human and mouse. Database 2015, bav095 (2015).
Marbach, D. et al. Tissue-specific regulatory circuits reveal variable modular perturbations across complex diseases. Nat. Methods 13, 366 (2016).
Subramanian, A. et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl Acad. Sci. USA 102, 15545–15550 (2005).
Phan, R. T., Saito, M., Basso, K., Niu, H. & Dalla-Favera, R. BCL6 interacts with the transcription factor Miz-1 to suppress the cyclin-dependent kinase inhibitor p21 and cell cycle arrest in germinal center B cells. Nat. Immunol. 6, 1054–1060 (2005).
Christian, S. L. et al. Suppression of IFN-induced transcription underlies IFN defects generated by activated Ras/MEK in human cancer cells. PLoS One 7, e44267 (2012).
Thomas, E. et al. HCV infection induces a unique hepatic innate immune response associated with robust production of type III interferons. Gastroenterology 142, 978–988 (2012).
Duncan, C. J. A. et al. Human IFNAR2 deficiency: Lessons for antiviral immunity. Sci. Transl. Med. 7, 307ra154 (2015).
Tsoi, L. C. et al. Hypersensitive IFN Responses in Lupus Keratinocytes Reveal Key Mechanistic Determinants in Cutaneous Lupus. J. Immunol. 202, 2121–2130 (2019).
Sirois, M. et al. TRAF6 and IRF7 control HIV replication in macrophages. PLoS One 6, e28125 (2011).
Hernandez, N. et al. Life-threatening influenza pneumonitis in a child with inherited IRF9 deficiency. J. Exp. Med 215, 2567–2585 (2018).
Steiger, J. et al. Imatinib Triggers Phagolysosome Acidification and Antimicrobial Activity against Mycobacterium bovis Bacille Calmette-Guérin in Glucocorticoid-Treated Human Macrophages. J. Immunol. 197, 222–232 (2016).
Mehraj, V. et al. Monocyte responses in the context of Q fever: from a static polarized model to a kinetic model of activation. J. Infect. Dis. 208, 942–951 (2013).
Nograles, K. E. et al. Th17 cytokines interleukin (IL)-17 and IL-22 modulate distinct inflammatory and keratinocyte-response pathways. Br. J. Dermatol 159, 1092–1102 (2008).
Smith, J. A. et al. Gene expression analysis of macrophages derived from ankylosing spondylitis patients reveals interferon-gamma dysregulation. Arthritis Rheum. 58, 1640–1649 (2008).
Kang, K. et al. IFN-γ selectively suppresses a subset of TLR4-activated genes and enhancers to potentiate macrophage activation. Nat. Commun. 10, 1–14 (2019).
Acknowledgements
We appreciate the critical discussions within the CoSI group, UMC Utrecht, which significantly improved this manuscript. This work was supported by China Scholarship Council (CSC) No. 201606300050, Netherlands Organization for Scientific Research (NWO) No. 016.Veni.178.027.
Author information
Authors and Affiliations
Contributions
W.T. and A.P. conceptualized the idea and designed the methodology. W.T. designed the algorithmic solutions and wrote the code. A.P. supervised the research. A.P. and T.R.D.J.R. provided the resources for the research. W.T., A.P., and T.R.D.J.R. wrote the original draft, and reviewed and edited the manuscript. All authors contributed to conceiving the idea. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics statement
According to the original studies of the datasets of the primary human cell/tissue samples, informed consent was obtained from study participants.
Competing interests
The authors declare no competing interests.
Peer review information
Communications Biology thanks Mariano Alvarez, Jackie (Jiekun) Yang and the other, anonymous, reviewers for their contribution to the peer review of this work. Primary Handling Editors: Eirini Marouli and George Inglis.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tao, W., Radstake, T.R.D.J. & Pandit, A. RegEnrich gene regulator enrichment analysis reveals a key role of the ETS transcription factor family in interferon signaling. Commun Biol 5, 31 (2022). https://doi.org/10.1038/s42003-021-02991-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-021-02991-5
- Springer Nature Limited
This article is cited by
-
Distinct patterns of proteostasis network gene expression are associated with different prognoses in melanoma patients
Scientific Reports (2024)
-
Leukocyte-associated immunoglobulin-like receptor-1 blockade in combination with programmed death-ligand 1 targeting therapy mediates increased tumour control in mice
Cancer Immunology, Immunotherapy (2024)