
Abstract
Autoimmune related kidney diseases (ARKDs), including minimal change nephropathy (MCN), membranous nephropathy (MN), IgA nephropathy (IgAN), and lupus nephritis (LN), significantly affect renal function. These diseases are characterized by the formation of local immune complexes and the subsequent activation of the complement system, leading to kidney damage and proteinuria. Despite the known patterns of glomerular injury, the specific molecular mechanisms that contribute to renal tubular damage across ARKDs remain underexplored. Laser capture microdissection and liquid chromatography–tandem mass spectrometry (LC–MS/MS) were used to conduct a comparative proteomic analysis of renal tubular tissues from formalin-fixed paraffin-embedded samples. The cohort comprised of 10 normal controls (NC), 5 MCN, 4 MN, 17 IgAN, and 21 LN patients. Clinical parameters and histopathological assessments were integrated with proteomic findings to comprehensively investigate underlying pathogenic processes. Clinical evaluation indicated significant glomerular damage, as reflected by elevated urinary protein levels and reduced plasma albumin levels in patients with ARKD. Histological analyses confirmed varying degrees of tubular damage and deposition of immune complexes. Proteomic analyses identified significant changes in protein expression, particularly in complement components (C3, C4A, C4B, C8G, CFB, and SERPINA1) and mitochondrial proteins (ATP5F1E and ATP5PD), highlighting the common alterations in the complement system and mitochondrial proteins across ARKDs. These alterations suggest a novel complement–mitochondrial–epithelial–mesenchymal transition (EMT) pathway axis that contributes to tubular damage in ARKDs. Notably, significant alterations in CFB in tubular ARKD patients were revealed, implicating it as a therapeutic target. This study underscores the importance of complement activation and mitochondrial dysfunction in the pathogenesis of ARKDs, and proposes CFB as a potential therapeutic target to inhibit complement activation and mitigate tubular damage. Future research should validate the complement-mitochondrial-EMT pathway axis and explore the effects and mechanisms of CFB inhibitors in alleviating ARKD progression.
Similar content being viewed by others

Introduction
Autoimmune related kidney diseases (ARKDs), including minimal change nephropathy (MCN), membranous nephropathy (MN), immunoglobulin A nephropathy (IgAN), and lupus nephritis (LN), are conditions in which the immune system mistakenly attacks healthy kidney tissue1. Autoimmunity can lead to inflammation, damage, and impaired kidney function. ARKDs can affect various parts of the kidney, including the glomeruli, tubules, and interstitial tissue, resulting in a range of symptoms that often progress to chronic kidney disease (CKD) or kidney failure2.
A key player in the pathogenesis of ARKDs is the complement system, a component of the innate immune response3. Dysregulation of the complement system contributes to these diseases by recruiting inflammatory cells to the site of immune complex deposition, exacerbating glomerular inflammation and damage4. However, although the impact of complement activation on glomerular injury has been well studied, its effects on renal tubular injury remain poorly explored.
Renal tubules, particularly proximal tubules, are essential for reabsorption and secretion and are rich in mitochondria, which provide the necessary energy for these processes5,6. The critical importance of mitochondrial function in maintaining normal tubular function is underscored by the abundance of mitochondria in renal tubules7. Mitochondria rely on the respiratory chain, which consists of four complexes (I, II, III, and IV) and adenosine triphosphate (ATP) synthase (complex V) to produce ATP8. Oxidative phosphorylation is the most important process during ATP generation9, and is accompanied by reactive oxygen species (ROS) generation9. ROS play vital roles in cell signaling and defense against pathogens, however, their excessive production leads to oxidative stress8,10,11. This disruption can cause mitochondrial DNA damage, impaired oxidative phosphorylation, reduced ATP production, and increased ROS generation in a damaging feedback loop12. Lack of ATP supply results in tubular epithelial cell injury13. Additionally, ROS are implicated in promoting the epithelial-to-mesenchymal transition (EMT)14, which is a common pathological feature of renal tubular damage in end-stage renal disease15.
A recent study reported that complement activation induces podocyte pyroptosis in MN by regulating mitochondrial dysfunction16. However, the relationship between the complement system and mitochondrial damage in regulating renal tubular injury and EMT in ARKDs remains unclear. This study aimed to use laser capture microdissection (LCM) of tubular tissues from ARKDs for proteomic analysis to elucidate the potential common mechanisms of tubular damage in patients with MCN, MN, IgAN, and LN.
Methods
Samples collection
The participants in this retrospective study were recruited from the Nine-hundred Twenty-Fourth Hospital and First Affiliated Hospital of Jinan University between 2013 and 2023. Formalin-fixed paraffin-embedded (FFPE) samples from renal biopsy tissues and clinical data were collected from each participant. Participants were selected based on the renal histology results. The study included six patients with MCN, nine patients with MN, 18 patients with IgAN, and 21 patients with LN. Additionally, zero-time kidney biopsy tissues from 12 donors for kidney transplantation were used as normal controls (NC). Informed consent was obtained from all participants and the study was approved by the Clinical Research Ethics Committees of the Nine-hundred Twenty-Fourth Hospital and the First Affiliated Hospital of Jinan University.
LCM glomerular and tubular
FFPE samples were cut into slides and stained with periodic acid-Schiff (PAS). Frozen tissue sections from the same patient were subjected to immunofluorescent staining. After confirming the type of renal pathology, FFPE samples from the same person were used to isolate glomerular and tubular tissues via microdissection. Sections of FFPE samples were cut at a thickness of 10 µm, deparaffinized in chloroform, and collected on LCM-compatible slides. Microdissection was performed using an LMD 7000 microscope (Leica Microsystems, Wetzlar, Germany) with a UV laser (power set to 38, aperture of 14, and speed of 4). Glomerular and tubular tissues were manually selected and placed in cap tubes under gravity. Each tube from one case, containing 5–8 target glomerular or renal tubules, was stored at − 80 °C for subsequent liquid chromatography–mass spectrometry (LC–MS) analysis.
Protein extraction and digestion
Each sample tube from the last step was centrifuged at 20,000×g for 10 min. The samples were reduced by adding 10 mM dithiothreitol to 50 mM ammonium bicarbonate and heating at 95 °C for 30 min. Subsequently, the samples were alkylated with 55 mM iodoacetamide and left to stand in the dark at room temperature for 30 min. The samples were subsequently sonicated for 20 min in a water-bath sonicator. For proteolysis, each sample was incubated with 2.5 µg of trypsin at 37 °C for 16 h. The resulting digested peptides were desalted using a Strata X column by washing with 1 mL of 0.1% formic acid three times and finally dried under vacuum.
Data-dependent acquisition (DDA) and data-independent acquisition (DIA) analysis by LC–MS/MS
Dried peptide samples were reconstituted with mobile phase A (800 µL of 75% acetonitrile and 1 mL of 0.1% formic acid) and centrifuged at 20,000×g for 10 min. The supernatant was used for injection. Separation was performed using a Thermo UltiMate 3000 UHPLC system (Thermo Fisher Scientific, Waltham, Massachusetts, USA). Samples were enriched, desalted, and processed using a self-packed C18 column. Further separation was performed at a flow rate of 500 nL/min.
For both DDA and DIA analyses, peptides were ionized by nanoESI and injected into a Fusion Lumos tandem mass spectrometer (Thermo Fisher Scientific, Waltham, Massachusetts, USA) in DDA and DIA detection modes, respectively, according to previously described parameters.
Data processing
DDA data were analyzed using the Andromeda search engine MaxQuant (version 2.5.2.0, https://www.maxquant.org). Oxidation of methionine and protein N-terminal acetylation were used as modifications. The minimum peptide length was seven amino acids, and at least one unique peptide was required for protein identification. The false discovery rate (FDR) threshold for proteins and peptides was set to 0.01. Protein and peptide quantitative values were obtained using MSstats, with log2-transformed and quantile-normalized intensity data. Differentially expressed proteins (DEPs) were identified based on a |log twofold change (Log2FC)| ≥ 1.5 and a P-value < 0.05, with comparisons made using two-sided Welch’s t-tests.
Bioinformatic analysis
Functional analysis of DEPs was performed using Gene Ontology (GO) and the Kyoto Encyclopedia of Genes and Genomes (KEGG)17. Venn diagrams and flower plots were generated using Xiantao Xueshu (https://www.xiantaozi.com/), whereas word clouds, Pearson correlation analyses, and heatmaps were generated using Gene Denovo (https://www.omicshare.com/tools/Home/Soft/wordcloud). Gene set variation analysis (GSVA) was conducted using the GSVA R package (version 1.30.0, https://bioconductor.org/packages/release/bioc/html/GSVA.html), and gene set enrichment analysis (GSEA) was performed using the GSEA online tool (http://software.broadinstitute.org/gsea/index.jsp). Bar and scatter plots were created using GraphPad Prism (version 9.5.1, GraphPad Software Inc., San Diego, California, USA). Protein–protein interaction (PPI) networks and minimal common oncology data element (MCODE) components were identified using the Metascape online database (https://metascape.org).
Immunofluorescence (IF) of kidney tissue
Frozen kidney tissue sections were blocked with 5% BSA in PBS for 1 h at room temperature and then incubated overnight at 4 °C with primary antibodies for IgA and IgG (rabbit anti-CD79a, 1:100, and rabbit anti-IgG1(Fc), 1:100, both from ABclonal Technology, Wuhan, China). After washing with PBS (3 × 5 min), sections were incubated for 1 h at room temperature in the dark with a fluorescently labeled secondary antibody (FITC Goat Anti-Rabbit IgG (H + L), 1:100, ABclonal Technology, Wuhan, China). After further PBS washes (3 × 5 min), the tissue sections were mounted on glass slides with mounting medium containing an anti-fade agent, coverslips were sealed, and the sections were observed using an Olympus BX63 fluorescence microscope (Olympus Corporation, Tokyo, Japan).
Immunohistochemistry (IHC) of kidney tissue
Paraffin-embedded sections (2 µm) were deparaffinized and rehydrated using standard protocols before blocking with normal goat serum diluted 1:5 in PBS (pH 7.4) for 20 min. The sections were then incubated overnight at 4 °C with primary antibodies against Complement factor B (CFB) and SERPINA1 (rabbit anti-CFB, 1:100, and rabbit anti-SERPINA1, 1:100, both from ABclonal Technology, Wuhan, China). After further washing with PBS (3 × 5 min), biotinylated goat anti-rabbit secondary antibody (1:100, Servicebio, Wuhan, China) was applied for 30 min, followed by processing with DAB substrate, according to the manufacturer’s instructions. The sections were counterstained with hematoxylin, dehydrated, and mounted for observation under a Leica DM2000 LED upright microscope (Leica Microsystems, Wetzlar, Germany).
Electron microscopy (EM) of kidney tissue
Electron microscopy was performed according to standard protocols. Kidney tissue sections were cut into slides (60–90 nm) and stained with lead citrate (Sigma-Aldrich, Burlington, Massachusetts, United States). Observations were performed using an electron microscope (EMSIS, Phurona, Münster, Germany).
Statistical analysis
Statistical comparisons of the differences in the relative expression of DEPs and the mean density of SERPINA1 and CFB between the four ARKDs and control groups were performed using one-way ANOVA (version 9.5.1, GraphPad Software Inc., San Diego, California, USA). Comparisons of the differences in the relative expression of DEPs and mean density of SERPINA1 and CFB between tubular and glomerular tissues within each group were performed using two-way ANOVA. Statistical significance was set at P < 0.05.
Ethics declarations
The study was registered on 03, August 2023, and the study protocol was reviewed and approved by the Clinical Research Ethics Committees of both the Nine-hundred Twenty-Fourth Hospital and the First Affiliated Hospital of Jinan University (approval number No. [2023] 70, and KY-2020-034. This study was conducted in accordance with the principles of the Declaration of Helsinki. Written informed consent was obtained from the living donors to participate in the study.
Results
Clinical parameters of subjects included in this study
Kidney biopsy samples were collected from 12 donors for kidney transplantation: 6 patients with MCN, 9 patients with MN, 18 patients with IgAN, and 21 patients with LN, after screening based on renal histological results (Supplementary Fig. 1). The clinical and laboratory parameters of the study participants are summarized in Table 1. Blood urea nitrogen (BUN) and serum creatinine (Scr) levels did not differ significantly between patients with MCN, MN, IgAN, LN, and controls. However, plasma cystatin C (Cys-C) levels are elevated in LN patients. 24-h urinary protein levels were significantly increased in patients with MN, IgAN, and LN compared to controls, indicating damage to the glomeruli. Correspondingly, the plasma albumin levels decreased in these groups.
Histological examination of kidneys in ARKD patients
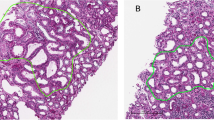
A flowchart outlining the proteomic process of laser-captured renal tissues and the subsequent bioinformatics analysis is shown in Fig. 1A. Histological examination using PAS staining, electron microscopy, and immunofluorescence is shown in Fig. 1B and Supplementary Fig. 2A–C. PAS staining showed minor tubular damage in the MCN, moderate damage in the MN, and severe damage in the IgAN and LN (Fig. 1B). Tubular injuries in patients with MCNs include wall sclerosis and lumen narrowing. Patients with MN exhibit more severe sclerosis and stenosis, with partial shedding of tubular epithelial cells. The tubular lumen was damaged in patients with IgAN, and protein casts were visible. Patients with LN displayed stiff, narrow tubule lumens with partially destroyed structures.

Proteomic analysis of laser-captured tubular tissue of different immune-related kidney disease. (A) Flowchart depicting the proteomic analysis of renal tubules isolated by laser capture microdissection. (B) PAS staining illustrates the renal tubules of NC, MCN, MN, IgAN, and LN, respectively. (C) Bar plots showing DEPs in renal tubules across various immune-related kidney diseases identified by proteomics, with comparison to NC for up and down regulation. (D) Heatmap showing the top 100 identified DEPs from each immune-related kidney disease comparing with NC.
Electron microscopy (EM) revealed disruption of the glomerular filtration barrier (GFB) in all ARKD patients, including podocyte foot process fusion, thickened basement membranes, and endothelial cell damage (Supplementary Fig. 2B). Immune complex deposition was observed in the mesangial areas of the patients with MN, IgAN, and LN (Supplementary Fig. 2B,C). Immunofluorescence revealed IgG deposition in the glomeruli of MN and LN patients, and IgA deposition in IgAN patients (Supplementary Fig. 2C).
DEPs identified in renal tissues of patients with ARKDs
FFPE samples stained with PAS were used to isolate glomerular and renal tubular tissues via LCM (Supplementary Fig. 2D). Proteins were extracted and analyzed using LC–MS/MS (Fig. 1A). Quality control filtered out samples with fewer than 1000 peptides (Supplementary Table 1), resulting in a total of 46 samples from glomeruli for analysis: 8 controls, 3 with MCN, 5 with MN, 11 with IgAN, and 19 with LN, and 57 tubular samples for analysis: 10 controls, 5 with MCN, 4 with MN, 17 with IgAN, and 21 with LN (Supplementary Fig. 1).
Proteomic analysis identified DEPs in the tubular and glomerular tissues (Fig. 1C). MCN patients had 303 tubular DEPs (237 upregulated and, 66 downregulated) compared to controls. MN patients had 662 DEPs (634 upregulated and 28 downregulated), IgAN patients had 365 DEPs (179 upregulated and 186 downregulated), and LN patients had 424 DEPs (313 upregulated, 111 downregulated). Meanwhile, MCN patients had 320 DEPs in the glomerulus (265 upregulated and 55 downregulated) (Supplementary Fig. 3A), MN patients had 1,130 DEPs (775 upregulated and 355 downregulated), IgAN patients had 2075 DEPs (1950 upregulated and 125 downregulated), and LN patients had 2285 DEPs (1525 upregulated and 760 downregulated).
A heatmap of the top 100 DEPs from tubular tissues in each group compared to the controls highlighted significant changes in proteins related to the complement system and mitochondria, including C3 and C1QC, in patients with ARKD (Fig. 1D). Upregulation of immunoglobulin components (IGHV1-8, IGKV3-20, and IGHG3) in LN indicates immune complex deposition and activation of the complement system. These findings, along with the significantly altered circulating levels of C3, C4, IgA, and IgM (Table 1), underscore the role of a dysregulated immune system, particularly the complement system and immunoglobulins, in ARKD pathogenesis. Additionally, the dysregulated expression of mitochondrial metabolic proteins (MECR, MT-CO2, ACSM2B, and GLS) and ROS-protective proteins (NQO1, GLS, GSTA2, and TXNRD2) suggests that mitochondrial dysfunction contributes to tubular damage.
Potential common molecular mechanisms of renal tubular damage in ARKDs by functional enrichment analysis of DEPs
GO and KEGG17 enrichment analyses of DEPs from renal tubular tissue in MCN, MN, IgAN, and LN compared to controls revealed overlapping biological process (BP), cellular component (CC), molecular function (MF), and KEGG signaling pathways. Venn diagrams showed 37 common BP, 32 CC, 20 MF, and 11 common KEGG pathways (Fig. 2A–D), indicating shared biological functions and signaling pathways among ARKD patients.

Function analysis of DEPs in tubular tissue of patients with ARKDs compared to NC. (A–D) Intersection analysis of enriched terms based on BP (A), CC (B), MF (C), and KEGG pathways (D) from enrichment analyses on DEPs in patients with ARKDs compared to NC. (E) Word cloud diagram depicting DEPs in each ARKD subjected to GO and KEGG analyses compared to NC. The diagram summarizes, ranks by frequency, and illustrates the top 100 proteins involved in common biological functions or signaling pathways across the four ARKDs. Notably, proteins related to mitochondrial ATP synthase F0/F1 subunits and mitochondrial respiratory chain complex I show the highest frequency. (F) Fold changes in the expression of mitochondrial function-related proteins in the four ARKDs compared to NC. *Indicates a significant difference in the log2FC of expression of each protein in each ARCKD group compared to the NC group (P < 0.05).
Word cloud diagrams emphasize DEPs related to mitochondrial function, including subunits of 1,4-Dihydronicotinamide adenine dinucleotide (NADH): ubiquinone oxidoreductase (complex I) and ATP synthase F0/F1 (Fig. 2E). These mitochondrial proteins were predominantly downregulated, as shown in the heatmap (Fig. 2F), particularly ATP synthase subunit alpha (ATP5F1E) and delta (ATP5PD).
Enrichment analysis of DEPs in glomerular tissues from patients with ARKDs highlighted the top-ranked KEGG pathways related to immune response and metabolism, such as “antigen processing and presentation”, “complement and coagulation cascades”, and various other metabolic pathways (Supplementary Fig. 3B). This suggests a crucial role of mitochondrial and complement system dysfunction in both tubular and glomerular tissues.
Given the importance of mitochondrial respiratory chain dysfunction in the processes of “oxidative phosphorylation” and the “ROS pathway” and its role in causing tubular damage in patients with ARKD, we further compared the activities of these pathways in tubular and glomerular tissues using GSVA analysis. The “ROS pathway” was not significantly enriched in glomerular tissues of ARKD patients. The activity of the “oxidative phosphorylation” pathway was significantly decreased in the tubular tissues of patients with IgAN and LN compared to that in NC (Supplementary Fig. 3C). Conversely, the activity of this pathway did not significantly differ in the glomerular tissues of patients with ARKDs compared to that in NC. In kidney tissues, the activity of this pathway was significantly different only in glomerular tissues compared to tubular tissues in the NC group.
We identified 81 core DEPs enriched in the “oxidative phosphorylation” pathway by GSVA analysis, expressed in tubular and glomerular tissues in patients with MCN, MN, IgAN, and LN compared to NC (Supplementary Fig. 3D). The heatmap revealed contrasting expression trends of these DEPs in tubular and glomerular tissues in patients with MCN, IgAN, and LN, whereas expression trends were similar in both tissues in patients with MN (Supplementary Fig. 3E), indicating that although there were shared molecules in tubular and glomerular tissues across ARKDs, the specific mechanisms underlying tubular and glomerular damage were different.
Pathway analysis and DEPs in ARKD: complement and coagulation cascades and mitochondrial dysfunction
GSEA analysis identified “complement and coagulation cascades” as the only significantly upregulated pathway across all ARKD tubular tissues, with normalized enrichment scores (NES) ranging from 2.16 to 3.76 (Fig. 3A–E). In glomerular tissues, GSEA for KEGG pathways revealed “complement and coagulation cascades” and “chemokine signaling pathway” as common pathways among patients with ARKD (Supplementary Fig. 4A). Sixteen shared DEPs were identified between the tubular and glomerular tissues of patients with ARKD (Supplementary Fig. 4B). These included central components of the complement system, such as C3; classical activation pathway components, such as C1QC, C4A, and C4B; and membrane attack complex (MAC) components, including C6, C8G, C8B, and C9. These complement components were found to be more profoundly activated in tubular tissues than in the glomerular tissues in all four diseases. In addition, SERPINA1 and CFB were upregulated in the tubular tissues of all ARKDs but were differentially expressed in the glomeruli (Supplementary Fig. 4C, Supplementary Table 2).

Comparative analysis of immune related KEGG signaling pathways in tubular tissues of patients with ARKDs through GSVA and GESA analysis. (A) Venn diagram showing the overlap of KEGG signaling pathways enriched by GSEA analysis among patients with ARKDs. (B–E) Results of GSEA analysis for complement and coagulation cascades in patients with MCN (B), MN (C), IgAN (D), and LN (E), respectively. (F) Heatmap displaying the GSVA scores of various immune-related KEGG signaling pathways. *Indicates a significant difference in the GSVA score of each signaling pathway in each ARCKD group compared with the NC group (P < 0.05).
The significant activation of the “complement and coagulation cascades” in ARKDs, especially MN and LN, was further confirmed by GSVA analysis (Fig. 3F). Additionally, the activation of other immune-related KEGG pathways, such as “B cell receptor signaling” in MCN and LN, “hematopoietic cell lineage” in IgAN, was observed.
IHC was used to validate the renal expression of SERPINA1 and CFB. The results showed that SERPINA1 was consistently upregulated in tubular tissues of ARKDs (Fig. 4A,B), consistent with the proteomic analysis results. CFB was significantly upregulated in MCN and IgAN but significantly downregulated in MN (Fig. 4A,C). Additionally, the expression of SERPINA1 and CFB was significantly higher in tubular tissues than in glomerular tissues, and their levels were not significantly altered in each group (Fig. 4A–C, Supplementary Fig. 5A). This suggests a possible in situ generation of SERPINA1 and CFB in tubular tissue.

Immunohistochemical staining and comparative semi-quantitative analysis of SERPINA1 and CFB in tubular and glomerular tissues from patients with ARKDs. (A) Immunohistochemical staining of SERPINA1 and CFB in tubular (top) and glomerular (bottom) tissue from patients with NC, MCN, MN, IgAN, and LN, respectively. (B,C) Comparative semi-quantitative analysis of immunohistochemical staining of SERPINA1 (B) and CFB (C) in tubular and glomerular tissue from patients with ARKDs. #Indicates a significant difference in the relative expression of SERPINA1 and CFB in each ARCKD group compared to the NC group, respectively (P < 0.05). *Indicates a significant difference between tubular and glomerular tissue in each group (P < 0.05).
Furthermore, SERPINA1 and CFB expressions closely correlated with various clinical parameters (Supplementary Fig. 5B). For example, CFB expression was negatively correlated with plasma Ca2+ in MCN, positively correlated with 24-h urinary protein and serum creatinine in MN, and showed various correlations in IgAN and LN, indicating that complement system activation, such as SERPINA1 and CFB, evolves the pathology in patients with ARKDs.
Pearson’s correlation analysis revealed numerous interactions between complement and mitochondrial DEPs in patients with ARKDs (Fig. 5). For instance, in patients with MCN, SERPINA1 was closely correlated with NDUFA7, NDUFA13, and ATP5F1B, whereas CFB was correlated with NDUFA2 and NDUFA12. In patients with MN, SERPINA1 correlated with NDUFS3, NDUFB10, NDUFB4, NDUFA13, NDUFA4, ATP5PO, ATP5PD, ATP5F1E, ATP5F1D, ATP5F1C, and ATP5F1A. In patients with LN, SERPINA1 was correlated with ATP5F1A and CFB was correlated with NDUFB11 and NDUFB9. In patients with IgAN, although there were interactions between SERPINA1 and CFB and complex I and ATP synthase F0/F1 proteins, the correlations were not significant. These results underscore the potential contribution of complement activation to mitochondrial respiratory chain damage in ARKDs.

Heatmap showing the relationships between mitochondrial-related proteins and proteins of the complement and coagulation cascades by Pearson correlation analysis. *Indicates a significant Pearson correlation coefficient between the expression levels of the complement and coagulation cascades related proteins and the expression levels of the mitochondrial-related proteins (P < 0.05).
Mitochondrial dysfunction and epithelial-mesenchymal transition in tubular damage of ARKD patients
Mitochondria generate ATP through oxidative phosphorylation, a critical process that is accompanied by ROS production18. Tubules with high ATP demands rely heavily on mitochondrial function to maintain the integrity and normal function of the tubular epithelial cells19. GSVA comparing selected KEGG pathways between each ARKD and control revealed a downward trend in “oxidative phosphorylation” and “ROS pathway” in patients, particularly those with IgAN and LN (Fig. 6A–J). GSEA confirmed a significant decrease in “oxidative phosphorylation” in MN, IgAN, and LN (Supplementary Fig. 6A). Conversely, “ROS pathway” activity showed an upward trend in MCN and MN but significantly decreased in IgAN and LN, indicating impaired mitochondrial function in tubular tissues of patients with ARKDs.

Enrichment and GSVA analysis of KEGG pathways in ARKD patients compared to NC. (A) Heatmap matrix displaying the selected KEGG pathways enriched by GSVA analysis of identified DEPs in patients with ARKDs compared to NC. (B–J) Bar plots showing GSVA scores of various KEGG pathways, including Apoptosis (B), Epithelial Mesenchymal Transition (C), Hypoxia (D), Notch Signaling (E), Oxidative phosphorylation (F), Reactive oxygen species Pathway (G), TGF-β signaling (H), TNF-α signaling via NFκB (I), and Wnt/β-catenin signaling (J) in NC and patients with ARKDs. *Indicates a significant difference in the GSVA score of each signaling pathway in each ARCKD group compared to the NC group (P < 0.05).
Mitochondrial damage leads to decreased ATP generation and increased oxidative stress, further exacerbating mitochondrial and tubular damage15. Plasma superoxide dismutase (SOD) levels, which protect against ROS-induced injury20, were significantly decreased in patients with MCN, MN, and LN, indicating reduced ROS inhibition (Table 1). Additionally, plasma Ca2+ levels, which are crucial for ATP production9, were significantly reduced in patients with MCN, MN, and LN compared with controls (Table 1).
EMT is the primary mechanism underlying tubulointerstitial fibrosis in CKD21. GSVA and GSEA analyses showed significantly upregulated EMT pathway activity in patients with IgAN and LN who exhibited the most severe renal tubular damage (Fig. 6A,C, Supplementary Fig. 6A). However, pathways involved in EMT, such as transforming growth factor-β (TGF-β) signaling and tumor necrosis factor (TNF)-α signaling via nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), Wnt β-catenin signaling (Fig. 6A,H–J), mammalian target of rapamycin complex 1 (mTORC1), and phosphoinositide 3-kinases (PI3K)/protein kinase B (AKT)/mammalian target of rapamycin (mTOR) signaling (Fig. 6A–J, Supplementary Fig. 6A), did not change significantly in patients with ARKD, suggesting that other mechanisms may drive EMT in ARKDs.
Mitochondrial dysfunction has been identified as a critical driver of EMT and is increasingly being considered as a therapeutic target to combat tubulointerstitial fibrosis19. In ARKDs, significant disruptions in “oxidative phosphorylation” and the “ROS pathway” were highlighted by our GSVA and GSEA analyses (Fig. 6F,G, Supplementary Fig. 6A). Glutathione peroxidase 4 (GPX4), identified in our study and another, is a key regulator of “oxidative phosphorylation” and the “ROS pathway” in ARKDs (Supplementary Fig. 6B)22. GPX4 plays an essential role in maintaining cellular redox homeostasis and antioxidant defense, and alterations in its expression have been linked to the EMT process either directly or indirectly22,23. Our Pearson correlation analysis demonstrated strong associations between GPX4 and mitochondrial respiratory chain proteins in ARKDs (Supplementary Fig. 6C–L), indicating that GPX4 may serve as a critical intermediary connecting mitochondrial dysfunction and EMT processes.
PPI analysis elucidated the relationships among the mitochondrial respiratory chain, complement-coagulation cascades, and EMT pathways, identifying significant interactions and nine MCODE modules among 103 proteins (Fig. 7). These results indicate that mitochondrial impairment may be linked to in situ complement activation, resulting in EMT in the tubular tissues of patients with ARKD (Fig. 8).

Protein–protein interaction network analysis of mitochondria-related proteins, complement and coagulation cascade-related genes, and proteins associated with epithelial-mesenchymal transition.

Schematic diagram of the hypothesis of this study. In this study, proteins were extracted from renal tubule tissues via microdissection of FFPE samples from kidney biopsy tissues of patients with ARKDs—including MCN, MN, IgAN, and LN, followed by proteomic analysis. A series of bioinformatics analyses were conducted to investigate the common molecular mechanisms between patients with ARKDs and normal kidney tissues. The findings revealed abnormal activation of the in situ coagulation-complement cascade in the renal tubules of ARKDs patients. This abnormal activation may cause damage to the mitochondrial respiratory chain, leading to reduced ATP production and increased generation of ROS. Additionally, the reduction in antioxidant products such as SOD induces oxidative stress, further exacerbating mitochondrial dysfunction. Mitochondrial damage in the renal tubules may be one of the mechanisms causing tubular injury and leading to EMT. There may be multiple interactions between the complement-coagulation cascades, mitochondrial dysfunction (oxidative phosphorylation and ROS pathway), and tubular EMT, which collectively contribute to the exacerbation of tubulointerstitial injury and fibrosis in ARKDs patients.
Discussion
ARKDs, including MCN, MN, IgAN, and LN, are often linked to the formation of local immune complexes1,2,3,4,24 that trigger the complement system of the innate immune system, leading to MAC formation. This process results in kidney and podocyte damage, which ultimately manifests as proteinuria24. Despite common immunological mechanisms in patients with ARKDs, different pathological backgrounds should be considered.
MCN primarily involves podocyte injury with minor or absent immune complex deposition in glomeruli25. A recent study reported the presence of autoantibodies in patients with MCN26, indicating that MCN can be classified as an ARKD. MN is characterized by subepithelial immune complex deposits, resulting in thickening of the glomerular basement membrane27. IgAN is marked by the deposition of IgA immunoglobulins in the mesangium28,29. LN is a result of systemic lupus erythematosus and features complex immune mechanisms and widespread immune complex deposition29. These differences in etiology indicate that the pathogenic processes driving each disease may have similarities and variations.
Previous studies, including ours, have highlighted the dysregulated activation of the complement and coagulation cascades in the glomeruli of affected patients1,2,3,4,24,30. Overactivation of complement factors can also contribute to tubular damage, an aspect that has been comparatively underexplored.
Using laser capture microdissection coupled with LC–MS/MS, we provide a comprehensive proteomic profile of renal tubular tissues from patients across a spectrum of ARKDs. Involving samples from five MCN, four MN, 17 IgAN, and 21 LN patients, we uncovered significant molecular pathways and DEPs, deepening our understanding of the pathophysiological mechanisms driving ARKDs.
Clinical evaluations revealed no significant differences in BUN or serum creatinine levels between the patient and control groups. However, LN patients exhibited elevated plasma cystatin C levels, and those with MN, IgAN, and LN showed significantly increased 24-h urinary protein levels and decreased plasma albumin levels, indicating significant glomerular damage. This was corroborated by the varying degrees of tubular damage and immune complex deposition observed upon histopathological examination. Histology also revealed considerable structural disruption in the renal architecture, particularly in the IgAN and LN cases.
Proteomic analysis revealed extensive alterations, particularly in the complement system and in mitochondrial proteins. Notably, upregulation of C3 and C1QC, along with immunoglobulin components such as IGHV1-8, IGKV3-20, and IGHG3 in patients with LN, emphasizes the active role of the immune system, specifically the complement pathway, in ARKD pathogenesis. This finding is consistent with elevated systemic levels of complement components and immunoglobulins and supports the literature linking C1QC and C3 dysregulation with deteriorating renal function and progression to glomerulonephritis3,4,31. A recent study has also underscored the role of complement activation in promoting renal inflammation and fibrosis via anaphylatoxin production3.
Moreover, our comparative analysis of DEPs in the glomerular and tubular tissues of patients with ARKDs indicated that the “complement and coagulation cascade” pathways are ubiquitously involved in renal injury across all ARKD types. Sixteen shared DEPs in the complement system, including C3, C1QC, C4A, C4B, C6, C8G, C8B, and C9, were identified. The upregulated central component C3, along with the MAC components C6, C8G, C8B, and C9, provided evidence of complement system activation in the tubular tissues of patients with ARKD, potentially contributing to tubular damage.
The upregulation of C1QC, C4A, and C4B indicates complement activation via the classical pathway, which is commonly reported as the main pathway of complement activation in MN and LN32,33. Although MCN is not typically associated with complement system activation, complement activation in IgAN is reported to rely more on the lectin and alternative pathways29. Our findings of C1QC, C4A, and C4B activation in the tubular tissues of patients with MCN and IgAN highlight the importance of the classical pathway in complement activation.
Alteration of CFB, a key rate-limiting enzyme of the alternative pathway34 in patients with MN, IgAN, and LN has been documented34,35,36,37,38,39. Chebotareva et al. proposed that increased urinary CFB is correlated with upregulated CD44 expression in IgAN, which contributes to kidney fibrosis35. Elevated urinary CFB levels are associated with IgAN severity and have been proposed as biomarkers for the disease36,37. Increased serum CFB levels have also been found to correlate with disease activity in MN and IgAN, suggesting it as a marker for monitoring these conditions38. Our study showed significant alterations in the CFB in tubular tissues across all four diseases. Furthermore, decreased CFB levels have been reported to have a protective effect against IgAN development in humans39 and animal models of LN34. These findings, together with ours, provide insight into how the dysregulated alternative pathway contributes to tubular damage and highlight the therapeutic potential of targeting CFB in ARKD patients. However, our study lacks evidence of complement activation via the lectin pathway in each ARKD.
SERPINA1, which is a serine protease inhibitor that plays a crucial role in immunological regulation by inhibiting neutrophil extracellular trap (NET) formation and complement activation40,41,42, was found to be consistently upregulated in ARKD patients. Alterations in urinary SERPINA1 levels have been proposed as biomarkers for various renal diseases43,44,45. To date, no previous reports have documented changes in SERPINA1 levels in the circulation and renal tissues of patients with ARKD. Our study found distinct expression trends of SERPINA1 by proteomic analysis and IHC validation in different renal compartments: upregulated in tubular tissues of ARKD patients, but variable in glomeruli. Besides the altered expression, SERPINA1 was closely correlated with various clinical parameters in the renal tissues in patients with ARKD. These observations suggest that SERPINA1 mitigates protease-mediated tissue damage and reduces inflammation with disease-specific roles.
Pearson’s correlation revealed close interactions between the complement components, including SERPINA1 and CFB, with clinical features in tubular tissues of ARKD patients, indicating their critical roles in evolving tubular damage.
We also observed that the expression of CFB and SERPINA1 was more pronounced in tubular tissues than in glomeruli in both the control and ARKD patients. The fact that higher expression of SERPINA1 and CFB in tubular tissues compared to glomeruli was already present in the control group suggests local complement factor production within renal tubules46. However, due to a lack of references describing the serum levels of SERPINA1 and CFB in patients with ARKDs, we could not determine whether the upregulated SERPINA1 and CFB resulted from in-situ activation or deposition from the circulation. Additionally, there was a shortage of blood samples for measuring these factors in the current retrospective study. Further studies are needed to uncover the circulation levels of complement components, especially SERPINA1 and CFB, to enhance our understanding in this area.
The DEPs identified between ARKDs and controls such as metabolic proteins (MECR, MT-CO2, ACSM2B, and GLS), and ROS-defensive proteins (NQO1, GLS, GSTA2, and TXNRD2), pointed that potential mitochondrial dysfunction in tubular in patients with ARKDs8. Additionally, through the analysis of GO, KEGG, GSEA, and GSVA of DEPs between the disease group and the normal group in both tubular and glomerular, we identified the DEPs which consist of ATP synthase F0/F1 subunit alpha (ATP5F1E) and subunit delta (ATP5PD), as core proteins in the oxidative phosphorylation pathway, and were significantly altered in both the glomerular and tubular tissues of ARKDs patients. This suggests that these molecules play a crucial role in mitochondrial and renal tissue in ARKDs6, especially more pronounced in tubules of patients with IgAN and LN.
PPI analysis revealed interactions among the activated complement components with the downregulation of ATP synthase F0/F1 subunits and EMT signaling pathway in tubular tissues in ARKDs, suggesting a potential mechanism of mitochondrial respiratory chain damage induced by complement activation and the consequent tubular damage and EMT (Fig. 8). This hypothesis was supported by results from two independent studies, recently. Wang et al,. reported the interaction between the activation of complement and mitochondrial dysfunction was reported in MN causing podocytes pyroptosis in vitro and in vivo16. Another study by Goetzl et al.47, which reported decreased mitochondrial electron transport proteins and increased complement mediators in plasma neural-derived exosomes of patients with early psychosis. However, this theoretical model of renal tubular damage in ARKD requires further verification by in vitro and in vivo experiments.
Conclusion
Our research elucidates significant molecular mechanisms in ARKD pathogenesis, particularly focusing on mitochondrial dysfunction and complement system activation. Identifying these pathways provides a cohesive framework for understanding these diseases and highlights the potential of targeted therapies, such as using CFB inhibitors, to enhance mitochondrial function and modulate immune responses, ultimately aiming to mitigate renal damage in ARKDs. However, this study has limitations, including this retrospective designed study has a relatively small number of patients and lack of gender diversity in control group, which limit the generalizability of the conclusions. Additionally, the limited number of FFPE and blood samples from patients restricted further analyses, such as transcriptomics and post-translational modification analyses on kidney samples, and ELISA on blood samples. Therefore, further prospective studies with larger sample sizes are needed, incorporating multi-omics approaches to deeply investigate the common and unique molecular mechanisms underlying ARKDs.
Data availability
The data supporting the findings of this study are available in additional files and from the corresponding authors.
References
Gong, J., Tamhaney, A., Sadasivam, M., Rabb, H. & Hamad, A. R. A. Chapter 68—Autoimmune diseases in the kidney. In The Autoimmune Diseases 6th edn (eds Rose, N. R. & Mackay, I. R.) 1355–1366 (Academic Press, 2020).
Sharma, V. & Patial, V. Peroxisome proliferator-activated receptor gamma and its natural agonists in the treatment of kidney diseases. Front. Pharmacol. 13, 991059 (2022).
Kovala, M. et al. Diagnostic and prognostic comparison of immune-complex-mediated membranoproliferative glomerulonephritis and C3 glomerulopathy. Cells 12, 5 (2023).
Kronbichler, A., Bajema, I., Geetha, D. & Säemann, M. Novel aspects in the pathophysiology and diagnosis of glomerular diseases. Ann. Rheum. Dis. 82(5), 585–593 (2023).
Qu, L. & Jiao, B. The interplay between immune and metabolic pathways in kidney disease. Cells 12, 12 (2023).
Kayhan, M. et al. Intrinsic TGF-β signaling attenuates proximal tubule mitochondrial injury and inflammation in chronic kidney disease. Nat. Commun. 14(1), 3236 (2023).
Chang, L. Y., Chao, Y. L., Chiu, C. C., Chen, P. L. & Lin, H. Y. Mitochondrial signaling, the mechanisms of AKI-to-CKD transition and potential treatment targets. Int. J. Mol. Sci. 25, 3 (2024).
Sharma, L. K., Lu, J. & Bai, Y. Mitochondrial respiratory complex I: Structure, function and implication in human diseases. Curr. Med. Chem. 16(10), 1266–1277 (2009).
Osellame, L. D., Blacker, T. S. & Duchen, M. R. Cellular and molecular mechanisms of mitochondrial function. Best Pract. Res. Clin. Endocrinol. Metab. 26(6), 711–723 (2012).
Daehn, I. et al. Endothelial mitochondrial oxidative stress determines podocyte depletion in segmental glomerulosclerosis. J. Clin. Investig. 124(4), 1608–1621 (2014).
Kurian, P., Obisesan, T. O. & Craddock, T. J. A. Oxidative species-induced excitonic transport in tubulin aromatic networks: Potential implications for neurodegenerative disease. J. Photochem. Photobiol. B 175, 109–124 (2017).
Miwa, S., Kashyap, S., Chini, E. & von Zglinicki, T. Mitochondrial dysfunction in cell senescence and aging. J. Clin. Investig. 132, 13 (2022).
Li, Z., Lu, S. & Li, X. The role of metabolic reprogramming in tubular epithelial cells during the progression of acute kidney injury. Cell Mol. Life Sci. 78(15), 5731–5741 (2021).
Liu, L. et al. Epithelial-mesenchymal transition in organ fibrosis development: Current understanding and treatment strategies. Burns Trauma 10, 011 (2022).
Kriz, W., Kaissling, B. & Le Hir, M. Epithelial-mesenchymal transition (EMT) in kidney fibrosis: fact or fantasy? J. Clin. Investig. 121(2), 468–474 (2011).
Wang, H. et al. Complement induces podocyte pyroptosis in membranous nephropathy by mediating mitochondrial dysfunction. Cell Death Dis. 13(3), 281 (2022).
Kanehisa, M., Furumichi, M., Sato, Y., Kawashima, M. & Ishiguro-Watanabe, M. KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res. 51(D1), D587–D592 (2023).
Patergnani, S., Bouhamida, E., Leo, S., Pinton, P. & Rimessi, A. Mitochondrial oxidative stress and “mito-inflammation”: Actors in the diseases. Biomedicines 9, 2 (2021).
Fontecha-Barriuso, M. et al. Tubular mitochondrial dysfunction, oxidative stress, and progression of chronic kidney disease. Antioxidants 11, 7 (2022).
Wang, Y., Branicky, R., Noë, A. & Hekimi, S. Superoxide dismutases: Dual roles in controlling ROS damage and regulating ROS signaling. J. Cell Biol. 217(6), 1915–1928 (2018).
Loeffler, I. & Wolf, G. Transforming growth factor-β and the progression of renal disease. Nephrol. Dial. Transplant. 29, i37–i45 (2013).
Ren, Y. et al. Ferroptosis and EMT: Key targets for combating cancer progression and therapy resistance. Cell Mol. Life Sci. 80(9), 263 (2023).
Jinesh, G. G. & Brohl, A. S. Classical epithelial-mesenchymal transition (EMT) and alternative cell death process-driven blebbishield metastatic-witch (BMW) pathways to cancer metastasis. Signal Transduct. Target. Ther. 7(1), 296 (2022).
Bruno, V., Mühlig, A. K., Oh, J. & Licht, C. New insights into the immune functions of podocytes: The role of complement. Mol. Cell Pediatr. 10(1), 3 (2023).
Kim, S. H. et al. Pathogenesis of minimal change nephrotic syndrome: An immunological concept. Korean J. Pediatr. 59(5), 205–211 (2016).
Watts, A. J. B. et al. Discovery of autoantibodies targeting nephrin in minimal change disease supports a novel autoimmune etiology. J. Am. Soc. Nephrol. 33(1), 238–252 (2022).
Ronco, P. et al. Membranous nephropathy. Nat. Rev. Dis. Primers 7(1), 69 (2021).
Wu, L. et al. Immunofluorescence deposits in the mesangial area and glomerular capillary loops did not affect the prognosis of immunoglobulin a nephropathy except C1q:a single-center retrospective study. BMC Nephrol. 22(1), 43 (2021).
Poppelaars, F., Faria, B., Schwaeble, W. & Daha, M. R. The contribution of complement to the pathogenesis of IgA nephropathy: Are complement-targeted therapies moving from rare disorders to more common diseases? J. Clin. Med. 10, 20 (2021).
Dong, J. et al. Global-feature of autoimmune glomerulonephritis using proteomic analysis of laser capture microdissected glomeruli. Front. Immunol. 14, 1131164 (2023).
van Schaik, M. et al. Clinical remission and reduction of circulating nephritic factors by combining rituximab with belimumab in a case of complement factor 3 glomerulopathy. Kidney Int. Rep. 9, 1919 (2024).
Seifert, L. et al. The classical pathway triggers pathogenic complement activation in membranous nephropathy. Nat. Commun. 14(1), 473 (2023).
Weinstein, A., Alexander, R. V. & Zack, D. J. A review of complement activation in SLE. Curr. Rheumatol. Rep. 23(3), 16 (2021).
Chen, K. et al. Complement factor B inhibitor LNP023 improves lupus nephritis in MRL/lpr mice. Biomed. Pharmacother. 153, 113433 (2022).
Chebotareva, N. et al. CD44 expression in renal tissue is associated with an increase in urinary levels of complement components in chronic glomerulopathies. Int. J. Mol. Sci. 24, 8 (2023).
Wang, D. et al. Urinary complement profile in IgA nephropathy and its correlation with the clinical and pathological characteristics. Front. Immunol. 14, 1117995 (2023).
Niu, X. et al. Urinary complement proteins in IgA nephropathy progression from a relative quantitative proteomic analysis. PeerJ 11, e15125 (2023).
Ji, F. P., Wen, L., Zhang, Y. P., Liu, E. P. & Wen, J. G. Serum complement factor B is associated with disease activity and progression of idiopathic membranous nephropathy concomitant with IgA nephropathy. Int. Urol. Nephrol. 54(6), 1287–1294 (2022).
Shi, D. C. et al. Functional variant rs12614 in CFB confers a low risk of IgA nephropathy by attenuating complement alternative pathway activation in Han Chinese. Front. Immunol. 13, 973169 (2022).
Silva, V. & Radic, M. COVID-19 pathology sheds further light on balance between neutrophil proteases and their inhibitors. Biomolecules 13, 1 (2022).
Kelly-Robinson, G. A. et al. The serpin superfamily and their role in the regulation and dysfunction of serine protease activity in COPD and other chronic lung diseases. Int. J. Mol. Sci. 22, 12 (2021).
Majewski, P. et al. Inhibitors of serine proteases in regulating the production and function of neutrophil extracellular traps. Front. Immunol. 7, 1 (2016).
Pérez, V. et al. Urinary peptide profiling to differentiate between minimal change disease and focal segmental glomerulosclerosis. PLoS ONE 9(1), e87731 (2014).
Choi, Y. W. et al. Potential urine proteomics biomarkers for primary nephrotic syndrome. Clin. Proteom. 14, 18 (2017).
Anania, V. G. et al. Discovery and qualification of candidate urinary biomarkers of disease activity in lupus nephritis. J. Proteome Res. 18(3), 1264–1277 (2019).
Li, D. et al. Complement factor B production in renal tubular cells and its role in sodium transporter expression during polymicrobial sepsis. Crit. Care Med. 44(5), e289–e299 (2016).
Goetzl, E. J. et al. Decreased mitochondrial electron transport proteins and increased complement mediators in plasma neural-derived exosomes of early psychosis. Transl. Psychiatry 10(1), 361 (2020).
Acknowledgements
The visualization of Supplementary Fig. 6 by Hao Tian is acknowledged.
Funding
This study was supported by the Guangdong Provincial Key Field R&D Program (No. 2023b0101200010).
Author information
Authors and Affiliations
Contributions
Conceptualization: M.X., J.D., Y.D., L.Y.; data collection: M.X., J.D., X.C., X.Z., Z.X., Y.Z., Y.P., S.L.; visualization: M.X., J.D., X.C., X.Z.; writing—original draft preparation: M.X.; writing—review and editing: X.C., X.Z., Z.X., Y.Z., Y.P., S.L., J.D., Y.D., L.Y.; supervision: J.D., Y.D., L.Y.; project administration: X.C., Y.Z., L.Y.; funding acquisition: L.Y. All authors have read and agreed to the published version of the manuscript. All authors have read and approved the manuscript and declare no potential conflicts of interest. All authors have agreed to publish this paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Xiao, M., Chi, X., Zhu, X. et al. Proteomic analysis of laser captured tubular tissues reveals complement activation and mitochondrial dysfunction in autoimmune related kidney diseases. Sci Rep 14, 19311 (2024). https://doi.org/10.1038/s41598-024-70209-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-70209-9
- Springer Nature Limited