
Abstract
The team aimed to explore the possible functional significance of M6A regulation in Pan-programmed cell death (PCD) among patients with bladder cancer (BLCA). In BLCA patients, the analysis was conducted on the13 patterns of programmed cell death (PCD) and the regulation of M6A. Transcriptome, genomics, and clinical data were collected from TCGA-BLCA, GEO32548, and IMvigor210. Consensus clustering analysis, functional enrichment analysis, and other prognostic tools were used to validate the Pan-PCD. Finally, in vitro experiments and transcription sequencing were performed to understand the potential influence of the PI3K pathway on Pan-PCD in BLCA patients. Diverse PCD patterns were simultaneously activated, and M6A regulators exhibited significant variability in bladder malignant tissues. The machine learning algorithm established an 8-gene M6A-related Pan-PCD signature. This signature was validated in three independent datasets, and BLCA patients with higher risk scores had worse prognosis. An unsupervised clustering approach identified activated and suppressed Pan-PCD subgroups of BLCA patients, with distinct responses to immunotherapy and drug sensitivity. In addition, the PI3K pathway was identified as a key mechanism for various forms of programmed cell death, encompassing apoptosis, pyroptosis, autophagy, and cell death dependent on lysosomes. This research revealed that the Pan-PCD model was a more promising approach for BLCA patients under M6A regulation. A new signature from M6A-related Pan-PCD was proposed, with prognostic value for survival or drug sensitivity. The PI3K pathway was a key mechanism for multiple PCDs in BLCA patients.
Similar content being viewed by others


Introduction
Bladder cancer research has typically focused on distinct pathways of programmed cell death (PCD) like apoptosis or autophagy1. However, with the continuous advancement of science, the Pan-programmed cell death model has emerged as a more rational and comprehensive perspective for understanding the progression mechanisms of bladder cancer2. This model is based on a thorough investigation of the interactive dynamics among various PCD pathways. In comparison to individual PCD, the Pan-PCD model better reflects the diversity of PCD mechanisms within cancer cells and emphasizes the interplay among these pathways. In bladder cancer, cells may simultaneously activate multiple PCD pathways, and the intricate regulatory relationships between these PCDs are crucial for the development of malignancies. Therefore, the introduction of the Pan-PCD model enables us to better understanding the overall regulatory network of PCD in bladder cancer. In this research, 13 different PCD patterns were enrolled into further analysis, including ferroptosis, apoptosis, parthanatos, necroptosis, Cuproptosis, pyroptosis, netotic cell death, entotic cell death, lysosome-dependent cell death, oxeiptosis, autophagy-dependent cell death, and alkaliptosis. The model enables a more precise forecast of prognosis and drug responsiveness in individual with bladder cancer and could aid in the advancement of more efficient therapeutic approaches.
Bladder cancer research greatly relies on the significant involvement of M6A regulation, which is a widely used regulatory model for programmed cell death. For example, FTO modifies the m6A level of MALAT and promotes bladder cancer progression; m6A writer WTAP targets NRF2 to accelerate bladder cancer malignancy via m6A-dependent ferroptosis regulation3. To begin, a comprehensive understanding of the functions and mechanisms of M6A regulation in cells is fundamental. M6A is a type of methylation modification that occurs on adenine in RNA, primarily on mRNA. This modification exerts profound effects on gene expression by regulating processes such as RNA stability, transport, and translation4. In the process of programmed cell death, M6A regulation has been found to participate in various crucial regulatory steps. M6A modification can influence the stability of RNA, thereby regulating the expression levels of genes associated with PCD. This adds a layer of complexity to the regulatory network of bladder cancer progression and suggests potential therapeutic targets for the treatment of bladder cancer patients.
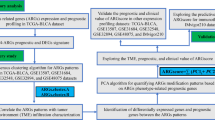
This study examined the interplay between Pan-programmed cell death and M6A regulation and their impact on bladder cancer. Through a series of investigations and experiments, researchers have discovered that: (1) Bladder cancer exhibit the characteristics of multiple concurrent programmed cell death pathways, and the Pan-programmed cell death model is a more rational analytical perspective than individual PCD. Additionally, there is a close interactive relationship between M6A regulation and Pan-PCD; (2) Developing a gene set based on Pan-PCD under M6A regulation can differentiate bladder cancer with different prognostic outcomes and guide subsequent drug treatments; (3) The PI3K pathway is a crucial pathway in the occurrence of Pan-PCD in bladder cancer. These discoveries improve our comprehension of the regulatory system of bladder cancer advancement and propose potential targets for therapy in patients with bladder cancer. The Fig. 1 presented the flow chart for the whole research.

The flow chart of the whole research.
Methods
The data collection
This study utilized three bladder cancer datasets, including the TCGA-BLCA dataset, GEO32548 dataset, and IMvigor210 dataset. All three datasets contained comprehensive clinical information, prognostic outcomes, and mRNA expression profiles. The TCGA-BLCA data was acquired from the GDC website, while the GEO32548 dataset was obtained from the GEO website. The IMvigor210 dataset was retrieved using the R package “IMvigor210CoreBiologies”5. All three datasets are freely accessible to any visitor for data downloading. This study adheres to the Helsinki Declaration (revised in 2013) and does not require ethical approval as the data we utilized was derived from public databases. In the light of the study’s retrospective nature, the requirement for informed consent was waived.
Cell line
The bladder cancer human cell line (5637 cells) were obtained from the Authenticated Cell Culture Compilation of China.
The genes associated with PCD consisted of the primary governing genes of the 13 PCD patterns previously mentioned. The genes were acquired from gene sets in GSEA, databases of KEGG pathways, and articles for review6. The final gene list consisted of the concatenated regulatory genes of all 13 PCD patterns. In the analysis, a total of 1437 genes associated with PCD were included, as shown in Supplementary Table S1. Additionally, based on relevant literature, 24 M6A regulation genes were also collected7.
Identification of the different expression and variation levels of PCD related genes.
Preparation was done for raw transcriptome count data of 406 BLCA patients and 19 normal tissues in the TCGA-BLCA dataset. The limma R package was then utilized to detect genes that were expressed differently (DEGs) using the criteria of an adjusted P-value less than 0.05 and an absolute log2FC greater than 18. The ‘maftools’ package in R was utilized to investigate concealed somatic mutation data in BLCA patients. The CNV values of genes associated with PCD were examined, and values exceeding 0.2 were categorized as ‘gains,’ whereas values below 0.2 were classified as ‘losses’.
Functional enrichment analysis
A cluster profiler package for R was used in this study to evaluate enrichment using the Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG). Gene Set Variation Analysis (GSVA) was also used to analyze the correlation between genes and pathway scores using the “GSVA” R package9.
Screening for M6A-associated PCD relative DEGs by three steps
The signature associated with M6A-associated PCD was identified through three methods: (1) The expression of DEGs related to PCD identified in this study and M6A regulators was correlated by Pearson correlation analysis. The M6A-associated PCD-related DEGs were filtered based on correlation coefficients (R and P) > 0.4 and < 0.05. (2) In order to identify prognostic DEGs associated with PCD, we conducted a univariate Cox proportional hazard regression. (3) A multivariate Cox proportional hazard model was performed using the results from step 2. In conclusion, we have identified candidate prognostic differentially expressed genes (DEGs) associated with M6A and overlapping with PCD10.
Construction of the signature and its risk score
By utilizing the highest lambda value selected through 1000 cross-validations, an L1-penalized regression was implemented using the LASSO Cox regression algorithm (referred to as ‘1-se’ lambda). This assisted in further determining candidates for PCD-related DEGs associated with M6A. Afterwards, a model for predicting the risk of survival was created using the LASSO coefficients (β), as shown below.
Prognostic analysis
BLCA patients in the TCGA cohort were collected for clinical data (age, T, N, and stage), which were analyzed along with risk scores through Analyses of Cox regression with univariate and multivariate data. By combining clinical characteristics such as age, T, N, and stage, along with the risk score, we developed a prognostic nomogram through the utilization of multivariable Cox regression and stepwise regression analysis. We generated the nomogram plot using the “regplot” package. The effectiveness of R package ‘caret’ and ‘rmda’ was assessed through the utilization of calibration plots and decision curve analysis (DCA). The “timeROC” R package was used to analyze Receiver Operating Characteristics (ROC). The construction of a dynamic nomogram was achieved by utilizing the packages ‘rsconnect’ and ‘DynNom’11.
The process of consensus clustering analysis and the examination of functional enrichment were conducted.
In order to examine the biological roles of M6A-related prognostic DEGs in BLCA patients, we employed the R software package ‘ConsensusClusterPlus’ to categorize patients into distinct subgroups according to the expression levels of M6A-related prognostic DEGs in the TCGA dataset. The task was completed using 50 rounds, an 80% resampling rate, and a Pearson correlation coefficient. In order to gain a deeper comprehension and interpretation of the correlation among M6A-related prognostic subcategories, we utilized the R package ‘clusterProfiler’ to conduct GO pathway analysis and GSEA for the functional annotation of genes exhibiting differential expression across various subcategories12.
Analysis of infiltration by immune cells and evaluation of the immune response against tumors
In BLCA, the estimation of immune cell composition in tumor-infiltrating cells was calculated using the CIBERSORT bioinformatic deconvolution algorithm, which relies on related-gene expression profiles. Using the Wilcoxon signed-rank test, we compared the fractions of immune cells, the expressions of genes related to exhausted T cells, genes involved in presentation of antigens, genes associated with activity of the interferon, genes related to cytolytic activity, kinase genes and integrin genes among various subtypes and subgroups13.
Thorough examination of the advantages of ICI treatment
In order to investigate the predictive significance of M6A-related pseudogene in BLCA patients following immunotherapy, we conducted a comparison of various subtypes using online calculations for a TIDE score, an MSI, disfunction of the T cell, a TAM_M2, a T cell exclusion score, a MASC, and a CAF score. Furthermore, the TIS score was determined by averaging the normalized expression for log2 scales of specific genes and then compared across various subtypes. Moreover, the purity of the tumor was estimated using four different methods (ABSOLUTE, IHC, CPE, and ESTIMATE), which provided a deeper insight into the advantages of ICI therapy.
Using predictive analysis to identify potential therapeutic agents for patients with either activated or suppressed Pan-PCD subgroups of BLCA patients.
Cancer Therapeutics Response Portal (CTRP v.2.0, https://portals.broadinstitute.org/ctrp/) and Profiling Relative Inhibition Simultaneously in Mixtures (PRISM, https://depmap.org/portal/prism/) were the sources for downloading cancer cell line drug sensitivity profiles (CCLs)14. Initially, compounds with a data loss of over 20% were excluded. After performing imputation, the K-nearest neighbor (k-NN) method was utilized to replace the absent AUC values. Two distinct processes were applied to identify potential agents with increased efficacy of drugs in the Activated Pan-PCD subgroup of BLCA patients, based on drug response profiles obtained from CTRP and PRISM. The variances in mRNA and protein expression levels of the targets of drugs for potential agents were computed between tumor and normal tissue. Higher values of differential (fold change) suggest increased potential and improved effectiveness of the potential agent.
The vitro experiments
For cultivation purposes, the 5637 cells from the Cell Bank of the Chinese Academy of Sciences, which are a human cell line for bladder cancer, were utilized. In order to examine the possible involvement of the PI3K pathway in BLCA, a compound (HY-10108) that inhibits the PI3K pathway at a concentration of 1 μM was introduced into the cell culture medium. This led to the formation of two cell groups: Group A, which received the PI3K pathway inhibitor, and Group B, the control group. Transcriptional profiling was performed by a Chinese comPany named Lian Chuan.
Ethical approval
Ethical review and approval were waived in accordance with the local legislation and institutional requirements. Written informed consent for participation was not required for this study in accordance with the national legislation and institutional requirements.
Results
Landscape present for Pan-programmed cell death (PCD) and the M6A mediated regulation on the bladder cancer patients
The Pan-programmed cell death (PCD) model, rather than the signal PCD model, is a significant mechanism contributing to oncogenesis and progression in bladder cancer. To identify the genes that are differently expressed in bladder cancer tissue compared to bladder benign tissue, we analyzed the TCGA-BLCA dataset, which revealed 2362 genes with differential expression, including 925 up-regulated and 1437 down-regulated genes (Fig. 2A, Table S2). Notably, 184 of the 1437 PCD-related genes (Table S1, S3) were found to be among the DEGs in bladder cancer. Gene Ontology (GO) enrichment analysis showed that the Pan-PCD model undergoes significant alterations from bladder benign tissue to malignant tissue. Autophagy, extrinsic apoptotic, and intrinsic apoptotic pathways were among those that exhibited marked changes (Fig. 2B). Similar observations were made from the perspective of gene mutation and copy number variation (CNV). The top 20 mutated PCD-related genes are presented in Fig. 2C, indicating multiple PCD models undergoing simultaneous change. Specifically, 14% of BLCA patients had ERBB3 mutations (a key gene for apoptosis), while 9% had CDKN2A mutations (cuproptosis), 5% had FANCD2 mutations (ferroptosis), and 6% had SYNPO2 mutations (autophagy). Moreover, the most widespread deletion CNVs encompassed CLU (apoptosis), CDKN2A (cuproptosis), and SREBF1 (autophagy), whereas E2F1 (apoptosis), SQLE (ferroptosis), and TP53INP2 (autophagy) exhibited the most significant copy number amplifications (Fig. 2D).

Landscape Present for Pan-programmed cell death (PCD) and the M6A mediated regulation on the bladder cancer patients. (A) The different expression of PCD relative genes in the BLCA patients, (B) the GO analysis for the different expression of PCD relative genes in the BLCA patients, (C) the TOP 20 mutation gene for PCD relative genes, (D) the CNV results for 157 PCD genes in the BLCA patients, (E) the M6A regulation genes’ expression between bladder cancer tissue and benign tissue, F: the interaction network of M6A regulation genes in the BLCA patients.
The mediation of M6A-mediated regulation also played a significant role in bladder cancer patients. Compared to bladder benign tissue, the three key M6A-mediated components (writers, readers, and erasers) displayed significant variations in bladder malignant tissue. Previous studies have increasingly identified functional interactions between M6A genes that contribute to oncogenesis and progression. This study also observed similar results in BLCA patients (Fig. 2E). As indicated in the STRING Database (string-db.org), a complex network of interactions was present between the M6A regulators in the TCGA-BLCA dataset (Fig. 2F).
Identification of a M6A-associated PCD related signature in TCGA dataset
Fourteen M6A-associated PCD-related genes were identified through a series of analytical steps. The first step was to use Pearson correlation analysis to analyze the expression of all PCD-related genes and a list of one-to-one gene pairs was filtered using 24 M6A genes, including 7168 pairs of genes and M6A genes with an absolute correlation coefficient (R) of ≥ 0.4 and a P-value of < 0.05. This process resulted in a list of 700 genes (Table S4) that had significant correlation with multiple M6A genes. The second step was to screen 259 pseudogenes with a P-value of 0.05 using the univariate Cox proportional hazard regression analysis. Thirdly, the intersection of 2362 DEGs, 700 genes, and 259 genes resulted in the identification of 14 genes (ABL1, CAV1, HIP1R, ITPRIP, JUN, KITLG, SLC9A3R1, THBS1, GSDMB, SQLE, ZEB1, ATP13A2, TP53INP2, and STXBP1) (Fig. 3A). The Pearson correlation analysis between the expression levels of individual M6A genes and the 14 PCD-related DEGs modified by M6A was conducted, and the results confirmed strong and universal co-expression and interaction (Fig. 3B). The forest plot of the 14 PCD-related DEGs modified by M6A showed that the hazard ratio (HR) of HIP1R and GSDMB was less than 1, suggesting that these two genes may be factors with good prognosis. Conversely, Other 12 genes with HRs greater than 1 may be associated with poor prognosis (Fig. 3C).

Identification of a M6A-associated PCD related signature in TCGA dataset. (A) the intersection of three genes sets, and identification of 14 key PCD genes; (B) the interaction between 14 PCD genes and M6A regulation genes, (C) the univariate Cox proportional hazard regression showed that all the 14 PCD genes had the significant survival value (P < 0.05); (D) the Lasso regression analysis was performed, and a 8-gene signature was identified, (E) the expression level for 8 genes between bladder cancer tissue and benign tissue, (F) the potential PCD pathways and TNM stages were compared and found by the risk score of this novel signature.
A M6A-associated PCD-related signature was further identified. An analysis of Lasso cox regression was performed on PCD-related DEGs (Fig. 3D). A M6A-associated PCD-related signature with 8 PCD-related DEGs (HIP1R, JUN, SLC9A33R1, GSDMB, SQLE, ZEB1, ATP13A2, TP53INP2) was constructed. The 8 genes included 3 apoptosis genes (HIP1R, JUN, SLC9A33R1), 1 pyroptosis gene (GSDMB), 2 ferroptosis genes (SQLE, ZEB1), and 2 autophagy genes (ATP13A2, TP53INP2) (Fig. 3E). The risk score of this signature was constructed according to the formula below. Each TCGA-BLCA case was divided into two groups (one with a low risk score and one with a high risk score). All 13 PCD models were compared between the two groups using the R package "GSEA", and the results showed significant differences in multiple PCD models: the top 5 PCD models included apoptosis, ferroptosis, Lysosome-dependent cell death, autophagy, and pyroptosis (Fig. 3F).
The M6A-associated PCD related signature is an independent and significant factor in BLCA patients’ prognosis
The 8 M6A-associated PCD-related genes exhibit remarkable prognostic value for BLCA patients. By utilizing the TCGA-BLCA dataset as a test case and demonstrating the proposed power estimation procedure in a real data application through Kaplan–Meier (KM) analysis, all 8 genes demonstrated significant prognostic value for overall survival (OS) and discriminative ability. Specifically, two genes (SLC9A33R1, JUN) exhibited a P value less than 0.001, five genes (HIP1R, GSDMB, SQLE, ZEB1, ATP13A2) had a P value less than 0.01, and one gene (TP53INP2) had a P value less than 0.05 (Supplementary Fig. 1). These results suggest that the 8 M6A-associated PCD-related genes may assist as promising predictive markers for BLCA patients.
The prognostic value of the M6A-associated PCD-related signature was further validated using the TCGA-BLCA dataset as the training set and two BLCA datasets (GSE32548 and IMvigor210) as validation cohorts. The risk score effectively distinguished the results from clinical trials of BLCA patients in both the training dataset and the two validation datasets. Firstly, higher risk scores were strongly associated with worse survival outcomes in BLCA patients (Fig. 4A). Secondly, principal component analysis (PCA) analysis showed that the risk score had the ability to clearly distinguish BLCA patients into two groups (Fig. 4B). Thirdly, Kaplan–Meier (KM) analysis confirmed that higher risk scores were strongly correlated with worse survival outcomes (P = 2.45 × 10−5 in the TCGA-BLCA dataset, P = 0.0173 in the GSE32548 dataset, and P = 0.00531 in the IMvigor210 dataset; Fig. 4C). Finally, these trends were observed in all three datasets, fully validating the prognostic value of the M6A-associated PCD-related signature.

Verify the survival value of this signature in three BLCA patients’ datasets. (A, B) the risk score of this signature could distinguish the bladder cancer patients in three different datasets, (C) the BLCA patients with high score would have the worse survival results in three datasets. (D) The tome dependent ROC analysis showed the potential value for risk score in the following 1/3/5 year.
The risk score was found to have a stable association with other clinical prognostic factors, as shown by the following observations: The deceased patients had higher risk scores (P = 5.51 × 10−11). The higher pathological T stage correlated with higher risk scores (P = 0.0033).
The higher pathological M stage correlated with higher risk scores (P = 0.0012). The higher pathological stage correlated with higher risk scores (P = 6.6 × 10−5, Supplementary Fig. 2).
Cox regression analysis further confirmed the signature’s independent prognostic value for BLCA patients. Univariate Cox regression analysis identified four key factors (risk score, age, N stage, and pathology; all four factors with P < 0.001, Fig. 5A) that were associated with survival outcomes in BLCA patients. However, only two factors (risk score and N stage; both factors with P < 0.001, Fig. 5B) continued to exhibit significant survival value in the multivariate Cox regression. Notably, the risk score presented a stronger prognostic value compared with N stage. These results suggest that the risk score derived from the M6A-associated PCD-related signature was a reliable and independent predictor of survival outcomes in BLCA patients, providing valuable prognostic information beyond traditional clinical factors.

This signature is an independent and significant factor in BLCA patients’ prognosis. (A) Cox regression analysis further confirmed the signature’s independent prognostic value for BLCA patients, risk score is better than any other indexed, including TNM stage, and age. (B) The nomogram also presented the risk score’s potential prognostic value in the BLCA patients (P < 0.001), (C–E) Calibration curves (C), KM analysis (D), and DCA (E) were used to present the signature’s significant potential prognostic value.
The nomogram, which integrated four key prognostic factors (risk score, age, N stage, and pathology), displayed excellent survival predictive capabilities in both short- and long-term follow-up periods. The nomogram model established in the TCGA-BLCA cohort was effective in estimating the 1-, 3-, and 5-year overall survival (OS). The model’s C-index value was 0.702 (95% CI 0.663–0.741, Fig. 5C). Calibration curves demonstrated the accuracy of this model in predicting the 1-, 3-, and 5-year survival rates (Fig. 5D). KM analysis further confirmed the nomogram score’s predictive ability for BLCA patients (P = 1.78 × 10−12, Fig. 5E). Additionally, DCA analysis showed that the nomogram score was superior to any other predictor for estimating the 1-, 2-, 3-, 4, and 5-year survival rates of BLCA patients (Fig. 5F and Supplementary Fig. 3A–B). Taken together, these results suggest that the nomogram integrating the risk score, age, N stage, and pathology provides a powerful and accurate tool for predicting survival outcomes in BLCA patients.
Using Q-PCR detection, the expression levels of the above gene set in bladder cancer cell lines were further verified. The results suggested that there were significant differences in the expression of eight genes in bladder cancer cell lines compared with bladder epithelial cells (Supplementary Fig. 3c).
Activated or suppressed Pan-PCD subgroups of BLCA patients had different immune components and function, with distinct responses to immunotherapy and drug sensitivity
The activated and suppressed Pan-PCD subgroups of BLCA patients were identified based on the expression profiles of the 8 M6A-associated PCD-related DEGs. To analyze the potential value of the Pan-PCD model for BLCA patients, Consensus clustering analysis was performed on the TCGA-BLCA dataset. It was found that k = 2 was the best selection when clustering stability increased from 2 to 10. The cases from the TCGA-BLCA dataset were divided into two subgroups (cluster 1 and cluster 2) by setting k as 2 (Fig. 6A,B). The activities of the 13 PCD pathways were analyzed by ssGSEA in the two subgroups. The cluster 1 with higher multiple PCD pathway activities was named as the activated Pan-PCD subgroup (Fig. 6C), while the cluster 2 with lower multiple PCD pathway activities was named as the suppressed Pan-PCD subgroup. This classification method was reliable and stable, which was demonstrated by the other two BLCA datasets (GSE32548 and IMvigor210). Furthermore, the activated Pan-PCD subgroup was strongly associated with worse survival outcomes (P = 0.00479 in the TCGA-BLCA, P = 0.00927 in the GSE32548, P = 0.0372 in the IMvigor210, Fig. 6D and Supplementary Fig. 3) and higher risk scores. Additionally, the activated Pan-PCD subgroup exhibited a higher degree of M6A regulation, further highlighting the strong correlation between M6A regulation and Pan-PCD (Fig. 6E).

activated or suppressed Pan-PCD subgroups of BLCA patients had different survival outcomes, with distinct responses to drug sensitivity. (A, B) The BLCA patients were divided into the activated or suppressed Pan-PCD subgroups by the Consensus clustering analysis, the differences between two subgroups were clear. (C) The activated subgroup had a worse survival result. (D) The M6A regulation level between two subgroups was different. (E) The Pan-PCD pathways between two subgroups were clearly different, including 10 PCD pathways with significant differences (P < 0.05). F: the potential small molecule drugs for two subgroups.
Further investigating the correlation between the expression patterns of the activated and suppressed Pan-PCD subgroups and Immune response to tumors in BLCA, we employed CIBERSORT to estimate an analysis of the infiltration of immune cells and the expression of T cell exhaustion, genes involved in antigen presentation, interferon activity, kinases, cytolytics, and integrins in tumor tissues between these two subgroups. The activated Pan-PCD subgroup was found to exhibit immune-suppressive characteristics, while the suppressed Pan-PCD subgroup maintained favorable immune activities (Supplementary Fig. 4). Similar trends were observed in higher- or lower risk score groups (Supplementary Fig. 5).
One purpose of this study is to explore the potential benefits of ICI therapy in the activated and suppressed Pan-PCD subgroups, a series of immune therapy tools, including TIDE, MSI score, Exclusion, and TAM-M2 score, were employed. All results consistently indicated that the suppressed Pan-PCD subgroup was more amenable to ICI therapy. Furthermore, BLCA patients with lower risk scores also appeared to exhibit a more satisfactory response to ICI therapy (Supplementary Figs. 6–7).
In order to discover possible therapeutic substances, we utilized two distinct approaches (CTRP and PRISM datasets) to validate potential medications that exhibit increased sensitivity to drugs in the activated and suppressed Pan-PCD subcategories. At first, we utilized methods for differential drug response to discover substances that had AUC calculations with a lower value in the group with a high-risk score. This was done by setting a log2FC threshold of greater than 0.10. In order to screen compounds, Spearman correlation calculations were performed between the risk score and AUC value of each compound, using an inverse calculation of correlation coefficients (Spearman’s R − 0.1 for CTRP and PRISM).
From the aforementioned examinations, a total of six compounds were obtained from CTRP (specifically PI-103, AZD7762, dasatinib, pluripotin, birinaPant, and RITA), while twelve compounds were derived from PRISM (namely mixtoxantrone, tosedostat, EPZ-5676, NVP-AUY922, atorvastatin, LY2606368, romidepsin, GZD824, topotecan, MK-2461, vinblastine, and rubitecan). The identified compounds exhibited reduced AUC calculations in the high-risk score subtype and showed an inverse relationship with the risk score in both CTRP and PRISM datasets. The activated Pan-PCD subgroup (Fig. 6F and Supplementary Fig. 8) could benefit from these compounds, which have the potential to be used as therapeutic agents for BLCA patients.
PI3K pathway was the key mechanism of Pan-programmed cell death for BLCA patients
The PI3K pathway represented the most significant functional variation among Pan-PCD genes in BLCA patients. Firstly, KEGG functional analysis of the 157 M6A-associated Pan-programmed cell death DEGs identified the PI3K pathway as the most significant functional variation (Fig. 7A). Secondly, when compared to the activated or suppressed Pan-PCD subgroups of BLCA patients, KEGG functional analysis once again showed that the PI3K pathway was the most striking difference (Fig. 7B). Previous in vivo and in vitro experiments have established that bladder cancer proliferation or invasion is assessed through activation of the PI3K pathway15,16. These results suggest that the PI3K pathway might promote bladder cancer proliferation or invasion via Pan-programmed cell death.

PI3K pathway is the key mechanism of Pan-programmed cell death for BLCA patients. (A) KEGG functional analysis of the 157 M6A-associated Pan-programmed cell death DEGs identified the PI3K pathway as the most significant functional variation, (B) when compared to the activated or suppressed Pan-PCD subgroups of BLCA patients, KEGG functional analysis once again showed that the PI3K pathway was the most striking difference, (C) the whole transcription sequencing’s results contained a total number of 344 DEGs, including 9 PCD genes, (D) 9 PCD genes had different expression when the PI3L inhibitor was added, (E) PI3K pathway was involved in four PCD models, including apoptosis, pyroptosis, autophagy, and lysosome-dependent cell death in the BLCA human cell line, (F) 13 PCD pathways’ score were analyze by the COX regression, and Apoptosis and autophagy presented significant survival values (P < 0.05), (G) the potential mechanism for PI3K pathway involved the bladder cancer progression, including four PCD pathways, and 9 PCD key genes.
It was verified that the PI3K pathway caused multiple PCDs in BLCA patients in vitro. To analyze the potential function of the PI3K pathway on BLCA, a bladder cancer human cell line (5637 cells) was cultured and a PI3K pathway inhibitor was added to the cell culture medium, resulting in two groups of cells (A: with the PI3K pathway inhibitor, and B: control group). Transcription sequencing was then performed. Table S6 provided the whole transcription sequencing’s results, and a total number of 344 DEGs (P < 0.05 and |log2FC|> 1, Table S7 and Fig. 7C) were found. The comparison of M6A-related genes between the two groups revealed that only HNRNPD exhibited downregulation in the PI3K-inhibited group (P = 0.03), while there were no significant differences observed in the other genes (Table S8). The results indicated that the PI3K pathway was involved in four PCD models, including apoptosis, pyroptosis, autophagy, and lysosome-dependent cell death in the BLCA human cell line (Fig. 7D,E). Additionally, nine PCD-related genes had statistical differences (P < 0.05). Among these nine genes, five genes (BMP4, CSF2, IFI27, IL6, and SOGA3) had more significant expression level differences. Three genes (BMP4, CSF2, IFI27) were key regulators of apoptosis, while IL6 (pyroptosis) and SOGA3 (autophagy) also provided similar potential function.
Furthermore, COX regression analysis was performed on the TCGA-BLCA dataset for 13 PCD scores, which were scored by the, ssGSEA method. The results showed that the signal apoptosis (P = 0.044) or autophagy (P = 0.022) relative scores had potential prognosis for BLCA patients (Fig. 7F). The interaction among the Pan-PCD models for BLCA patients’ potential prognosis was worth further analysis.
Based on published articles, only apoptosis and autophagy were verified to be associated with the PI3K pathway in BLCA patients through in vitro experiments. However, three genes (IL6, BMP4, and BLK) were reported to have a correlation with the PI3K pathway in cancer-related studies. This research first revealed that inhibiting the PI3K pathway could regulate two new PCD models (pyroptosis and lysosome-dependent cell death) and six PCD key genes (CSF2, IFI27, SOGA3, TRIML1, CPLX2, and NAPSA) in BLCA patients (Fig. 7G).
Discussion
Programmed cell death (PCD) is a crucial concept employed by researchers to comprehend the abnormal growth and widespread metastasis of cancer. In recent years, novel forms of PCD, such as ferroptosis and cuproptosis, have been discovered and proven to play crucial roles in the initiation and progression of cancer. The emergence of these novel modes of PCD not only supplements the intricate and dynamic signaling pathways and pathogenic mechanisms of cancer but also paves the way for the development of innovative targeted drugs and addresses a novel direction to counter drug-resistant cancer strains.
Furthermore, researchers have also observed phenomena of mutual interaction and reciprocal influence among various forms of PCD within cancer tissues. For example, It is a close connection between apoptosis, necroptosis, and pyroptosis and can cross-regulate each other17. Among the earliest connections between different types of cell death was the role of caspase-8 as a mediator of apoptotic and necroptotic pathways18. A further example of how different cell death pathways are linked by RNA viruses is how NLRP3 is activated in a RIPK1/RIPK3-dependent and MLKL-independent manner19. These findings suggest that targeting multiple forms of PCD simultaneously may be a more effective approach to combat cancer. Improved understanding of molecular mechanisms underlying the crosstalk between different forms of PCD in cancer tissues will pave the way for Innovative therapeutic approaches aimed at reversing drug resistance and improving patient outcomes.
This research has demonstrated that bladder cancer exhibits the characteristics of a Pan-PCD model, as evidenced by the following observations: 1) A total of 159 PCD-related genes were identified as DEGs in bladder cancer tissues, including 10 different PCD models that impact the progression of bladder cancer. 2) From the perspective of gene mutation and CNV, the top genes were derived from distinct PCDs within the Pan-programmed cell death model, rather than the signal-programmed cell death model, highlighting the more significant role of the Pan-PCD model in the oncogenesis and progression of bladder cancer. 3) By analyzing the activated and suppressed Pan-PCD subgroups, it was observed that bladder cancer exhibits stronger internal interactions among 13 different PCDs compared to other cancers. Specifically, the activated subgroup of BLCA patients displayed significant activation of Pan-PCD pathways, including 7 PCD pathway scores with P < 0.001, 1 PCD pathway score with P < 0.01, and 3 PCD pathway scores with P < 0.05.
It is most commonly found on mRNAs and noncoding RNAs as an epigenetic modification, M6A methylation plays a crucial role in influencing RNA metabolism, thereby exerting negative control over protein expression of PCD. The focus of this study was on the regulation of M6A in the Pan-PCD model20. Firstly, an increasing body of research has demonstrated that M6A modification regulates the expression and function of key proteins involved in multiple PCD pathways. For instance, M6A induction promotes apoptosis through four mechanisms: regulation of genes associated with apoptosis, methylating and demethylating enzymes, and YTHDF2-mediated transcription reduction21. Ulk1, a protein kinase activated during autophagy, abrogates the distribution of M6A within transcripts by FTO, leading to an increase in Ulk1 protein production and signifying the initiation of autophagy22. Secondly, this research also revealed the close connection between the M6A regulation of the Pan-PCD model in BLCA patients. A total of 7168 one-to-one pairs of PCD genes and M6A genes were identified. Additionally, a rich interaction network of 24 M6A regulators in BLCA patients was presented in Fig. 2F.
This research proposed a new signature based on M6A-associated PCDs. The risk score of this signature exhibited significant prognostic value for BLCA patients, which was verified by three datasets (TCGA-BLCA, GEO32548, and IMvigor210) through Kaplan–Meier (KM) analysis. Additionally, further analysis including time-dependent ROC, nomogram, Calibration curves, and DCA curves demonstrated the prognostic value of the signature with significant statistical significance. Moreover, the BLCA patients were clustered into Activated or suppressed Pan-PCD subgroups based on the Consensus clustering analysis of the signature’s 8 key genes. These two subgroups exhibited distinct immune components and functions, along with different responses to immunotherapy and drug sensitivity. These observations suggested that the activation or suppression of Pan-PCD might be the key factors accounting for the distinct responses of BLCA patients to immunotherapy and drug sensitivity.
The PI3K pathway is a crucial mechanism of Pan-programmed cell death in BLCA patients. This research has identified the PI3K pathway as a potential key mechanism of Pan-PCD in BLCA patients, which regulates four PCD models, including Apoptosis, Pyroptosis, Autophagy, and Lysosome-dependent cell death, as well as nine key PCD genes. In addition, a series of studies have shown that the PI3K pathway plays a role in multiple PCDs. For example, during necroptosis, activation of the PI3K/AKT pathway inhibits MLKL activation and necroptosis23. Additionally, activation of the PI3K/AKT pathway reduces the expression levels of caspase-9 and -3, which inhibit apoptosis24. Furthermore, in vitro and in vivo experiments have demonstrated that bladder cancer proliferation or invasion is promoted by activation of the PI3K pathway25,26. The correlation between the PI3K pathway and Apoptosis27 or Autophagy28 in ovarian cancer has also been verified. Therefore, there is an urgent need for further research to explore the potential hypothesis that the PI3K pathway via multiple PCDs promotes bladder cancer progression, and multiple PCDs exist and cross-regulate each other in BLCA.
This was the first step taken by the researcher’s team in understanding the Pan-PCD mechanism in BLCA patients. Future research will focus on three main areas: (1) The M6A-associated PCD signature contains 8 key genes, each with its upstream M6A regulators. The specific mechanism and function of the “M6A regulator-PCD gene-cell phenotype” pathway in bladder cancer will be deeply researched. (2) The results from the activated or suppressed Pan-PCD subgroups suggest a strong correlation among different PCD pathways, indicating a possible complex cross-regulation between them. The team will focus on understanding the specific functions and mechanisms of individual PCD pathways. (3) The PI3K pathway has been verified as a key mechanism regulating four different PCD pathways in BLCA patients. In the future, the PI3K pathway will be a focal point of research. Overall, this research provides a foundation for further exploring the role of PCDs in BLCA patients and opens up new avenues for potential therapeutic interventions.
Conclusion
This research has revealed that the Pan-PCD model, regulated by M6A, holds great promise as an insight for BLCA patients. A new signature derived from M6A-related Pan-PCD has been proposed, which carries prognostic value for survival and drug sensitivity. Furthermore, the PI3K pathway has been identified as a key mechanism in Pan-PCD for BLCA patients.
Data availability
The original data presented in this study are included in the article. All data were retrieved from public databases. This study utilized three bladder cancer datasets, including the TCGA-BLCA dataset, GEO32548 dataset, and IMvigor210 dataset. All three datasets contained comprehensive clinical information, prognostic outcomes, and mRNA expression profiles. The TCGA-BLCA data was acquired from the GDC website, while the GEO32548 dataset was obtained from the GEO website. The IMvigor210 dataset was retrieved using the R package “IMvigor210CoreBiologies”. All three datasets are freely accessible to any visitor for data downloading.
References
Ou, S. L. et al. Safety and efficacy of programmed cell death 1 and programmed death ligand-1 inhibitors in the treatment of cancer: An overview of systematic reviews. Front. Immunol. 13, 953761 (2022).
Peng, F. et al. Regulated cell death (RCD) in cancer: Key pathways and targeted therapies. Signal Transduct. Target. Ther. 7(1), 286 (2022).
Chen, J. et al. Novel insights into the interplay between m6A modification and programmed cell death in cancer. Int. J. Biol. Sci. 19(6), 1748–1763 (2023).
He, L. et al. Functions of N6-methyladenosine and its role in cancer. Mol. Cancer 18(1), 176 (2019).
Necchi, A. et al. Atezolizumab in platinum-treated locally advanced or metastatic urothelial carcinoma: Post-progression outcomes from the phase II IMvigor210 study. Ann. Oncol. 28(12), 3044–3050 (2017).
Zou, Y. et al. Leveraging diverse cell-death patterns to predict the prognosis and drug sensitivity of triple-negative breast cancer patients after surgery. Int. J. Surg. 107, 106936 (2022).
Garbo, S., Zwergel, C. & Battistelli, C. m6A RNA methylation and beyond—The epigenetic machinery and potential treatment options. Drug Discov. Today 26(11), 2559–2574 (2021).
Shen, J. et al. Prognostic significance and mechanisms of CXCL genes in clear cell renal cell carcinoma. Aging (Albany NY) 15(16), 7974–7996 (2023).
Wang, R. et al. Fatty acid metabolism-related signature predicts survival in patients with clear cell renal carcinoma. Aging (Albany NY) 14(24), 9969–9979 (2022).
Tan, L. et al. N6-methyladenosine-associated prognostic pseudogenes contribute to predicting immunotherapy benefits and therapeutic agents in head and neck squamous cell carcinoma. Theranostics 12(17), 7267–7288 (2022).
Song, M. et al. The advanced lung cancer inflammation index is the optimal inflammatory biomarker of overall survival in patients with lung cancer. J. Cachexia Sarcopenia Muscle 13(5), 2504–2514 (2022).
Zhang, B. et al. Construction and validation of a cuproptosis-related prognostic model for glioblastoma. Front. Immunol. 14, 1082974 (2023).
Yu, M. et al. TREM2 is associated with tumor immunity and implies poor prognosis in glioma. Front. Immunol. 13, 1089266 (2022).
Lv, Y. et al. Identification and characterization of aging/senescence-induced genes in osteosarcoma and predicting clinical prognosis. Front. Immunol. 13, 997765 (2022).
Hao, J., Zhang, W. & Huang, Z. Bupivacaine modulates the apoptosis and ferroptosis in bladder cancer via phosphatidylinositol 3-kinase (PI3K)/AKT pathway. Bioengineered 13(3), 6794–6806 (2022).
Zhu, S. et al. Synergistic antitumor activity of pan-PI3K inhibition and immune checkpoint blockade in bladder cancer. J. Immunother. Cancer 9(11), 22 (2021).
Bertheloot, D., Latz, E. & Franklin, B. S. Necroptosis, pyroptosis and apoptosis: An intricate game of cell death. Cell Mol. Immunol. 18(5), 1106–1121 (2021).
Newton, K. et al. Cleavage of RIPK1 by caspase-8 is crucial for limiting apoptosis and necroptosis. Nature 574(7778), 428–431 (2019).
Heneka, M. T. et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 493(7434), 674–678 (2013).
Liu, L. et al. Insights into N6-methyladenosine and programmed cell death in cancer. Mol. Cancer 21(1), 32 (2022).
Chen, J. et al. Epitranscriptomic m6A modification in the stem cell field and its effects on cell death and survival. Am. J. Cancer Res. 9(4), 752–764 (2019).
Jin, S. et al. m(6)A RNA modification controls autophagy through upregulating ULK1 protein abundance. Cell Res. 28(9), 955–957 (2018).
Liu, L. et al. 1,8-cineole alleviates bisphenol A-induced apoptosis and necroptosis in bursa of Fabricius in chicken through regulating oxidative stress and PI3K/AKT pathway. Ecotoxicol. Environ. Saf. 226, 112877 (2021).
Chen, Y., Wang, B. & Zhao, H. Thymoquinone reduces spinal cord injury by inhibiting inflammatory response, oxidative stress and apoptosis via PPAR-γ and PI3K/Akt pathways. Exp. Ther. Med. 15(6), 4987–4994 (2018).
Chi, M. et al. TEAD4 functions as a prognostic biomarker and triggers EMT via PI3K/AKT pathway in bladder cancer. J. Exp. Clin. Cancer Res. 41(1), 175 (2022).
Yu, H. et al. STIL promotes tumorigenesis of bladder cancer by activating PI3K/AKT/mTOR signaling pathway and targeting C-Myc. Cancers (Basel) 14(23), 5777 (2022).
Chen, Y. H. et al. Metformin induces apoptosis and inhibits migration by activating the AMPK/p53 axis and suppressing PI3K/AKT signaling in human cervical cancer cells. Mol. Med. Rep. 23(1), 1–1 (2021).
Zhou, J. et al. Tanshinone I attenuates the malignant biological properties of ovarian cancer by inducing apoptosis and autophagy via the inactivation of PI3K/AKT/mTOR pathway. Cell Prolif. 53(2), e12739 (2020).
Acknowledgements
We thank Home for Researchers editorial team (www.home-for-researchers.com) for language editing service.
Funding
This study was financially supported by the public welfare projects of Huzhou science and technology bureau (2022GZ62), and Zhejiang provincial natural science foundation (LTGY24H160039).
Author information
Authors and Affiliations
Contributions
W.R.J. wrote the draft manuscript; S.J.W. edited the manuscript and finished the experiments; L.Z.J. analyzed the data and finished the images. All authors revised the manuscript and approved the submitted version.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, R., Li, Z. & Shen, J. Predicting prognosis and drug sensitivity in bladder cancer: an insight into Pan-programmed cell death patterns regulated by M6A modifications. Sci Rep 14, 18321 (2024). https://doi.org/10.1038/s41598-024-68844-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-68844-3
- Springer Nature Limited