
Abstract
Hepatocellular carcinoma (HCC) associated with viral or metabolic liver diseases is a growing cancer without effective therapy. AMPK is downregulated in HCC and its activation diminishes tumor growth. Alpha lipoic acid (ALA), an indirect AMPK activator that inhibits hepatic steatosis, shows antitumor effects in different cancers. We aimed to study its putative action in liver-cancer derived cell lines through AMPK signaling. We performed cytometric studies for apoptosis and cell cycle, and 2D and 3D migration analysis in HepG2/C3A and Hep3B cells. ALA led to significant inhibition of cell migration/invasion only in HepG2/C3A cells. We showed that these effects depended on AMPK, and ALA also increased the levels and nuclear compartmentalization of the AMPK target p53. The anti-invasive effect of ALA was abrogated in stable-silenced (shTP53) versus isogenic-TP53 HepG2/C3A cells. Furthermore, ALA inhibited epithelial-mesenchymal transition (EMT) in control HepG2/C3A but not in shTP53 nor in Hep3B cells. Besides, we spotted that in patients from the HCC-TCGA dataset some EMT genes showed different expression patterns or survival depending on TP53. ALA emerges as a potent activator of AMPK-p53 axis in HCC cells, and it decreases migration/invasion by reducing EMT which could mitigate the disease in wild-type TP53 patients.
Similar content being viewed by others


Introduction
Hepatocellular carcinoma (HCC) is one of the main causes of death by cancer and current surgical or chemotherapeutic treatments fail in improving survival in most of the patients1. HCC is generally associated with an underlying chronic liver disease such as viral hepatitis, alcoholic cirrhosis or metabolic associated fatty liver disease. The latter is an increasing cause of pathology in occidental countries2. Carcinogenic transformation develops on these inflammatory diseases by producing dysplastic nodules that evolve into neoplastic foci with great molecular heterogeneity, to which a high intra and extra hepatic metastatic capacity is added making HCC a very aggressive cancer3,4. In this regard, to find new treatments that reduce or even prevent the rapid progression of HCC is a pressing matter of research. AMP activated kinase (AMPK) is a hetero trimer constituted by α, β and γ subunits, whose catalytic α subunit, in response to energy stress, is phosphorylated and activated by upstream kinases like LKB1 and it is stabilized by increased levels of AMP that bounds to γ subunit5. AMPK activation is both a hierarchical and a compartmentalized process that depends on the stress level, and where different isoforms of AMPK subunits form active complexes with different proteins in lysosomes, mitochondria and nucleus6,7. Activation of AMPK in different types of cancer cells induces distinct antitumor effects including decrease in cell survival or reduction of invasiveness8,9,10,11, which support it as an eligible target for therapy, at least in the initial states of the transformation process when AMPK would act as a tumor suppressor12,13. Moreover, given that during energy stress the effects of the tumor suppressor p53 are intertwined with the LKB1-AMPK axis as both a target and activator, the role of p53 in the therapeutic activation of this pathway should be considered, for a review see14,15.
In HCC patients, active AMPK is decreased in tumor in comparison to neighboring tissue, and AMPK activation has been suggested as an effective antitumor strategy in mice16,17. In previous reports, we found that in HCC-derived cells different ways of activating AMPK, such as glucose restriction18 and metformin, not only induce cell death but they early inhibit migration and invasion19.
Alpha lipoic acid (ALA), a natural compound that is a physiological cofactor of mitochondrial pyruvate dehydrogenase (PDH), was studied as a possible anticancer drug in different types of cancer cells20,21. ALA induces mitochondrial dysfunction and cytotoxicity in various tumor cells where it is able to activate AMPK and, in some cases, ALA also decreases invasion through AMPK-p53 axis20,22. It is attractive to determine if this AMPK activator leads to any AMPK mediated anticarcinogenic actions in HCC cells, given that former studies show induction of apoptosis by ALA in HCC cell lines without clarifying other possible antitumor effects23. Interestingly, ALA activates hepatic AMPK in rat steatosis, and it decreases lipogenesis24,25. We hypothesize that ALA can induce different antitumor events through AMPK signaling in HCC cells as well. This therapeutic scenario would be very promising because of the putative multiple actions of ALA in metabolic or neoplastic liver disease.
The control of epithelial-mesenchymal transition (EMT) in the carcinogenic and pre carcinogenic liver is a potential target to stop HCC progression26. p53 has been postulated as an unequivocal regulator of the stability of EMT transcription factors in diverse tumors27,28. Therefore, it is interesting to explore if ALA could modulate these functions in HCC cells via p53.
In this study we tried to analyze the effect of ALA not only in the viability but also in the invasiveness of HCC cells, and to investigate if AMPK and its target p53 could be involved. For this purpose, we studied the response to ALA in different liver-cancer derived cell lines with different AMPK and p53 status.
Methods
Cell culture and genetic manipulations
Liver cancer cell lines C3A (HepG2/C3A, a clonal derivative of HepG2, ATCC, Manassas, VA), and Hep3B (Hep 3B2.1–7, ATCC) were grown with 4.5 g/L glucose DMEM (Gibco, Thermo Fisher Scientific, Waltham, MA), plus 10% FBS and 100 U/ml penicillin and streptomycin (Invitrogen, Waltham, MA). Cells were cultured in a humidified incubator at 37º℃ with 5% CO2, and tested periodically for mycoplasma by PCR.
When indicated, alpha lipoic acid (0.5 or 1 mM, Santa Cruz Biotechnology Inc., Santa Cruz, CA) was added.
For reducing AMPKα1 protein expression in C3A cells, 21 nucleotide duplexes targeting two specific sequences and a scrambled control were designed and synthesized by Ambion SilencerTMsiRNA (Ambion, Thermo Fisher Scientific). The target sequences were AMPKα1 1842–1864: AACATTTCTGCATATTAGGCTCCTGTCTC and 2659–2681: AAGAGCTGAGTTGCATATACTCCTGTCTC, as we previously reported19. Experiments with silenced cells were carried out 24–48 h after transfection using siPORT NeoFX (Ambion, Thermo Fisher Scientific) with the Negative Control siRNA (Ambion) or the synthesized siRNAs against AMPKa1, and decrease (60 to 80%) in AMPKα expression for both duplexes was confirmed by immunoblotting.
For stable knockdown of p53 in C3A cells, transduction with retrovirus-based plasmids harboring the following specific and control shRNAs were carried out: shp53 (GACUCCAGUGGUAAUCUAC), shp53-3′UTR (GGUGAACCUUAGUACCUAA), and control shLacZ (GUGACCAGCGAAUACCUGU), as we previously described29. Plasmid DNA was prepared using the Wizard DNA purification kit (Promega, Madison, WI). p53 diminution (80–90%) was confirmed by qRT-PCR.
Colony formation assay
Low density cell inoculum (1000 cells) was seeded in 35 mm dishes of 6-well plates and grew up to 15 days to allow the formation of visible colonies. After washing the colonies were fixed with cold methanol and toluidine blue staining was performed for 10 min. After removing the excess of colorant, the dishes were photographed and the number of colonies were counted and compared.
MTT assay
HCC cells were plated at the same density by triplicate in 96-well microplates grown for 16, 24, 48 and 72 h, and incubated 2 h with methylthiazolyldiphenyl-tetrazolium bromide (MTT, Sigma Chemical Co., St Louis, MO). The metabolite produced from viable cells was dissolved in DMSO and detected at 540 nm in a microplate reader (Beckman Coulter LD400), as we described19. Results were expressed as percentage of absorbance in cells at 0 time.
Annexin V/propidium iodide assay
Annexin V assay (BD Biosciences, San José, CA) was used following the manufacturer recommendations and the samples were prepared as we previously described18. Briefly, after 48 h of treatments, homogenization in the medium and harvest, 100,000 cells were gently re-suspended and phosphatidylserine externalization and cell death was detected by staining with Annexin V-FITC and PI, respectively. The stained cells were subjected to flow cytometric analysis (Cell Sorter BD FACSAria II, BD Biosciences).
Cell cycle analysis
The proportion of cells in each cell cycle population at 48 h of culture in different conditions was analyzed by determining the cellular DNA content by flow cytometry (Cell Sorter BD FACSAria II, BD Biosciences), as we previously described18. In brief, 1 × 106 cells were fixed in 70% ethanol, washed and stained with 50 μg/ml propidium iodide (Sigma Chemical Co.) in a solution containing 0.1% sodium citrate, 0.02 mg/ml RNAse, and 0.3% NP-40. WinMDi and Cylchred softwares were used for data analysis.
Wound healing assay
Collective cell migration was determined by wound healing assay as we previously described30, with slight modifications. Briefly, cells were plated (6-well plates) at 1.5 × 106 cells/ml and growth for 24 h. Monolayers were wounded by dragging a 200 μl pipette tip, washed and treated. Captures of microscopy images (Zeiss Axiovert 25) of wounds were obtained at 0 and 24 h after wounding. Lineal advance of the wound front (μm) was assessed by using ImageJ.
Invasion studies
For invasion assays, transwell chambers (Biofil, Beijing, China) with 8 μm pore size polyester membrane filters were coated with 250 μg/ml Matrigel (Corning, Corning, NY). Suspension of cells (1 × 105) in DMEM containing 1% FBS were plated into the upper chamber, and the lower chamber was filled with complete DMEM medium containing 10% FBS. Cells were treated for 48 h and after this period those cells that reached the lower chamber were fixed with methanol, stained with 1% toluidine blue-1% borax for 5 min, and counted by microscopy captures, as we previously described19.
Immunofluorescence microscopy
Cells were grown on coverslips, treated for 24 h and, at the end of experiments, fixed, permeabilized and blocked with 0.3% Triton X-100–1% albumin. Then, cell samples were incubated with anti p53 (sc-126, Santa Cruz Biotechnology), washed, incubated with anti-mouse secondary antibody (Invitrogen) and then incubated for nuclei staining with 4′,6-diamidino-2-phenylindole (DAPI, Molecular Probes, Eugene, OR), and mounted with ProLong (Molecular Probes). Fluorescence was detected by a confocal microscopy (Nikon C1SiR with inverted microscope Nikon TE200). Nuclear intensity of p53 staining was assessed in the Z-stack projections by using ImageJ tools.
Nuclear and cytosolic subcellular preparations
Nuclear and cytosolic fractionation was carried out from C3A and Hep3B cells after culture in the indicated conditions by sequential lysis with cytosol and nuclear extraction buffer solutions. Briefly, cells cultured in 10 cm plates were washed with saline and scrapped with 500 μl of a buffer solution containing 10 mM HEPES (pH 7.8), 10 mM KCl, 0.1 mM EDTA, 0.4% NP-40, 1 mM DTT and protease inhibitors. The lysates were centrifuged at 15,000 ×g and 4° for 3 min and the supernatants (cytosolic fraction) were stored in new tubes. The pellets were resuspended in 150 μl of a buffer solution containing 20 mM HEPES (pH 7.8), 0.4 M NaCl, 1 mM EDTA, 10% glycerol, 1 mM DTT and protease inhibitors, and incubated at 0° for 2 h with 15 s-cycle of vortexing every 15 min. The resulting lysates were centrifuged at 15,000 ×g and 4° for 5 min and the supernatants (nuclear fraction) were stored in new tubes.
Western blotting
Cell lysates were obtained after scrapping and washing the cultured cells subjected to the indicated treatments during the indicated time. The harvest cells were incubated in 100 μl of ice cold RIPA buffer with protease and phosphatase inhibitors for 30 min and sonicated. Protein concentration was determined by the Lowry’s method. Equal amounts of total protein (30–50 μg) were loaded, resolved by electrophoresis on SDS–polyacrylamide gels31, and transferred to polyvinylidene difluoride membranes (Perkin Elmer Life Sciences, Boston, MA, USA). Molecular weights were estimated using “rainbow” molecular weight markers. The membranes were blocked with 5% non-fat milk/0.3% Tween/PBS, washed and incubated O.N. with each primary antibody [α tubulin, β actin (AC-74) (T5168, A2228 Sigma Chemical Co.); cyclin D1 (EPR2241) (ab 134175 Abcam, Waltham, MA 02453); p53(DO-1), GAPDH (FL-335), histone H1 (AE-4) (sc-126, sc-25778, sc-8030 Santa Cruz Biotechnology); AMPKα, Phospho-AMPKα (Thr172), SNAIL/C15D3 (2532, 2535, 3879 Cell Signaling Technology); or E-cadherin (610182 BD Biosciences)]. After incubation of the membranes with the appropriate secondary antibody, the bands were detected by chemiluminescence (Amersham Pharmacia Biotech, Piscataway, NJ).
Real-time quantitative PCR
cDNA was obtained from Trizol (Invitrogen) extracts by retrotranscription with retrotranscriptase M-MLV (Promega) following the manufacturer conditions, and real-time PCR was performed using SYBR Green PCR master mix (Promega) according to the following conditions: 2 min at 95º℃ for one cycle; and 30 s at 95º℃, 20 s at 60º℃, 30 s at 72º℃ for 40 cycles. Results were analyzed using the comparative Ct method. Values were normalized to GAPDH expression. The following real-time PCR forward and reverse primers were used, respectively: TP53: CTCCTCTCCCCAGCCAAAGA, GGAACATCTCGAAGCGCTCA; CDH1: AAGAAGCTGGCTGACATGTACGGA, CCACCAGCAACGTGATTTCTGCAT; VIM: CCCTGCAATCTTTCAGACAG, ATCTGAGCCTGCAGCTCCT; SNAI1: CCAGAGTTTACCTTCCAGCAGCC, CAGGACAGAGTCCCAGATGAGCAT; GAPDH: TCTCTGCTCCTCCTGTTC, GCCCAATACGACCAAATCC.
Bioinformatic analyses
RNAseq data from HCC patients (371 tumor and 50 non tumor samples) were obtained from TCGA datasets accessible from http://gdac.broadinstitute.org/. Data were organized and processed for differential gene expression (DGE) analysis for a selected group of EMT related genes with EDAseq and edgeR, respectively. Other packages from tydiverse were used for data management. Mean log2 fold change (log2FC), p, and false discovery rate (FDR) values were obtained. Values of log2FC > 1 or < 1, and −log2FDR > 1 (FDR < 0.01) were set as cutoffs. A heatmap plot from the 50 patients with paired tumor and non-tumor samples was performed for the indicated genes using complexHeatmap, and the results were split in two groups by the status of TP53 gene. In the same dataset a comparison of the distribution of frequencies of log2FC values for each gene was performed between those patients with wild type (n = 37) or mutant TP53 (n = 13) using the Kolmogorov–Smirnov test. This analysis was performed with SAS Studio, and a p-value < 0.05 was considered statistically significant.
Further expression profiles of the genes studied from the same TCGA patients were analyzed through the tools of the UALCAN data portal (http://ualcan.path.uab.edu). These tools match expression and clinical data allowing comparison of expression levels in different groups of patients, as cancer stages and others. Data processing and statistical analysis were described by the authors32. In addition, expression plus survival assays of these genes from the same dataset of TCGA were also analyzed with the visualization tools of The Human Protein Atlas (www.proteinatlas.org).
For those groups of patients with low or high expression (below or above the median) of the selected genes with wild type or mutant TP53 survival analysis were performed using survival and survminer R tools. p < 0.05 was set as a significant difference among the Kaplan-Meier curves. When it was necessary, hazard ratios (HR) were calculated and represented as a forest plot and the p value for HR between the reference group versus each of the others was obtained.
Statistical analysis
In all the studies except for bioinformatics, data were expressed as mean ± S.E.M. and statistical comparisons were made by using Student t test. p < 0.05 was considered statistically significant.
Results
Changes in cell viability and growth in response to ALA
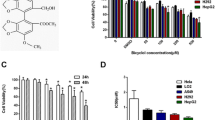
ALA is well established as an oxidative stress and apoptosis inductor in HCC cells at a dose of 0.5 mM23, as occurs in different cancer cells20. We explored the effects of this low millimolar level of ALA in HCC derived cells: Both 0.5 and 1 mM concentrations led to decreases in the number of viable cells, but the lower concentration had no significant effect below 48 h. At 48 the viability of cells treated with 0.5 mM ALA decreased by 20% compared to untreated cells, as observed by MTT assay in C3A cells (Fig. 1A), and similarly in Hep3B cells (data not shown). A marked diminution in colony formation was induced by ALA already at 0.5 mM suggesting a loss in the ability of treated cells to form clonal colonies (Fig. 1B). To better characterize these phenomena, we performed cell cycle and cell death analyses in C3A and Hep3B cells. A 48-h treatment with 0.5 mM ALA arrested cell cycle at G0/G1 (Fig. 1C), which was in accordance with a significant decrease in the levels of cyclin D1 in both cell lines (Fig. 1E). In addition, cytometric analysis of apoptotic populations by AnnexinV/PI staining in HCC derived cells showed that total apoptotic cells (Annexin V positive) significantly increased by 30 or 80% after 48 h treatments with 0.5 or 1 mM ALA, respectively. Those cells dead by apoptosis (double positive) reached nearly 8 or 12% of the cells in the presence of 0.5 or 1 mM ALA after 48 h, respectively (Fig. 1D). Altogether, results corroborated that ALA had diverse antitumor effects in HCC cells: a proapoptotic and a considerable cytostatic action reflected in the inhibition of anchorage independent growth.

Viability and cell cycle progression in HCC cells during ALA treatment. (A) MTT assay was performed, as detailed in Methods, in HepG2/C3A cells cultured in DMEM medium alone (C) or in the presence of 0.5 or 1 mM ALA for 0, 16, 24, 48 and 72 h. Results were expressed as % of the absorbance at 0 time. The plots represent mean values of three-four independent experiments ± S.E.M. * p < 0.05 vs. (C) (B) Captures of stained wells after typical experiments of colony formation assay for HepG2/C3A cells in DMEM medium (control, C), or treated with 0.5 or 1 mM ALA, respectively (upper panel). The bars represent mean values of the number of colonies for three independent experiments ± S.E.M. * p < 0.05 vs. control. (D) Cytometric analysis of PI-stained cells for cell cycle evaluation was performed in Hep3B cells after 48 h cultures in DMEM alone (control, C) or with 0.5 mM ALA. Bars represent the mean percentages of each cell population from three independent experiments ± S.E.M. * p < 0.05 vs. control. D. Annexin V and propidium iodide (PI) staining were performed in HepG2/C3A cells after 48 h culture in DMEM alone (control, C) or with ALA, and the cells were prepared for flow cytometry assay. Single Annexin V stained and Annexin V plus PI-stained cells were analyzed. Representative outputs of the cytometric assay are shown (upper panel). The percentage of each stained population was obtained. Bars represent mean values ± S.E.M of three independent experiments for Annexin V stained cells (total apoptosis) in control, 0.5 mM and 1 mM ALA treated cells as a percentage of the control group. * p < 0.05 vs. control. (E) Cell lysates were obtained after 24 h treatment from HepG2/C3A or Hep2B cells cultured in DMEM (control, C) or in the presence of 0.5 mM (0.5) or 1 mM (1) ALA, and 30 μg of protein were loaded in mini-gels for immunodetection of cyclin D1. β Actin was used as loading control. Immunoblots show an experiment representative of three independent ones. Selected lanes for each detection are in their original order and correspond to the same gel, and they are shown after cropping, aligning and separating them by white space. Full-length blots are available in Supplementary Dataset. Bars represent mean values of band density relativized to load and expressed as percentage of the control from three independent experiments ± S.E.M. * p < 0.05 vs. control.
ALA impaired migration and invasion in wild type p53-HCC cells
When migratory capacity and invasiveness of C3A and Hep3B cells were analyzed at a dose of 0.5 mM, which showed minor effects in viability, we found that, in HepG2/C3A cells (wild type TP53), 2D migration in the wound front along 24 h decreased by 40% in ALA treated versus control cells. Conversely, migration in Hep3B cells (TP53 deletion) was the same irrespectively of ALA treatment (Fig. 2A). In addition, invasion studies showed that invasion through matrigel coated transwells was significantly diminished by a half of the control values in C3A cells treated with 0.5 mM ALA, whilst under this treatment invasion of Hep3B cells was maintained without significant changes (Fig. 2B). These results suggested that a distinct response of the migratory function to ALA could be attributed to the different genetic background of the cells.

Effect of ALA on migration and invasion in HepG2/C3A and Hep3B cells. (A) Wound healing assays were performed in HCC cells in DMEM medium alone (control, C) or with 0.5 mM ALA (ALA), and the distance migrated by the front cells, i.e. the difference of the respective wound widths in micra, was calculated through image analysis from captures at 0 and 24 h. Bars represent mean values ± S.E.M of three independent experiments expressed as percentages of the controls in HepG2/C3A and Hep3B cells, respectively. (B) Invasion in matrigel coated transwells was compared for control (C) and 0.5 mM ALA treated (ALA) HepG2/C3A and Hep3B cells after 48 h. After staining, images were obtained and cells were quantified. Captures of typical experiments are shown (right panel). Bars represent mean values (as a percentage of the control groups) of three independent experiments ± S.E.M. * p < 0.05 vs. control.
The anti-migratory effect of ALA was completely dependent on AMPK activation
In order to deepen in the mechanism of the striking effect of ALA in migration and invasion of HCC cells that express the AMPK target p53, we evaluated the role of AMPK signaling in the decrease of these migratory functions. First, we detected AMPK activation in these HCC cells subjected to ALA treatment. ALA increased the levels of pAMPK in both HepG2/C3A and Hep3B cells (Fig. 3A), and this in the case of cells with wild type p53 was accompanied by a prominent augment of total p53 protein levels and also of its nuclear localization (Fig. 3A,B). In fact, nuclear p53, detected by fluorescent microscopy or biochemically, increased after ALA treatment around fifty percent, in contrast to its cytosolic diminution, as depicted in Fig. 3B (right), this suggesting an augment of the activation and translocation of the protein.

Activation of AMPK and p53 during ALA treatment in wild type p53-HCC cells signals migration/invasion processes. (A) HepG2/C3A or Hep3B cells cultured in DMEM (C, control) or in DMEM with 0.5 mM ALA (ALA) and after 24 h cell lysates were obtained, as stated in Methods, and 20–30 μg of protein were loaded in mini-gels for immunodetection of p-AMPK, AMPK catalytic subunit, and p53 (HepG2/C3A cells). α-tubulin (α tub) was used as loading control. Immunoblots show an experiment representative of three independent ones. Selected lanes for each detection are in their original order and correspond to the same gel, and they are shown after cropping, aligning and separating them by white space. Full-length blots are available in Supplementary Dataset. Band densities were quantified relativized to AMPK or loading and represented in bar charts. Bars represent the mean values, expressed as a percentage of the control group, ± S.E.M from three independent experiments. # p < 0.05 vs. control, # # p < 0.005 vs. control. (B) Left: C3A cells were incubated for 24 h in control DMEM (C, control) or in the presence of 0.5 mM ALA (ALA). Afterwards, cells were fixed and stained to visualize p53 (green) and nuclei (blue), red staining with Mitotracker Red was used for obtaining a signal of cell integrity. p53 intensity in the nucleus was relativized to total p53 signal. Bars represent the mean values, expressed as a percentage of the control group, ± S.E.M from 10 fields for three independent experiments. Right: Cell nuclear and cytosolic fractions of the same experimental groups were obtained as described in Methods, and 30 μg were loaded and subjected to electrophoresis in mini-gels followed by immunodetection of p53. For the assessment of nuclear p53 histone H1 was used as loading control, whereas β actin was used for cytosolic p53. Immunoblots show an experiment representative of three independent ones with the same considerations stated above in 3A. Nuclear and cytosolic p53 protein levels in the indicated groups were calculated and graphed in bar charts. Bars represent mean values of band density relativized to load and expressed as percentage of the control from three independent experiments ± S.E.M. * p < 0.05 vs. control. C and D. HepG2/C3A cells were transfected with AMPKα1 targeted (siAMPKa1) or scrambled siRNAs (sc) cultured for 48 h and subjected to additional 48 h (C) or 24 h (D) treatments. (C) Cell lysates were processed as stated in Methods and AMPK silencing was corroborated by detection of total AMPK by the same procedure and with the same considerations stated above in 3A. A capture of a typical blotting is shown (upper right). Wound healing assays were performed and data of cell migration were obtained as indicated in Fig. 2A. * p < 0.05 vs. control. (D) Cell lysates were obtained and processed for immunodetection of p53 as in 3A, with the same considerations. p53 protein levels in the indicated groups were calculated and graphed in bar charts. Bars represent the mean values, expressed as a percentage of the control group, ± S.E.M from three independent experiments. # # p < 0.005 vs. control.
In turn, we analyzed the effect of the knockdown of the catalytic subunit of AMPK in HepG2/C3A cells (Fig. 3C, upper left) on the migratory response and observed that this significantly blocked the anti-migratory effect of 0.5 mM ALA in C3A cells (Fig. 3C). In association with this, the silencing of AMPKa abrogated the increase in total p53 levels in ALA treated cells (Fig. 3D).
Migration-Invasion in cell lines with diminished expression of p53
To determine the precise contribution of wild type p53 in the reduction of migration/invasion in HCC cells by ALA, we constructed from HepG2/C3A cells a control (shLacZ) and two stable retroviral clones with impaired expression of p53 (shTP53 and shTP53-3′UTR). The silenced cell lines showed more than 80%-reduction of TP53 transcription checked by qPCR (Supp. Fig. 1A). ALA (0.5 mM) effect was analyzed in these cells at the level of migration in wound healing assay and invasion through matrigel coated transwells. ALA treatment decreased by 60% migration in control cells, and the knockdown of p53 partially blocked this anti migratory effect reducing migration in 30%. Besides, ALA reduced invasion around 50% in control cells and this effect was completely blocked in shTP53 cells (Fig. 4A,B), and in shpTP53-3′UTR cells (data not shown), thus confirming that p53 expression is needed for ALA anti migratory and anti-invasive actions.

ALA treatment does not affect migration/invasion capacity of cells with stable p53 knock down. Transformed cell lines were constructed from HepG2/C3A cells transduced with retroviral plasmids containing specific shRNAs targeted to TP53 or control sequences, named shTP53 and shLacZ cells, respectively. These cell lines were cultured in DMEM without (C) or with 0.5 mM ALA (ALA) for 24 h or 48 h to analyze (A) cell migration, or (B) cell invasion. Data from three independent experiments were obtained and expressed as indicated in Fig. 2A,B, respectively. (A) The mean absolute values in control shLacZ cells were 189.0 and 73.5 μm in C and cells, respectively. Yellow lines illustrate the widths measured in each capture. (B) The mean absolute values in control shLacZ cells were 11.3 and 6.3 cells in C and ALA cells, respectively. * p < 0.05 vs. each untreated group (C); # p < 0.05 vs. ALA treated shLacZ cells.
Analysis of EMT markers in HCC cells treated with ALA: role of wild type TP53
Migration and invasion in cancer cells depend mostly on the acquisition of mesenchymal properties which take place with the epithelial mesenchymal transition. Given that ALA reduced migration-invasion only in HCC cells that express wild type p53, a possibility existed that one or more proteins involved in EMT were affected via p53 in these cells thus inhibiting EMT and migration-invasion.
When we analyzed some of these EMT markers after ALA treatment, we detected that the epithelial protein E-cadherin significantly increased its levels in ALA treated C3A cells but remained unchanged in Hep3B, in opposition with the levels of its negative regulator SNAIL which showed significant reduction in ALA treated C3A cells (Fig. 5A). We confirmed similar ALA effects dependent on TP53 expression in the constructed cell clones: In fact, CDH1 mRNA levels increased after ALA treatment in control but the opposite occurred in shTP53 cells (Fig. 5B). Conversely, VIM decreased after ALA treatment in control cells only, although SNAI1 mRNA levels showed no significant changes at the time analyzed (Fig. 5B). Altogether, these data pointed out that ALA inhibited EMT in HCC cells mostly via p53.

ALA reduces EMT activation in HCC cells with wild type p53 expression. (A) Cell lysates were obtained after 24 h treatment from HepG2/C3A or Hep2B cells cultured in DMEM (control, C) or in the indicated conditions, and 20–30 μg of protein were loaded in minigels for immunodetection of E-cadherin (E-cadh) and SNAIL. GAPDH was used as loading control. Immunoblots show an experiment representative of three independent ones. Selected lanes for each detection are in their original order and correspond to the same gel, and they are shown after cropping, aligning and separating them by white space. Full-length blots are available in Supplementary Dataset. Bars represent mean values of band density relativized to load and expressed as percentage of the control from three independent experiments ± S.E.M. * p < 0.05 vs. control. (B) shLacZ (control) and shTP53 cells obtained from HepG2/C3A cells were cultured in DMEM alone (untreated) or with 0.5 mM ALA for 24 h. After that, Trizol extracts and total cDNA from each group were generated by retrotranscription for use as qPCR samples. Transcript levels from CDH1, VIM and SNAI1 were estimated by Ct calculation from duplicate assays. GAPDH was used as a housekeeping gene. Bars represent mean values of Ct from three independent experiments ± S.E.M expressed as fold change of each untreated group. # p < 5 × 10−3 vs. untreated control, # # p < 5 × 10−4 vs. untreated control, * p < 5 × 10−2 vs. untreated control, ** p < 1 × 10−3 vs. untreated control.
Variations in the expression of EMT related genes putatively regulated by p53 and the impact in survival in HCC patients
We then determined if in fact some EMT actors had different expressions and could conditionate the outcome of HCC patients depending on p53. We first evaluated if HCC-TCGA patients presented changes in the expression of EMT related genes putatively regulated by p53. We focused on a set of genes including the EMT markers CDH1, CDH2 and VIM; as well as EMT transcription factors demonstrated to be negatively regulated by p53 in vitro: ZEB1/2, TWIST1/2 and SNAI1/226,33. In addition, we included genes involved in ECM degradation as the metalloproteinases MMP2 and MMP9, also regulated by p5334. DGE analysis carried out in the complete dataset of HCC-TCGA patients showed that, of the selected genes, only VIM, MMP2, SNAI2, MMP9 and TWIST1 were differentially expressed and increased their expression in tumor versus non tumor samples, and no decreases (log2FC < 1) with statistically significance were detected (Fig. 6A). A heatmap representation of gene expression in those 50 patients with paired control and tumor samples was performed and the plots were filtered and split in two groups of patients by TP53 status: although the clustering of genes was discretely different depending on TP53, most of the patients had increased expression of MMP9 and SNAI2, and, in the opposite side, most of them presented a decreased expression of CDH1, as SNAI1 and ZEB2 which seemed counter-regulated with respect to their related transcription factors (Fig. 6B). In the same group we compared the values of relative expression levels between patients with wild type TP53 and patients with mutant TP53, and we observed that only in the case of CDH1 the distribution of values was significantly different in both populations: Patients with wild type TP53 had more preserved levels of CDH1 than those with mutant TP53 (Fig. 6C). We also analyzed this set of genes in the same HCC-TCGA patients using the data analysis tools accessible from UALCAN and we compared gene expression levels among each cancer stage: Significant decreases in CDH1, and increases in VIM, SNAI2, TWIST1, ZEB1 and MMPs in different stages were observed (Fig. 6D). Again, SNAI1, ZEB2 and TWIST2 had decreased expressions (omitted in the figure), which were non-significant in the case of SNAI1.

Evaluation of selected EMT related genes in HCC-TCGA patients: expression and survival analyses of groups with different mutation status of p53. (A) Differential gene expression analysis was performed in the complete HCC-TCGA dataset (tumor samples N = 371, non-tumor samples N = 50) for a selected group of EMT related genes. Those genes with significantly increased expression (log2FC > 1) are enlisted. Mean log2FC, p and FDR values are indicated. (B) Heatmaps for the differential gene expression of the studied genes (columns) in the 50 HCC-TCGA patients with paired tumor and control non tumor samples (rows), filtered and split by their TP53 status: wild type (WT, left) or mutant (MUT, right). CDH1 gene column in each group is underlined. (C) The distribution of the relative expression levels of CDH1 was compared by K-S test between patients with wild type (WT) TP53 (N = 37, blue line) and patients with mutant (MUT) TP53 (N = 13, red line) from the 50 HCC-TGCA patients. The densities of patients for the variable log2FC are graphed. Vertical lines indicate the median values in each population. p = 0.0497. (D) Comparison of the expression levels in TPM of the indicated genes from HCC-TCGA patients in non-tumor (NT, N = 50) and in the four cancer stages, S1 (N = 168), S2 (N = 84), S3 (N = 82), S4 (N = 6) obtained from UALCAN data portal. Figures were constructed by adapting box and whisker plots of gene expression in non-tumor and in the four cancer stages from UALCAN web. Red lines represent the median values. Significant p values are indicated. (E) Kaplan–Meier survival analyses for CDH1 (left) and MMP9 (right) in HCC-TCGA patients with low or high expression levels of each gene studied and wild type (WT) or mutant (MUT) TP53. In the case of MMP9 a forest tree illustrating hazard ratios (HR) in each group is added. p values are indicated.
In turn, we studied if these EMT related genes which are potential p53 targets led to any change in survival depending on the TP53 mutation status. In fact, TP53 expression levels were increased in most HCC patients from TCGA, which did not affect survival, however, survival did depend on mutation status: WT-TP53 patients showed a risk of death 65% lower than MUT-TP53 patients (Supp. Fig. 1B). We then analyzed if low or high levels of each selected gene in combination with the status of TP53, wild type or mutant, led to any change in survival. Neither CDH1 (Fig. 6E, left) nor VIM nor the EMT transcription factors expression levels in the presence of any of the two TP53 status were associated with significant changes in patient survival. Only in the case of MMP9 a significant difference among survival curves was found: In patients with low MMP9 expression and WT TP53 the longest median survival was registered, and the risk of death was less than half of the risk of those with high MMP9 plus mutant TP53 (Fig. 6E, right).
Discussion
The uncontrolled growth and proliferation of tumor cells are very dependent on their increased glycolytic rate35. PDH catalyzes the decarboxylation of pyruvate in the TCA cycle, and prevention of pyruvate entry into the TCA cycle favors glycolysis and would promote tumor metabolic phenotype and survival of cancer cells, which is associated with high expression of the kinases that negatively regulate PDH, PDKs36,37. In this connection, recent studies in HCC cells demonstrate that PDH activation through downregulation of PDK4 leads to energy stress, increased ROS production and apoptosis38. The drug we analyze in this work, ALA, acts as a bonafide PDK inhibitor that is an effective anticarcinogenic drug in different tumor cells39, however its actions in HCC cells are not completely studied, especially the putative activation of AMPK and p53 as a mediator of antitumor effects.
Considering this perspective, in this report we first tried to determine the effect of ALA in the whole viability and capacity of growth of liver-cancer derived cells, and to further evaluate ALA effect on the metastatic ability through migration and invasion studies. We used the HepG2/C3A subclone, derived from the widely studied HepG2 cell line, which harbors wild-type TP53. This subclone retains certain features related to energy metabolism40,41,42, making it suitable for testing liver antitumor and metabolic disease drugs, as we previously reported18,19. Additionally, we employed Hep3B cells, which lack TP53. We demonstrated that ALA had a cytotoxic effect, due to a well described pro apoptotic action. In addition, ALA treatment led to inhibition of cell cycle at G0 in the two different cell lines assayed. However, we observed that the anti-migratory and anti-invasive effects of ALA were significant in HepG2/C3A, but not appreciable in Hep3B cells. Given that the last cell line has a distinctive genetic background because of HBV gene inserts and p53 gene deletion, we aimed to better define the role of p53 in this response to ALA.
ALA induces AMPK activation in different tumor cells where it may mediate many anticarcinogenic effects21, so we determined AMPKa phosphorylation (T172) levels in HepG2/C3A and Hep3B cells treated with ALA: p-AMPK levels were significantly increased by ALA at low millimolar concentration in both cell lines. These results indicated that ALA’s cytostatic effects in Hep3B, also observed in HepG2/C3A cells, could be attributed to AMPK signaling. Notably, in Hep3B cells, these effects appeared to be p53-independent. In fact, AMPK regulates the cell cycle and apoptosis in cancer cells through p27 phosphorylation with ulterior cyclins downregulation without relying on p5343,44,45. Besides, we demonstrated that ALA increased total and nuclear levels of the target of AMPK p53 in HepG2/C3A cells, which could be associated with the activation of p53. These results agreed strongly with recent evidence describing both stabilization and nuclear translocation of p53 in HepG2 cells during energy stress by fasting46,47. Furthermore, we observed that interfering with the expression of the catalytic subunit of AMPK resulted in a complete blockage in both p53 increase and migration reduction after ALA treatment in C3A cells. These effects occurred despite an apparently higher basal level of p53 in cells with silenced AMPKα expression. In fact, p53 regulation is complex, and numerous interactions exist that could explain in our setting transient p53 changes through feedback loops and counter-regulations48. One proposed disbalanced loop –not explored here– could involve AMPK and mTOR stabilization of p5349, two opposing kinase activities that are altered in HCC16,50. Altogether, these findings indicated that ALA antimigratory effect is dependent on AMPK signaling and that p53 activation is a putative downstream event leading to this antitumor action.
Pioneer genomic studies in HCC patients show by the first that survival in patients with mutations in the tumor suppressor p53 is diminished51. In fact, mutation of TP53 occurs early and it is a driver of HCC progression52, whilst the conservation of wild type TP53 could represent a significant benefit in the outcome of HCC, and a possibility to stabilize or activate p53 through novel strategies with antitumor results53,54,55. Wild type p53 is involved in the negative regulation of different EMT transcription factors in HCC33,56,57, hence we postulate that the status of p53 could be a strong determinant of the migratory ability of HCC cells and that the increase of WT p53 could lead to anti migratory effects. In the results presented herein, the stable silencing of WT p53 in HepG2/C3A cells impeded the reduction of migration and invasion after ALA treatment attributed to AMPK activation. These findings corroborated those we obtained in the p53-null cell line Hep3B, and they indicated that the activity of p53 is necessary in the anti migratory effect of ALA in HCC cells.
We then tried to determine if EMT was inhibited by this axis after ALA treatment in HCC cells, thus explaining the reduction in migration and invasion observed. We evaluated the effect of ALA on EMT in C3A and Hep3B cells and in control and shTP53-C3A cells, and we found that ALA reduced EMT in cells with wild type p53. Of note, the expression patterns of EMT markers CDH1 and VIM in shTP53 cells treated with ALA were opposite to isogenic controls. This potential induction of EMT by ALA in shTP53 cells was not significant in Hep3B cells, although a tendency existed. In contrast, functional analyses showed ALA had a significantly lower anti-migratory effect in shTP53 cells, and no effect on migration in Hep3B cells, nor on invasion in both shTP53 and Hep3B cells, compared to their controls. In fact, the trends of ALA actions on migration, invasion and EMT markers in shTP53-C3A and Hep3B cells were comparable and their differences could be attributed to the different genetic background of these cells. Further study is needed to understand the curious ALA actions on some EMT mediators when p53 is suppressed. Nonetheless, our results strongly demonstrated that ALA reduced migratory and invasive functions in a p53-dependent manner, associated with EMT inhibition. Regarding the potential clinical considerations of these results, it must keep in mind that TP53 mutations occur in less than one-third of HCC patients, missense mutations conserve mRNA levels and only truncated mutations decrease expression compared with WT-TP53 patients58. Given most HCC patients maintain TP53 expression levels, our findings support ALA as a therapy to induce a p53-regulated reduction of EMT in these patients. Overall, this set of results demonstrated that ALA increases E-cadherin, probably via SNAIL reduction, in cells expressing wild type p53 only, which is in the vein with the anti-migratory effect of ALA in these cells. The results also suggested the gravitation of E-cadherin in HCC biology depending on the presence of wild type p53. In this connection, several findings support that E-cadherin expression diminishes in more metastatic HCC forms59,60,61, and that SNAIL or TWIST roles in E-cadherin downregulation and metastasis are crucial62. However, the impact of the variations of these EMT markers in relation with other tumor regulators as p53 is understudied in HCC patients. For these reasons, we analyzed if some of these actors of the EMT process potentially regulated by p53 changed its gene expression and conditioned survival in HCC patients depending on TP53 status. Of the genes selected, CDH1 significantly decreased its expression, whilst SNAI2, TWIST1, ZEB1, VIM, MMP2 and MMP9 significantly increased in tumors of HCC-TCGA patients in one or more analyses. Furthermore, CDH1 had a distinct pattern of change in the expression: lower diminutions in patients with wild type TP53 compared with patients with mutated TP53 was observed. Among all these genes only MMP9 resulted in significant survival increase depending on p53. Given that p53 may regulate not only gene expression but also protein stability of EMT transcription factors like SNAIL and TWIST1 in HCC and in diverse tumor cells56,63,64, and considering that protein escindition of E-cadherin by metalloproteinases like MMP9 is also a way of EMT regulation in different cancers65,66,67, the complete picture is missing in transcriptomic analyses. Proteomic analyses from patients organized in databases are still scarce, but further studies would be necessary in HCC to find actual EMT determinants of the disease progression.
In sum, in this study we showed strong evidence of ALA antitumor actions in HCC cells from cytotoxic to never described cytostatic and antimigratory effects. We found that the reduction of migration and invasion by ALA treatment were dependent on AMPK and p53 activations which reduced EMT. Our results confirmed that core genes of EMT regulable by p53 variate their expression in HCC patients, sometimes in a progressive stage-dependent or p53 status-dependent way. Our results constitute promising data on the putative therapeutic effect of ALA or similar mitochondrial drugs in HCC, specially by reducing EMT and cell migration in those patients with wild type p53.
Data availability
The full-length blots used for the figures during this study are included in this published article [and its supplementary information files]. The datasets generated and/or analyzed during the current study are available in http://gdac.broadinstitute.org/ and http://ualcan.path.uab.edu. All other data are available from the corresponding author on reasonable request.
References
Llovet, J. M. et al. Hepatocellular carcinoma. Nat. Rev. Dis. Primers 7, 6. https://doi.org/10.1038/s41572-020-00240-3 (2021).
Singal, A. G., Lampertico, P. & Nahon, P. Epidemiology and surveillance for hepatocellular carcinoma: New trends. J. Hepatol. 72, 250–261. https://doi.org/10.1016/j.jhep.2019.08.025 (2020).
Marquardt, J. U., Andersen, J. B. & Thorgeirsson, S. S. Functional and genetic deconstruction of the cellular origin in liver cancer. Nat. Rev. Cancer 15, 653–667. https://doi.org/10.1038/nrc4017 (2015).
Schulze, K., Nault, J. C. & Villanueva, A. Genetic profiling of hepatocellular carcinoma using next-generation sequencing. J. Hepatol. 65, 1031–1042. https://doi.org/10.1016/j.jhep.2016.05.035 (2016).
Hardie, D. G. AMP-activated/SNF1 protein kinases: Conserved guardians of cellular energy. Nat. Rev. Mol. Cell Biol. 8, 774–785. https://doi.org/10.1038/nrm2249 (2007).
Zong, Y. et al. Hierarchical activation of compartmentalized pools of AMPK depends on severity of nutrient or energy stress. Cell Res. 29, 460–473 (2019).
Carling, D. AMPK hierarchy: A matter of space and time. Cell Res. 29, 425–426 (2019).
Schaffer, B. E. et al. Identification of AMPK phosphorylation sites reveals a network of proteins involved in cell invasion and facilitates large-scale substrate prediction. Cell Metab. 22, 907–921. https://doi.org/10.1038/s41422-019-0171-6 (2015).
Yan, Y. et al. Augmented AMPK activity inhibits cell migration by phosphorylating the novel substrate Pdlim5. Nat. Commun. 6, 6137. https://doi.org/10.1038/ncomms7137 (2015).
Henry, W. S. et al. Aspirin suppresses growth in PI3K-mutant breast cancer by activating AMPK and inhibiting mTORC1 signaling. Cancer Res. 77, 790–801. https://doi.org/10.1158/0008-5472.CAN-16-2400 (2017).
Duan, W. et al. Desmoplasia suppression by metformin-mediated AMPK activation inhibits pancreatic cancer progression. Cancer Lett. 385, 225–233. https://doi.org/10.1016/j.canlet.2016.10.019 (2017).
Li, W., Saud, S. M., Young, M. R., Chen, G. & Hua, B. Targeting AMPK for cancer prevention and treatment. Oncotarget 6, 7365–7378. https://doi.org/10.18632/oncotarget.3629 (2015).
Vara-Ciruelos, D., Russell, F. M. & Hardie, D. G. The strange case of AMPK and cancer: Dr Jekyll or Mr Hyde?. Open Biol. 9, 190099. https://doi.org/10.1098/rsob.190099 (2019).
Gomes, A. S., Ramos, H., Soares, J. & Saraiva, L. p53 and glucose metabolism: An orchestra to be directed in cancer therapy. Pharmacol. Res. 131, 75–86. https://doi.org/10.1016/j.phrs.2018.03.015 (2018).
Lacroix, M., Riscal, R., Arena, G., Linares, L. K. & Le Cam, L. Metabolic functions of the tumor suppressor p53: Implications in normal physiology, metabolic disorders, and cancer. Mol. Metab. 33, 2–22. https://doi.org/10.1016/j.molmet.2019.10.002 (2020).
Lee, C. W. et al. AMPK promotes p53 acetylation via phosphorylation and inactivation of SIRT1 in liver cancer cells. Cancer Res. 72, 4394–4404. https://doi.org/10.1158/0008-5472.CAN-12-0429 (2012).
Cheng, J. et al. AMP-activated protein kinase suppresses the in vitro and in vivo proliferation of hepatocellular carcinoma. PLoS One. 9, e93256. https://doi.org/10.1371/journal.pone.0093256 (2014).
Ferretti, A. C. et al. AMPK and PKA interaction in the regulation of survival of liver cancer cells subjected to glucose starvation. Oncotarget 7, 17815–17828. https://doi.org/10.18632/oncotarget.7404 (2016).
Ferretti, A. C. et al. Metformin and glucose starvation decrease the migratory ability of hepatocellular carcinoma cells: Targeting AMPK activation to control migration. Sci. Rep. 9, 1–14. https://doi.org/10.1038/s41598-019-39556-w (2019).
Dörsam, B. & Fahrer, J. The disulfide compound α-lipoic acid and its derivatives: A novel class of anticancer agents targeting mitochondria. Cancer Lett. 371, 12–9. https://doi.org/10.1016/j.canlet.2015.11.019 (2016).
Farhat, D. & Lincet, H. Lipoic acid a multi-level molecular inhibitor of tumorigenesis. Biochim. Biophys. Acta Rev. Cancer 1873, 188317. https://doi.org/10.1016/j.bbcan.2019.188317 (2020).
Park, S., Choi, S. K., Choi, Y. & Moon, H. S. AMPK/p53 axis Is essential for α-lipoic acid-regulated metastasis in human and mouse colon cancer cells. J. Investig. Med. 63, 882–885. https://doi.org/10.1097/JIM.0000000000000233 (2015).
Simbula, G. et al. Increased ROS generation and p53 activation in alpha-lipoic acid-induced apoptosis of hepatoma cells. Apoptosis 12, 113–123. https://doi.org/10.1007/s10495-006-0487-9 (2007).
Park, K. G. et al. Alpha-lipoic acid decreases hepatic lipogenesis through adenosine monophosphate-activated protein kinase (AMPK)-dependent and AMPK-independent pathways. Hepatology 48, 1477–1486. https://doi.org/10.1002/hep.22496 (2008).
Yang, Y. et al. Alpha-lipoic acid improves high-fat diet-induced hepatic steatosis by modulating the transcription factors SREBP-1, FoxO1 and Nrf2 via the SIRT1/LKB1/AMPK pathway. J. Nutr. Biochem. 25, 1207–1217. https://doi.org/10.1016/j.jnutbio.2014.06.001 (2014).
Giannelli, G., Koudelkova, P., Dituri, F. & Mikulits, W. Role of epithelial to mesenchymal transition in hepatocellular carcinoma. J. Hepatol. 65, 798–808. https://doi.org/10.1016/j.jhep.2016.05.007 (2016).
Shih, J.-Y. & Yang, P.-C. The EMT regulator slug and lung carcinogenesis. Carcinogenesis 32, 1299–1304. https://doi.org/10.1093/carcin/bgr110 (2011).
Kim, J. et al. Cooperative actions of p21 WAF 1 and p53 induce Slug protein degradation and suppress cell invasion. EMBO Rep. 15, 1062–1068. https://doi.org/10.15252/embr.201438587 (2014).
Etichetti, C. B. et al. Isoprenylcysteine carboxy methyltransferase (ICMT) is associated with tumor aggressiveness and its expression is controlled by the p53 tumor suppressor. J. Biol. Chem. 294, 5060–5073. https://doi.org/10.1074/jbc.RA118.006037 (2019).
Tonucci, F. M. et al. Centrosomal AKAP350 and CIP4 act in concert to define the polarized localization of the centrosome and Golgi in migratory cells. J. Cell Sci. 128, 3277–3289. https://doi.org/10.1242/jcs.170878 (2015).
Laemmli, U. K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680–685. https://doi.org/10.1038/227680a0 (1970).
Chandrashekar, D. S. et al. UALCAN: A portal for facilitating tumor subgroup gene expression and survival analyses. Neoplasia 19, 649–658. https://doi.org/10.1016/j.neo.2017.05.002 (2017).
Wang, Z. et al. Critical roles of p53 in epithelial-mesenchymal transition and metastasis of hepatocellular carcinoma cells. PLoS ONE 8, e72846. https://doi.org/10.1371/journal.pone.0072846 (2013).
Powell, E., Piwnica-Worms, D. & Piwnica-Worms, H. Contribution of p53 to metastasis. Cancer Discov. 4, 405–414. https://doi.org/10.1158/2159-8290.CD-13-0136 (2014).
Pavlova, N. N. & Thompson, C. B. The emerging hallmarks of cancer metabolism. Cell Metab. 23, 27–47. https://doi.org/10.1016/j.cmet.2015.12.006 (2016).
Leclerc, D. et al. Oncogenic role of PDK4 in human colon cancer cells. Br. J. Cancer 116, 930–936. https://doi.org/10.1038/bjc.2017.38 (2017).
Pavlova, N. N., Zhu, J. & Thompson, C. B. The hallmarks of cancer metabolism: Still emerging. Cell Metab. 34, 355–377. https://doi.org/10.1016/j.cmet.2022.01.007 (2022).
Wu, J. et al. Loss of PDK4 switches the hepatic NF-κB/TNF pathway from pro-survival to pro-apoptosis. Hepatology 68, 1111–1124. https://doi.org/10.1002/hep.29902 (2018).
Anwar, S., Shamsi, A., Mohammad, T., Islam, A. & Hassan, M. I. Targeting pyruvate dehydrogenase kinase signaling in the development of effective cancer therapy. Biochim. Biophys. Acta Rev. Cancer 1876, 188568. https://doi.org/10.1016/j.bbcan.2021.188568 (2021).
Lee, J. I. et al. HepG2/C3A cells respond to cysteine deprivation by induction of the amino acid deprivation/integrated stress response pathway. Physiol. Genomics 33, 218–229. https://doi.org/10.1152/physiolgenomics.00263.2007 (2008).
Iyer, V. V., Yang, H., Ierapetritou, M. G. & Roth, C. M. Effects of glucose and insulin on HepG2-C3A cell metabolism. Biotechnol. Bioeng. 107, 347–356. https://doi.org/10.1002/bit.22799 (2010).
Zhu, J., Wu, Y., Tang, Q., Leng, Y. & Cai, W. The effects of choline on hepatic lipid metabolism, mitochondrial function and antioxidative status in human hepatic C3A cells exposed to excessive energy substrates. Nutrients 6, 2552–2571. https://doi.org/10.3390/nu6072552 (2014).
Hnit, S. et al. p27Kip1 signaling: Transcriptional and post-translational regulation. Int. J. Biochem. Cell Biol. 68, 9–14. https://doi.org/10.1016/j.biocel.2015.08.005 (2015).
Zhuang, Y. & Miskimins, W. Cell cycle arrest in Metformin treated breast cancer cells involves activation of AMPK, downregulation of cyclin D1, and requires p27 Kip1 or p21 Cip1. J. Mol. Signal. 3, 1–11. https://doi.org/10.1186/1750-2187-3-1 (2008).
Short, J. D. et al. AMPK-mediated phosphorylation of murine p27 at T197 promotes binding of 14–3-3 proteins and increases p27 stability. Mol. Carcinog. 49, 429–439. https://doi.org/10.1002/mc.20613 (2010).
Krstic, J. et al. Fasting improves therapeutic response in hepatocellular carcinoma through p53-dependent metabolic synergism. Sci. Adv. 8, eabh2635. https://doi.org/10.1126/sciadv.abh2635 (2022).
Galhuber, M. et al. Complementary omics strategies to dissect p53 signaling networks under nutrient stress. Cell. Mol. Life Sci. 79, 326. https://doi.org/10.1007/s00018-022-04345-8 (2022).
Cui, D. et al. The cross talk between p53 and mTOR pathways in response to physiological and genotoxic stresses. Front. Cell. Dev. Biol. 9, 775507. https://doi.org/10.3389/fcell.2021.775507 (2021).
Jung, S. H. et al. mTOR kinase leads to PTEN-loss-induced cellular senescence by phosphorylating p53. Oncogene 38, 1639–1650. https://doi.org/10.1038/s41388-018-0521-8 (2019).
Chen, J. S. et al. Involvement of PI3K/PTEN/AKT/mTOR pathway in invasion and metastasis in hepatocellular carcinoma: Association with MMP-9. Hepatol. Res. 39, 177–186. https://doi.org/10.1111/j.1872-034X.2008.00449.x (2009).
Honda, K. et al. p53 mutation is a poor prognostic indicator for survival in patients with hepatocellular carcinoma undergoing surgical tumour ablation. Br. J. Cancer 77, 776–782. https://doi.org/10.1038/bjc.1998.126 (1998).
Craig, A. J. et al. Tumour evolution in hepatocellular carcinoma. Nat. Rev. Gastroenterol. Hepatol. 17, 139–152. https://doi.org/10.1038/s41575-019-0229-4 (2020).
Tortelli, T. C. Jr. et al. Metformin-induced chemosensitization to cisplatin depends on P53 status and is inhibited by Jarid1b overexpression in non-small cell lung cancer cells. Aging (Albany NY) 13, 21914 (2021).
Song, S. et al. Reticulon 3-mediated Chk2/p53 activation suppresses hepatocellular carcinogenesis and is blocked by hepatitis B virus. Gut 70, 2159–2171. https://doi.org/10.1136/gutjnl-2020-321386 (2021).
Yao, Jy. et al. Novel CDK9 inhibitor oroxylin A promotes wild-type P53 stability and prevents hepatocellular carcinoma progression by disrupting both MDM2 and SIRT1 signaling. Acta Pharmacol. Sin. https://doi.org/10.1038/s41401-021-00708-2 (2021).
Lim, S. O., Kim, H. & Jung, G. p53 inhibits tumor cell invasion via the degradation of snail protein in hepatocellular carcinoma. FEBS Lett. 584, 2231–2236. https://doi.org/10.1016/j.febslet.2010.04.006 (2010).
Kim, T. et al. p53 regulates epithelial–mesenchymal transition through microRNAs targeting ZEB1 and ZEB2. J. Exp. Med. 208, 875–883. https://doi.org/10.1084/jem.20110235 (2011).
Donehower, L. A. et al. Integrated analysis of TP53 gene and pathway alterations in the cancer genome atlas. Cell Rep. 28, 1370–1384. https://doi.org/10.1016/j.celrep.2019.07.001) (2019).
Matsumura, T., Makino, R. & Mitamura, K. Frequent down-regulation of E-cadherin by genetic and epigenetic changes in the malignant progression of hepatocellular carcinomas. Clin. Cancer Res. 7, 594–599 (2001).
Fransvea, E., Angelotti, U., Antonaci, S. & Giannelli, G. Blocking transforming growth factor–beta up-regulates E-cadherin and reduces migration and invasion of hepatocellular carcinoma cells. Hepatology 47, 1557–1566. https://doi.org/10.1002/hep.22201 (2008).
Yang, B. et al. Dynamic network biomarker indicates pulmonary metastasis at the tipping point of hepatocellular carcinoma. Nat. Commun. 9, 678. https://doi.org/10.1038/s41467-018-03024-2 (2018).
Yang, M. H. et al. Comprehensive analysis of the independent effect of twist and snail in promoting metastasis of hepatocellular carcinoma. Hepatology 50, 1464–1474. https://doi.org/10.1002/hep.23221 (2009).
Yang-Hartwich, Y. et al. p53–Pirh2 complex promotes Twist1 degradation and inhibits EMT. Mol. Can. Res. 17, 153–164. https://doi.org/10.1158/1541-7786.MCR-18-0238 (2019).
Semenov, O., Daks, A., Fedorova, O., Shuvalov, O. & Barlev, N. A. Opposing roles of wild-type and mutant p53 in the process of epithelial to mesenchymal transition. Front. Mol. Biosci. 9, 928399 (2022).
Cowden Dahl, K. D. et al. Matrix metalloproteinase 9 is a mediator of epidermal growth factor–dependent E-cadherin loss in ovarian carcinoma cells. Cancer Res. 68, 4606–4613. https://doi.org/10.1158/0008-5472.CAN-07-5046 (2008).
Zuo, J. H. et al. Activation of EGFR promotes squamous carcinoma SCC10A cell migration and invasion via inducing EMT-like phenotype change and MMP-9-mediated degradation of E-cadherin. J. Cell. Biochem. 112, 2508–2517. https://doi.org/10.1002/jcb.23175 (2011).
Wang, J. R. et al. Orphan nuclear receptor Nur77 promotes colorectal cancer invasion and metastasis by regulating MMP-9 and E-cadherin. Carcinogenesis 35, 2474–2484. https://doi.org/10.1093/carcin/bgu157 (2014).
Acknowledgements
This work was supported by the Argentine Govern through CONICET: PIP 11220120100287CO and PIP 11220150100293CO, and Agencia I + D + i: PICT 2021-427. Authors specially thank Dr. M. Ojeda (CONICET) for her technical assistance in cytometric analyses.
Funding
CONICET, PIP 11220120100287CO, PIP 11220120100287CO, PIP 11220120100287CO, PIP 11220120100287CO, PIP 11220120100287CO, PIP 11220120100287CO, PIP 11220120100287CO, Agencia Nacional de Promoción Científica y Tecnológica, PICT 2021-427, PICT 2021-427.
Author information
Authors and Affiliations
Contributions
FH and ACF equally contributed to the study. Conceived and designed the experiments: FH, ACF, CF. Performed the experiments or data analysis: FH, ACF, CBE, EB, APP, TRM and JB. Supervised the experiments: MCL, JEG, and CF. Analyzed the results: FH, ACF, CF. Wrote the paper: CF. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Hidalgo, F., Ferretti, A.C., Etichetti, C.B. et al. Alpha lipoic acid diminishes migration and invasion in hepatocellular carcinoma cells through an AMPK-p53 axis. Sci Rep 14, 21275 (2024). https://doi.org/10.1038/s41598-024-72309-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-72309-y
- Springer Nature Limited