
Abstract
Cryopreservation is crucial for conserving genetic diversity in endangered species including the critically endangered group of sturgeons (Acipenseridae), but it can compromise sperm quality and protein profiles. Although cryopreservation with dimethyl sulfoxide (DMSO) and methanol (MeOH) results in the recovery of good post-thaw motility, DMSO-preserved sperm show reduced fertilization ability. This study was conducted in Siberian sturgeon as a model for Acipenserid fishes to explore the effects of DMSO and MeOH on the proteome of semen using advanced proteomics methods—liquid chromatography‒mass spectrometry and two-dimensional difference gel electrophoresis. We analyzed the proteomic profiles of fresh and cryopreserved spermatozoa and their extracellular medium and showed that cryopreservation decreases motility and viability and increases reactive oxygen species levels, membrane fluidity, and acrosome damage. Despite having similar post-thaw semen motility, sperm treated with DMSO had significantly lower fertilization success (6.2%) than those treated with MeOH (51.2%). A total of 224 and 118 differentially abundant proteins were identified in spermatozoa preserved with MeOH and DMSO, respectively. MeOH-related proteins were linked to chromosomal structure and mitochondrial functionality, while DMSO-related proteins impacted fertilization by altering the acrosome reaction and binding of sperm to the zona pellucida and nuclear organization. Additionally, cryopreservation led to alterations in the proacrosin/acrosin system in both cryoprotectants. This study provides the first comprehensive proteomic characterization of Siberian sturgeon sperm after cryopreservation, offering insights into how cryoprotectants impact fertilization ability.
Similar content being viewed by others


Introduction
Sturgeons, which belong to the family Acipenseridae, are ancient fish of significant ecological, evolutionary, and economic interest which face a dramatic decline of their populations worldwide. As a result, is the most highly threatened family of all vertebrates (IUCN 2011)1. Sturgeon are characterized by unique reproductive biology, including atypical testicular morphology and a distinct sperm maturation process. In addition, compared to those of teleost fish, the spermatozoa of sturgeon exhibit a complex structure with an acrosome, extended motility duration, and lower sperm concentration. The seminal plasma also has unique characteristics, such as lower osmolality and protein concentration2. These factors pose additional challenges for the control of sturgeon reproduction. Given these complexities, deciphering and enhancing sturgeon reproductive success has emerged as a pivotal approach for conservation and sustainable aquaculture advancement.
The cryopreservation of spermatozoa is a critical technique for the ex situ conservation of genetic resources, facilitating the preservation of genetic diversity and supporting the reproduction of these endangered species. This technique, which involves freezing sperm cells at ultralow temperatures, effectively pauses biological time, safeguarding genetic material for future controlled fertilization endeavors. Semen cryopreservation in sturgeons, as detailed by Alavi et al.3 and Kolyada et al.4, often employs extenders containing sucrose with KCl buffered by Tris–HCl or saline solutions enriched with permeating cryoprotectants such as dimethyl sulfoxide (DMSO), N,N-dimethylacetamide (DMA), ethylene glycol (EG), methanol (MeOH), or the nonpermeating cryoprotectant egg yolk5 to enhance post-thaw sperm viability and motility. However, the freeze‒thaw cycle entails sudden osmotic and temperature shifts, which detrimentally impact semen quality, resulting in reduced sperm viability, diminished motility, and low fertilization capacity3. Furthermore, cryopreservation may alter spermatozoa molecular integrity, impacting the composition and functionality of sperm proteins that are crucial for effective fertilization and early embryonic development.
DMSO and MeOH are widely used as cryoprotectants due to their ability to freely permeate cell membranes, which is facilitated by their low hydrophilicity and molecular weight. These properties enable them to interrupt ice crystal nucleation and growth by forming hydrogen bonds with water6. Although cryopreservation with DMSO and MeOH resulted in the recovery of good post-thaw motility in sturgeon7,8, the fertilization ability of DMSO-cryopreserved spermatozoa decreased, and these spermatozoa were characterized by higher amount of acrosome-reacted spermatozoa than MeOH-treated spermatozoa9. The exact nature of such disturbances is unknown and requires further investigation, especially regarding alteration in sturgeon-specific proteins. Such knowledge can potentially be important for developing recommendations concerning cryopreservation methods of sturgeon semen.
To explore the mechanism of cryodamage, proteomic techniques such as liquid chromatography‒mass spectrometry (LC‒MS) have been used to detect changes in sperm proteins of mammalian species10,11,12. In fish species, two-dimensional gel electrophoresis (2DE) and two-dimensional difference gel electrophoresis (2D-DIGE) have been used to analyze spermatozoa from common carp and sea bass13,14, as well as the extracellular medium (EM) from common carp and rainbow trout15,16, to monitor proteins released from sperm cells following cryopreservation. Only two studies have described the impact of cryopreservation on the sperm proteome in sterlet17,18. Despite the identification of a limited number of sturgeon proteins, these efforts underscore the need for further research to fully understand sturgeon sperm cryodamage mechanisms as a basis for the improvement of the method.
This study presents the hypothesis that cryopreservation with DMSO or MeOH differentially impacts the sperm proteome of the Siberian sturgeon, leading to specific molecular alterations. We also assumed that the DMSO variant would reveal changes in proteins critical for fertilization ability. The aim of this study was to bridge the gap in understanding sturgeon reproductive biology, the intricacies of sperm cryopreservation, and the roles of DMSO and MeOH as cryoprotectants. We used two complementary quantitative proteomic approaches, mass spectrometry-based (label-free LC‒MS) and gel-based (2D-DIGE) coupled with matrix-assisted laser desorption/ionization time of flight (MALDI-TOF/TOF) mass spectrometry and performed simultaneous analysis of spermatozoa and the EM.
Methods
Ethics statement
The Siberian sturgeons used for this study were sourced from a selectively bred broodstock at the Department of Sturgeon Fish Breeding in Pieczarki, Poland, in compliance with national aquaculture guidelines and veterinary requirements for activities in the aquaculture sector (veterinary identification number 28199201) and were under the supervision of the District Veterinary Officer. Our research adhered to the Polish Animal Protection Act (2023, Item 1580) and was exempt from additional ethics approval under the Polish Protection of Animals Used for Scientific Purposes Act (2015, Article 1.2, subparagraphs 1 and 5) https://isap.sejm.gov.pl/isap.nsf/download.xsp/WDU20230000465/T/D20230465L.pdf and aligned with Directive 2010/63/EU (Article 1, Paragraph 5) on animal protection for scientific purposes. All procedures followed the ARRIVE guidelines (https://arriveguidelines.org/).
Fish maintenance and gamete collection
Semen was collected from seven mature Siberian sturgeons (weighing 9.5 ± 2.9 kg and aged 7–14 years) maintained in the Department of Sturgeon Fish Breeding at the National Inland Fisheries Research Institute in Pieczarki, Poland. Twenty-four hours before semen collection, males were hormonally stimulated by an injection of 0.03 mg kg-1 of luteinizing hormone-releasing hormone (LH-RHa, Genscript, USA)19. Semen samples were collected via a syringe attached to a rigid tube inserted into the urogenital opening, ensuring no blood contamination. The semen was stored individually in open glass beakers at 8 °C.
Eggs were collected from two mature female sturgeons (aged 9 and 14 years, body weight 16.5 ± 2.7 kg) maintained in the Department of Sturgeon Fish Breeding at the National Inland Fisheries Research Institute. The females were hormonally stimulated with two doses totalling 0.1 mg kg-1 of LH-RHa (GenScript), which were administered 36 and 24 h prior to ovulation and egg collection20. Eggs were collected via a catheter21 in separate dry plastic containers for each female. Before gamete collection, the fish were anesthetized by spraying the gills with a Propiscin anesthetic (IFI, Poland) solution at a concentration of 15 mg L−1 (100 mL per fish).
Cryopreservation of semen
Semen samples were frozen individually (n = 7). The milt was diluted 1:1 with an extender containing 0.1 M glucose and 15% MeOH or 15% DMSO according to Judycka et al.19. This mixture was loaded into 250 μL plastic straws (IMV Technologies, L’Agile, France), which were placed on a floating rack (Minitübe GmbH, Tiefenbach, Germany). The straws were then frozen in liquid nitrogen vapor (3 cm above the liquid surface) for 5 min in a Styrofoam box with an isolating Neopor block (Minitübe GmbH) followed by placement in liquid nitrogen. Straws were thawed by immersion in a 40 °C water bath for 6 s and immediately used for the analysis of sperm quality parameters and proteomic analysis. The experimental design is shown in Fig. 1.

Experimental design and workflow for the proteomic analysis of cryopreserved Siberian sturgeon spermatozoa and extracellular medium using two complementary proteomic approaches: gel-based (2D-DIGE) and gel-free (label-free LC–MS) methods.
Measurement of semen quality parameters
Sperm motility parameters
The sperm movement characteristics were measured before and after cryopreservation via computer-assisted sperm analysis with the CEROS II system (Hamilton-Thorne, USA) using 12-well multitest slides (well diameter 4 mm, depth 30 μm; ICN Biomedicals Inc., Aurora, OH, USA). The semen was activated in 1:100 (for fresh semen) or 1:50 (for frozen/thawed semen) dilutions of activation solution (10 mM Tris, 20 mM NaCl and 2 mM CaCl2, 0.5% bovine serum albumin, pH 8.5)22 at 8 °C. The value for each sample represents the mean of two separate measurements of motility parameters of 200 spermatozoa. Eight representative motility parameters were chosen for further analysis: straight-line velocity (VSL, μm s−1), curvilinear velocity (VCL, μm s−1), average path velocity (VAP, μm s−1), linearity (LIN = 100 × VSL/VCL, %), amplitude of lateral head displacement (ALH, μm), percentage of motile sperm (MOT, %), beat-cross frequency (BCF, Hz) and progressive motility (PROG, %).
Flow cytometry analysis
Flow cytometry analyses of fresh and cryopreserved semen were performed using a Guava EasyCyte Flow Cytometer (Guava Technologies Inc., Luminex, Austin, TX, USA) cytometer. The data were acquired using GuavaSoft 3.2 software (Merck KGaA, Darmstadt, Germany). Sperm membrane fluidity, sperm viability, ROS+ levels and acrosomal membrane integrity were assessed. Calibration was performed using the EasyCyte Check Kit (Merck KGaA) according to the manufacturer's instructions.
Sperm concentration and viability
For the evaluation of sperm concentration and viability, semen was diluted 1:100 (for fresh semen) or 1:50 (for frozen/thawed semen) with immobilizing solution. The diluted semen was rediluted with ViaCount Reagent to obtain a final concentration of 3 × 105 cells per mL and measured according to the Guava ViaCount protocol (Luminex, USA).
Membrane fluidity
The spermatozoa membrane fluidity was assessed using a flow cytometric method incorporating both the lipophilic dye merocyanine-540 (M540) and the impermeable nucleic acid stain Yo-Pro-1 (YP), adapted from Guthrie et al.23. A stock solution of M540 (5 mM) was prepared in DMSO, and for analysis, a working solution was created at a 1:100 dilution in immobilizing buffer (1 L: 1 g NaCl, 0.2 g KCl, 0.05 g CaCl2, 0.05 g MgSO4, 0.15 g NaH2PO 4 × 2H2O, 0.38 g Na2HPO4 × 12H2O, 9.0 g glucose, pH 7.3–7.5)24. A 0.1 mM stock solution of YP in DMSO was diluted at a 1:10 ratio, and this diluted solution was added to 190 µL of the semen sample (1 × 106 cells per mL) and incubated for 10 min. at 37 °C. After this incubation, 5 µL of the M540 working solution was added to each sample, followed by a 2-min. incubation at 37 °C. The samples were then subjected to flow cytometry analysis, with green fluorescence (YP) detected using a 525-nm bandpass filter and red fluorescence (M540) detected using a 575-nm bandpass filter.
Oxidative stress (ROS-positive cell content)
Oxidative stress was measured using a portable flow cytometer (Muse Cell Analyzer, Millipore, Billerica, MA, USA). The ROS in the samples were measured using a Muse Oxidative Stress kit according to Mostek et al.25. Briefly, semen was diluted to a concentration of 1 × 107 spermatozoa per mL; then, 10 μL of the semen suspension was mixed with 190 μL of Muse Oxidative Stress Working Solution and incubated at 37 °C for 30 min. A positive control containing completely oxidized sperm cells was prepared by incubating sperm cells in 3% H2O2. The data were generated using the Muse Count and Viability Software Module (Millipore).
Acrosome integrity
The sperm acrosome status was assessed with the lectin PNA from Arachis hypogaea conjugated with Alexa Fluor® 488 (Life Technologies Ltd., Grand Island, NY, USA). The samples (3 × 105 cells per mL) were combined with 1 μL of PNA working solution (1 mg mL–1) and incubated for 5 min at room temperature in the dark. After incubation, 1 μL of propidium iodide (PI) (BD Biosciences, USA) was added before fluorescence analysis. The samples then underwent flow cytometry analysis, with green fluorescence (PNA) detected using a 515-nm bandpass filter, indicating acrosome-reacted spermatozoa and red fluorescence (PI) detected using a 617-nm bandpass filter, indicating dead spermatozoa.
Fluorescence microscopy analyses
The samples (1 × 106 cells per mL) were prepared as described above for the Acrosome Integrity Sect. “Acrosome integrity” with the same fluorochromes (PNA and PI). After staining, spermatozoa were fixed with 1% paraformaldehyde (PFA) for 5 min. Four microliters of the sperm suspension were placed on a clean glass slide and covered with a cover slip. The cells were observed under an Axio Observer Z1/7 fluorescence microscope (Carl Zeiss, Inc., Oberkochen, Germany) equipped with ZEN 2.3 blue edition software (Carl Zeiss).
Measurement of seminal plasma osmolality and protein concentration
Seminal plasma was obtained by centrifugation of fresh semen at 9000×g (4 °C, 10 min), and then the supernatant was centrifuged again at 10,000×g (4 °C, 10 min). Osmolality was measured using a Minitübe Abfüll-u Labortechnik Loser apparatus (Tiefenbach, Germany). The protein concentration was measured by the Bradford method using a Coomassie Plus Kit (Thermo Scientific, Rockford, IL, USA) with bovine serum albumin as a standard.
Fertilization
Fertilization was performed one year after semen cryopreservation using eggs collected from two mature females. Each variant of cryopreserved semen (n = 7 for each cryoprotectant) was used separately for each female. Immediately before fertilization, the straws were thawed in a water bath at 40 °C for 5 s. The fertilization procedure was conducted according to Judycka et al.19. Briefly, an adequate volume of frozen/thawed milt (100,000 spermatozoa/egg) was added to approximately 100 eggs, and then 5 mL of sperm activation solution (10 mM Tris, 20 mM NaCl and 2 mM CaCl2, pH 8.5) was added and left for 3 min. Next, the samples were washed with hatchery water. Egg samples were placed in separate baskets in an incubation apparatus. Fertilization rates were calculated as the percentage of hatched larvae after 7 days of incubation at 15 °C. All trials were replicated twice. An excess of fresh semen collected from two males was used exclusively to test the quality of the eggs.
Preparation of spermatozoa and extracellular medium for proteomic analysis
EM, which is defined as a fluid surrounding extended semen15,16, was obtained by centrifugation of fresh semen diluted 1:1 with immobilizing buffer (2.3.2.1) or semen cryopreserved with MeOH or DMSO at 9000×g (4 °C, 10 min) followed by a second centrifugation at 12,000×g (4 °C, 10 min).
The spermatozoa pellets were washed twice in immobilizing solution by centrifugation at 3000×g at 4 °C for 10 min. Spermatozoa proteins were extracted according to Dietrich et al.26 using lysis buffer (8 M urea, 2 M thiourea, 4% [w/v] 3-[(3-cholamidopropyl)-dimethylammonio]-1-propanesulfonate [CHAPS]), 0.1% (w/v) Triton X-100, 100 mM dithiotreitol (DTT), 2% (v/v) immobilized pH gradient (IPG) buffer (3–10 NL), and 2.5% (v/v) protease inhibitor cocktail (Sigma‒Aldrich, St. Louis, USA). Next, the samples were sonicated (5 s three times), kept on ice for 1 h, and centrifuged for 10 min at 14,000×g at 4 °C. The spermatozoa protein extracts were stored at − 80 °C until analysis.
Label-free LC‒MS
Protein digestion
Spermatozoa
The sperm protein extracts were processed using a Clean-Up Kit (GE Healthcare, Uppsala, Sweden) according to the manufacturer's protocol, and the pellets were reconstituted in 100 µL of lysis buffer containing 1% SDS, 6% trifluoroethanol and 100 mM ammonium bicarbonate buffer (ABC). After 15 min of sonication in an ultrasonic bath followed by 15 min of vortexing, the proteins were digested according to the FASP method as described in a previous publication with minor modifications27. In brief, the samples were vortexed for 60 min in 10 mM tris(2-carboxyethyl)phosphine (TCEP) at 60 °C to reduce cysteine residues and then were transferred onto a Vivacon 30 kDa molecular weight cutoff filter (Sartorius Stedim Biotech). Proteins were rinsed with urea solution (8 M urea in 100 mM ABC) before cysteine blocking with 25 mM chloroacetamide (CAA) for 30 min at room temperature. The filters were further washed three times with urea buffer and three times with ABC. After each addition, the samples were centrifuged for 30 min at 14,500 × g until the cutoff filter was dry. Digestion was performed overnight using 2.4 µg of trypsin (Promega GmbH) at 37 °C. Peptides were eluted from spin filters by two washes with ABC. Peptide concentrations were measured using a Pierce™ Quantitative Colorimetric Peptide Assay (Thermo Fisher Scientific). For analysis, 10 µg of peptide was suspended in 100 µL of Evosep solvent A (0.1% formic acid (FA)) in water.
EM
Three hundred microliters of each sample were subjected to protein precipitation and digestion. Proteins were precipitated by vortexing for 40 min with 80 µL of 90% trichloroacetic acid and 620 µL of acetonitrile (ACN). After centrifugation (30 min, 21,000×g, 4 °C), the protein pellets were washed three times with 1 mL of ice-cold ACN. Proteins were reconstituted in 40 µL of urea buffer (7.5 M urea, 10 mM tris(2-carboxyethyl)phosphine, 100 mM ABC) and vortexed for 60 min at 37 °C. Reduced cysteines were blocked by the addition of 30 mM methyl methanethiosulfonate (MMTS). Digestion was performed using 3 µg of trypsin/Lys-C mix (Promega GmbH) in two steps—30 min at 60 °C, followed by the addition of 250 µL of ABC buffer and overnight incubation at 37 °C. Peptide samples were acidified with FA to a final concentration of 0.1%.
Mass spectrometry
Spermatozoa and EM samples (n = 7 for each group) were measured using LC‒MS system composed of an Evosep One instrument (Evosep Biosystems) directly coupled to an Orbitrap Exploris 480 mass spectrometer (Thermo Fisher Scientific). One-third of the final sample volume was loaded onto disposable Evotips C18 trap columns (Evosep Biosystems) according to the manufacturer’s protocol. Chromatography was conducted at a flow rate of 220 nL min–1 using an 88 min (15 samples per day) gradient on an EV1106 analytical column (Dr Maisch C18 AQ, 1.9 m beads, 150 m ID, 15 cm long, Bruker). The data were acquired in positive mode with a data-dependent method using the following parameters. The MS1 resolution was set at 60,000 with a normalized AGC target of 300%, an auto maximum injection time and a scan range of 300 to 1600 mz. For MS2, the resolution was set at 15 000 with a standard normalized AGC target, auto maximum injection time and top 40 precursors within an isolation window of 1.6 mz considered for MS/MS analysis. Dynamic exclusion was set at 20 s with an allowed mass tolerance of 10 ppm, and the precursor intensity threshold was set at 5e3. Precursors were fragmented in HCD mode with a normalized collision energy of 30%. The spray voltage was set to 2.1 kV, the funnel RF level was 40, and the capillary temperature was 275 °C.
Data analysis
Two sets of calculations in Andromeda MaxQuant (version 2.1.1.0) were performed using Acipenser proteins derived from NCBI (version 13042023 for spermatozoa and version 10012023 for EM) supplemented with common contaminants. Label-free quantification (LFQ), along with the match between runs option, was enabled, and a target decoy strategy was used to determine the false discovery rate (FDR). Other search parameters were as follows: variable modification oxidation (M), fixed modification methylthio (C), enzyme trypsin, missed cleavages 2, PSM and protein FDR 0.01. The quantitative results from the global analysis were further analyzed in Perseus (version 1.6.15). Proteins identified only by site, contaminants and proteins from the reverse database were removed from the analysis. The intensity values were log2-transformed. Proteins with one or no quantitative value in one or more groups were treated separately.
2D-DIGE and protein identification by MALDI TOF/TOF
2D-DIGE
Two independent 2D-DIGE analyses were performed to compare the protein profiles of spermatozoa and EM before and after cryopreservation with different cryoprotectants (DMSO or MeOH; n = 7 for each group). Before analysis, the EM was concentrated using an Amicon ultracentrifuge filter with a cutoff of 3 kDa (Millipore). Aliquots containing approximately 800 μg of spermatozoa and EM proteins were processed using a Clean-Up Kit (GE Healthcare) according to the manufacturer's protocol. For each biological replicate, 50 μg of protein extract of each sample type (spermatozoa and EM from fresh and cryopreserved samples with MeOH or DMSO) was labeled with 400 pmol of Cy3 or Cy5 (GE Healthcare), with dye swaps to exclude dye bias. An internal standard was created by mixing equal amounts of each sample within the experiment and was labeled with Cy2. After incubation on ice and in the dark for 30 min, the reaction was terminated by the addition of 10 mM lysine. The three labeled samples were then combined within each experiment and diluted with rehydration buffer (8 M urea, 2% CHAPS, 18 mM dithiothreitol (DTT), 0.5% carrier ampholyte, pH 3–10 NL) to 340 μL. The combined samples were loaded on a pH gradient strip (24 cm, pH 3–10 NL) for isoelectric focusing (IEF) on an Ettan IPGphor system (Amersham Biosciences) as described by Dietrich et al.28. After IEF, the strips were first equilibrated in equilibration solution (50 mM Tris–HCl (pH 8.8), 6 M urea, 30% (v/v) glycerol, 2% (w/v) SDS, traces of bromophenol blue, and 1% (w/v) DTT) for 15 min and later in the same solution except that DTT was replaced by 4% (w/v) iodoacetamide for an additional 15 min. IPG strips were transferred onto 12.5% vertical polyacrylamide gels (with a gel size of 25.5 × 19.6 cm and a thickness of 1 mm) cast on low-fluorescence glass plates using an Ettan DALTsix system (GE Healthcare, Uppsala, Sweden). Electrophoresis was conducted overnight at a constant current of 1.5 W per gel. The Cy2-, Cy3-, and Cy5-labeled images were acquired on a Typhoon 9400 scanner (Amersham Biosciences) at excitation and emission wavelengths of 488/520, 532/580, and 633/670 nm, respectively. Intragel spot detection and quantification and Intergel matching and quantification were performed using differential in-gel analysis (DIA) and biological variation analysis (BVA) modules of DeCyder software version 6.5 (Amersham Biosciences) according to Dietrich et al.15.
MALDI-TOF/TOF protein identification
After 2D-DIGE analysis, the differentially abundant spots were manually cut off from the gels and prepared for digestion and protein identification using MALDI-TOF/TOF. Spots were digested and identified using a protocol described previously18. Briefly, MS and MS/MS spectra were searched using MascotServer (Matrix Sciences) in the Acipenser database, created on 05.07.2022 with the following MASCOT settings: cleavage enzyme, trypsin; max missed cleavages, 2; fragment ion mass tolerance, 0.7 Da; parent ion mass tolerance, 50 ppm; alkylation of cysteine by carbamidomethylation as a fixed modification; and oxidation of methionine as a variable modification. For the peptide mass fingerprinting (PMF) and MS/MS ion searches, statistically significant (p ≤ 0.05) matches identified by MASCOT were regarded as correct hits.
Functional classification of differentially abundant proteins (DAPs)
Ingenuity pathway analysis (IPA) (Qiagen, CA, US) of the identified proteins was used to interpret the identified proteins in the context of molecular and cellular functions and canonical pathways. The functional profiling of the identified proteins into three Gene Ontology (GO) categories, biological processes, cellular components, and molecular functions, was performed using ShinyGO (v. 0.8; http://bioinformatics.sdstate.edu/go/) and g:Profiler (https://biit.cs.ut.ee/gprofiler/gost, v. e111_eg58_p18_30541362). Protein–protein interaction network analysis of proteins associated with reproductive processes was performed using the Search Tool for the Retrieval of Interacting Genes (STRING) database (v. 12.0; http://string-db.org/) with a medium confidence score cutoff of 0.4. The search for interactions was restricted to Homo sapiens protein pairs. The reliability of the interactions between proteins was assessed by a combined score (edge score). Functional diagrams were visualized using SRplot tools (http://www.bioinformatics.com.cn/SRplot).
Western blot analysis
We used a western blot procedure using stain-free gels (V3 stain-free workflow, Bio-Rad, Hercules, CA, USA) to validate the mass spectrometry results, as previously described by Kodzik et al.29, with some modifications. This method eliminates the need for housekeeping proteins as loading controls for western blots30. The expression of seven proteins of interest (l-lactate dehydrogenase A chain (LDHA), creatine kinase B-type (CKB), enolase 3 (ENO3), fructose-bisphosphate aldolase A (ALDOA), histone (H2A.Z), mitochondrial fumarate hydratase (FH) and acrosin (ACR)) were evaluated. Equal amounts of protein (10 μg for LDHA, CKB, ENO3, ALDOA, and FH and 20 μg for H2A.Z and ACR in spermatozoa and 7 μg for LDHA, ENO3, and ALDOA, 10 μg for CKB, H2A.Z, and FH and 9 μg for ACR in EM) were applied to Mini-Protean TGX Stain-Free 4–20% gels (Bio-Rad, Hercules, CA, USA). The quality of protein separation was checked after gel activation on a ChemiDoc imager (Bio-Rad), and proteins were then transferred to nitrocellulose membranes (0.22 µm) using a Mini Trans Biol Cell (Bio-Rad) in 20 mM Tris–HCl (pH 8.2), 150 mM glycine, and 10% MeOH at 60 V for 90 min (4 °C). Nitrocellulose membranes were briefly rinsed in distilled water and blocked with 5% nonfat dried milk. Next, the membranes were incubated overnight at 4 °C with primary polyclonal antibodies against LDHA, CKB, ENO3, ALDOA, FH, H2A.Z and ACR. Details of the dilution of the tested antibodies are provided in Supplementary Table S1. After the membranes were rinsed to remove unbound primary antibodies, they were exposed to goat anti-rabbit antibodies (Sigma‒Aldrich) linked to alkaline phosphatase. The products were visualized by incubation in a solution of alkaline phosphate buffer containing nitro blue tetrazolium (Sigma‒Aldrich) and 5-bromo-4-chloro-3-indolyl phosphate (Sigma‒Aldrich) in the dark. The staining was stopped with 0.2 M EDTA. Antibody-bound proteins were detected by enhanced chemiluminescence using a ChemiDoc imaging system (Bio-Rad). The optical density of the protein bands detected on the membranes and the intensity of the protein bands on the TGX Stain-Free gels were analyzed using Image Lab 6 software (Bio-Rad). The image of the gel acquired before its transfer was used as a control for equal protein loading. The volume density of each target protein band was normalized to its respective total protein content, whereas the total protein band density was normalized to the total protein loaded into each lane using stain-free technology, with the data expressed in arbitrary units according to the manufacturer’s instructions (Bio-Rad) and Posch et al.30.
Statistical analysis
One-way ANOVA followed by Tukey’s test for post hoc comparison of means was used for the analysis of sperm quality parameters and western blotting. To analyze the differences in the hatching rates of semen cryopreserved with MeOH and DMSO for each female, two-way ANOVA with a Šidák post hoc test was used. The percentage data were normalized using an arcsine square root transformation prior to the statistical procedures. The results are presented as the mean ± standard deviation (SD). All the statistical analyses were performed at a significance level of p < 0.05 using GraphPad Prism software 9.0.0 (GraphPad Software Inc., San Diego, CA, USA).
Statistical analysis of changes in protein abundance after 2D-DIGE and label-free LC‒MS was performed using the BVA module of DeCyder Differential In-Gel Analysis v. 5.02 and Perseus software (version 1.6.15.0), respectively. Protein spots with a p value < 0.05 according to one-way ANOVA, which indicated an increase or decrease in relative intensity, were considered DAPs. For LC‒MS analysis of spermatozoa, paired t tests with Benjamini‒Hochberg correction for multiple testing were performed to compare the groups, with significance assigned to protein peptides with q values less than 0.05. For EM, two-sample t tests with permutation-based FDR were performed on proteins with at least three valid values in both tested groups.
Principal component analysis (PCA) and heatmaps for 2D-DIGE data of the protein abundance change between samples were generated using DeCyder Differential Analysis Software (v 5.0; GE Healthcare) in the BVA module, whereas LC‒MS data were generated using Perseus software (version 1.6.15).
Results
Parameters of fresh semen
The quality parameters of individual Siberian sturgeon semen samples, detailed in Supplementary Table 2, demonstrated high sperm motility (> 80%) and viability (> 96%). The sperm concentration varied between 0.85 and 2.50 × 109 spermatozoa mL–1, with seminal plasma osmolality ranging from 81 to 102 mOsm kg–1 and a protein concentration of 219.35 ± 72.80 µg mL–1.
Impact of cryopreservation with MeOH and DMSO on sperm quality parameters
Sperm movement characteristics
Cryopreservation significantly reduced MOT, VCL and PROG, regardless of the cryoprotectant used (Fig. 2). A decrease in ALH was observed for DMSO-cryopreserved semen, while the BCF increased in MeOH-cryopreserved semen compared to fresh semen. These changes in ALH and BCF were significantly different when comparing semen cryopreserved with MeOH to that cryopreserved with DMSO. Other motility parameters did not differ between MeOH- and DMSO-cryopreserved semen.

Impact of cryopreservation with the use of an extender containing either MeOH or DMSO on sperm movement parameters, including motility (MOT), curvilinear velocity (VCL), straight-line velocity (VSL), linearity (LIN), average path velocity (VAP), amplitude of lateral head displacement (ALH), beat cross frequency (BCF), and progressive motility (PROG). The results are expressed as the mean ± SDs. Different superscripts indicate significant differences (p < 0.05) between groups.
Flow cytometry parameters
Membrane fluidity
Flow cytometry analysis using M540 and YP staining revealed significant changes in sperm membrane fluidity between fresh and cryopreserved semen with DMSO and MeOH (Fig. 3A). The proportion of live sperm with destabilized membranes (YP–/M540+) increased from 1.5 ± 0.4% in fresh semen to 10.1 ± 4.1% and 12.3 ± 5.2% in semen cryopreserved with MeOH and DMSO, respectively, with DMSO treatment showing a more pronounced increase. Similarly, the percentage of live sperm with intact membranes (YP–/M540–) decreased from 90.7 ± 5.3% in the fresh samples to 29.8 ± 7.3% and 35.4 ± 8.3% in the MeOH- and DMSO-cryopreserved samples, respectively.

Flow cytometry evaluation of sperm membrane fluidity (M540/YP staining) (A), viability (Guava ViaCount reagent) (B) and ROS+ levels (C) in fresh and cryopreserved semen with DMSO and MeOH. (A) Representative plots showing viable sperm with intact membranes (YP-/M54–, lower left quadrant), viable sperm with destabilized membranes (YP-/M54+, lower right quadrant), dead sperm with intact membranes (YP+/M54–, upper left quadrant) and dead sperm with destabilized membranes (YP+/M54+, upper right quadrant). (B) Plots showing live cells in the upper-left quadrant of the first plot and in the lower-right corner of the second plot. (C) Histogram showing spermatozoa in the ROS- (marker M1) and ROS+ (marker M2) groups. Different superscripts indicate significant differences (p < 0.05) among groups. The data are presented as the means ± SDs. YP-Yo-Pro-1, M540- merocyanine 540.
Sperm viability
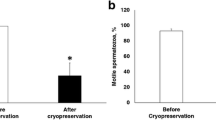
Cryopreservation resulted in a significant reduction in sperm viability, decreasing from 97.9% ± 1.3% in fresh semen to 61.1% ± 3.8% and 70% ± 3.2% in cryopreserved semen with MeOH and DMSO, respectively (Fig. 3B). Notably, the viability of sperm cryopreserved in DMSO was significantly greater than that of those cryopreserved in MeOH.
ROS+ level
Cryopreservation increased the percentage of sperm undergoing oxidative stress, with the proportion increasing from 10.7% ± 2.5% in fresh semen to 23.1% ± 8.6% and 23.7% ± 7.7% in cryopreserved semen with MeOH and DMSO, respectively (Fig. 3C). No significant differences in ROS production were detected between MeOH- and DMSO-cryopreserved semen.
Acrosome integrity
Flow cytometric analysis of acrosome membrane integrity for fresh and cryopreserved semen is shown in Fig. 4. Briefly, the proportion of live sperm with intact acrosomes (PI-/PNA-) decreased from 96.5 ± 2.3% in fresh semen to 57.5 ± 4.1% and 68.8 ± 7.5% in semen cryopreserved with MeOH and DMSO, respectively. Similarly, the percentage of live sperm with damaged acrosomes (PI-/PNA+) increased from 0.3 ± 0.5% in the fresh samples to 0.65 ± 0.54% and 1.00 ± 0.66% in the MeOH– and DMSO-cryopreserved samples, respectively (Fig. 4C). The percentage of dead spermatozoa with damaged acrosomes (PI+/PNA+) significantly increased from 0.3 ± 0.2% in the fresh samples to 24.8 ± 3.4% and 21.8 ± 4.7% in the MeOH- and DMSO-cryopreserved samples, respectively. No significant changes were observed between cryopreserved samples in any group (Fig. 4C). Representative flow cytometry plots and fluorescence microscopy images are shown in Fig. 4A and B, respectively. The number of spermatozoa with reacted acrosomes was negatively correlated with the variability of fresh and cryopreserved spermatozoa (Fig. 4D).

Flow cytometry analysis (PNA/PI labeling) of acrosome membrane integrity in fresh and cryopreserved spermatozoa treated with MeOH and DMSO. (A) Representative flow cytometry plots showing viable acrosome intact (PI–/PNA–, lower left quadrant), viable acrosome ruptured (PI–/PNA+, lower right quadrant), dead acrosome intact (PI+/PNA–, upper left quadrant) and dead acrosome-ruptured (PI+/PNA+, upper right quadrant) spermatozoa. (B) Fluorescence microscopy images showing unstained live sperm (indicated by bold-headed arrows for spermatozoa with intact acrosomes and asterisks for spermatozoa with damaged acrosomes) and dead sperm stained in red (indicated by fine-headed arrows for spermatozoa with intact acrosomes and arrowheads for spermatozoa with damaged acrosomes). (C) Graphs and (D) correlation between sperm viability and acrosome-reacted sperm in fresh and cryopreserved semen. Different superscripts indicate significant differences (p < 0.05) among groups (n = 7 in each group). The data are presented as the means ± SDs. PI propidium iodide; PNA peanut agglutinin.
Hatching rate
The use of semen cryopreserved with DMSO resulted in a significantly lower hatching rate (0.8 ± 1.6% for female #1 and 11.6 ± 13.3% for female #2) compared to semen cryopreserved with MeOH (35.5 ± 8.5% for female #1 and 66.9 ± 18.1% for female #2) for both females (p < 0.0001; Fig. 5). Significant differences in hatching rates were also observed among females under the same cryoprotectant conditions (p < 0.05). Fresh semen, used as a control to assess egg quality, achieved a hatching rate of 57% for female #1 and 75% for female #2.

Effects of the cryoprotectants MeOH and DMSO on the hatching rate of two females. The data are presented as the means ± SDs (n = 7). Different superscripts indicate significant differences (p < 0.0001) among the cryoprotectants for female #1 (a, b) and female #2 (x, y). The hatching rates for fresh semen were 57% for female #1 and 75% for female #2. Asterisks (*) indicate significant differences (p = 0.0052) between females under the same cryoprotectant conditions.
Label-free LC‒MS and 2D-DIGE identification of DAPs in spermatozoa and EM of fresh and cryopreserved semen with MeOH or DMSO
Spermatozoa
Using label-free LC‒MS, we identified 1109 proteins from at least four biological replicates across the fresh, CryoMeOH, and CryoDMSO groups. Among these, 160 proteins exhibited differential abundance in the comparison of MeOH vs. fresh groups, and 43 proteins were differentially abundant when comparing between CryoDMSO and fresh groups. A comparison between CryoMeOH and CryoDMSO spermatozoa identified four DAPs. The putative methyltransferase DDB_G0268948, which does not have specific human homologs, was identified in the MeOH group compared with the fresh group. These findings are detailed in Supplementary Table S3.
Further analysis employing the 2D-DIGE approach revealed significant changes in the abundance of spermatozoa proteins, with 163 and 144 protein spots altered following cryopreservation in the MeOH and DMSO groups, respectively, out of 2,469 matched spots. These corresponded to 93 and 86 unique proteins, with 35 and 28 proteins identified in multiple protein spots, likely representing proteoforms varying by isoelectric point (pI) and/or molecular weight (MW). Notable examples include pyruvate kinase PKM (PKM) (identified in 4 spots for MeOH and 8 for DMSO), LDHA (3 spots for MeOH and 4 for DMSO), glyceraldehyde-3-phosphate dehydrogenase, testis-specific (GAPDHS) (4 spots across both groups), phosphoglucomutase-1 (PGM1) (5 spots for MeOH and 3 for DMSO), ENO3 (4 spots for MeOH and 2 for DMSO) and puromycin-sensitive aminopeptidase (NPEPPS) (4 spots for MeOH and 3 for DMSO). Comparison of CryoDMSO and CryoMeOH groups revealed 19 DAPs. These findings are detailed in Supplementary Table S4, with representative 2D-DIGE images provided in Supplementary Figs. S1 and S2.
Additionally, 8 proteins (actin, cytoplasmic 1 (ACTB), actin-related protein T3 (ACTRT3), mitochondrial hydroxyacyl-coenzyme A dehydrogenase (HADH), muscle glycogen phosphorylase (PYGM), ropporin-1A (ROPN1), septin-11 (SEPTIN11), sperm acrosome-associated protein 9 (SPACA9) and tubulin beta-4B chain (TUBB4B)) in MeOH vs. fresh samples and 6 proteins (ACTB, isoaspartyl peptidase/l-asparaginase (ASRGL1), calcium-binding tyrosine phosphorylation-regulated protein (CABYR), PYGM, ROPN1L and SEPTIN11) in DMSO vs. fresh samples were identified as both up- and downregulated. These findings are detailed in Supplementary Fig. S3, with representative 2D-DIGE images.
The top downregulated proteins following cryopreservation with MeOH included vimentin (VIM), cilia- and flagella-associated protein 96 (CFAP96), peptidyl-prolyl cis–trans isomerase-like 3 (PPIL3), histone H2A.Z (H2A.Z) and ciliary microtubule-associated protein 2 (LEXM) via LC‒MS and CABYR, PYGM, TUBB4B, phosphoglucomutase-1 (PGM1) and ENO3 via 2D-DIGE, whereas nucleoporin nup85 (NUP85), H2A.Z1, acyl-CoA-binding protein (DBI), arylsulfatase A (ARSA) and leucine-rich repeat-containing protein 37A (LRRC37A) (LC‒MS method) and CABYR, PYGM, glycogen debranching enzyme (AGL), plasma kallikrein (KLKB1) and transmembrane protease serine 13 (TMPRSS13) (2D-DIGE method) were predominantly altered after cryopreservation with DMSO. A summary of the top significantly altered proteins is presented in Fig. 6.

List of the top proteins identified as downregulated in spermatozoa and upregulated in the EM for MeOH and DMSO, as determined by LC/MS and 2D DIGE analysis.
Combining the results from 2D-DIGE and LC–MS/MS yielded 224 and 118 DAPs in cryopreserved semen with MeOH and DMSO, respectively, compared to fresh semen, highlighting the proteomic alterations induced by cryopreservation (Fig. 7A–C).

Diagrams representing DAPs of spermatozoa (A–C) and upregulated proteins in the extracellular medium (EM) (D–F) following cryopreservation with MeOH or DMSO identified by two complementary approaches: LC‒MS and 2D-DIGE. Venn diagram illustrating the overlap of sperm leakage proteins identified as downregulated proteins in spermatozoa and upregulated proteins in the EM following cryopreservation with MeOH (G) and DMSO (H).
Extracellular medium
Label-free LC‒MS analysis of the EM revealed 1,900 proteins, with 330 and 349 proteins significantly upregulated following cryopreservation with MeOH and DMSO, respectively. Comparison of CryoDMSO and CryoMeOH samples revealed 11 DAPs. The detailed identifications are provided in Supplementary Table S5. The upregulation of proteins in the EM suggested protein leakage from the post-cryopreservation sperm. The 2D-DIGE analysis corroborated these findings, identifying 116 and 131 upregulated protein spots following cryopreservation in the MeOH and DMSO groups, respectively, out of 1,529 matched spots. These spots corresponded to 56 and 47 unique proteins, with 24 and 23 identified in multiple spots, implying proteoform diversity. Prominent among them were ENO3 (identified in 9 spots for MeOH and 10 for DMSO), LDHA (9 spots in both groups), PGK1 (6 spots for MeOH and 9 for DMSO), GAPDHS (8 spots in both groups), PKM (7 spots for MeOH and 8 for DMSO) and ALDOA (7 spots in both groups). Comparison of CryoDMSO and CryoMeOH groups revealed 4 DAPs. Representative 2D-DIGE images and detailed spot identifications are provided in Supplementary Figs. S4 and S5 and Supplementary Table S6, respectively.
The top upregulated proteins following cryopreservation in the EM included 60 kDa mitochondrial heat shock protein (HSPD1), mitochondrial alpha-aminoadipic semialdehyde synthase (AASS), mitochondrial isocitrate dehydrogenase [NADP] (IDH2), 2-oxoglutarate dehydrogenase complex component E1 (OGDH) and FH (LC‒MS method) and T-complex protein 1 subunit zeta (CCT6); ACTRT3, FH, mitochondrial creatine kinase U-type (CKMT1A) and ALDOA (2D-DIGE method) for the MeOH group; and HSPD1, AASS, protein disulfide-isomerase A3 (PDIA3), argininosuccinate synthase (ASS1), high affinity 3',5'-cyclic-AMP phosphodiesterase 7A (PDE7A) (LC‒MS method) and ACTRT3, CCT6, CKMT1A, profilin-3 (PFN3), and PKM (2D-DIGE method) for the DMSO group. Figure 6 presents a summary of the top significantly altered proteins identified in EM.
Overall, the integration of 2D-DIGE and LC–MS/MS facilitated the identification of 342 and 363 upregulated proteins in the EM from cryopreserved semen with MeOH and DMSO, respectively, compared to those in fresh semen (Fig. 7D–F).
Overlap of sperm leakage proteins identified by analysis of spermatozoa and EM following cryopreservation
The comparative analysis between downregulated proteins in spermatozoa and upregulated proteins in the EM after cryopreservation revealed a subset of overlapping proteins that were concurrently downregulated in spermatozoa and upregulated in the EM. Specifically, 77 proteins in the MeOH group and 53 proteins in the DMSO group exhibited this pattern, indicating an overlap of sperm leakage proteins due to cryopreservation effects (Fig. 7G,H).
Principal component analysis
PCA and hierarchical clustering of spermatozoa and the EM proteome after 2D-DIGE analysis revealed distinct clustering patterns, with fresh samples segregating from those cryopreserved with MeOH and DMSO. Specifically, PC1 accounted for 50.9% of the variance in the spermatozoa and 68.2% in the EM, whereas PC2 explained an additional 13.5% and 12.6% of the variation in the spermatozoa and EM, respectively (Fig. 8A,C). Heatmaps further illustrate the differences in the proteomes of fresh and cryopreserved semen (Fig. 8B,D). Additional PCA score plots and heatmaps derived from the LC‒MS data are provided in Supplementary Fig. S6.

PCA score plots and heatmaps of the 2D-DIGE data acquired for fresh (in gray) and cryopreserved (in blue; CryoMeOH, in green; CryoDMSO) spermatozoa (A,B) and extracellular medium (EM, C,D). PC1 (x-axis) was 50.9%; PC2 (y-axis) was 13.5% for spermatozoa. PC1 (x-axis) was 68.2%, and PC2 (y-axis) was 12.6% for the EM. The heatmap code is presented, with red areas representing greater amounts of protein and green areas representing less protein.
Functional insights into spermatozoa proteins altered by cryopreservation with MeOH and DMSO
For the combined set of proteins associated with sperm leakage (downregulated proteins in spermatozoa and upregulated proteins in the EM, totaling 376 proteins in the MeOH group and 379 in the DMSO group), key biological processes included metabolic processes, protein folding, positive regulation of the establishment of protein localization to telomeres, binding of sperm to the zona pellucida, and cellular detoxification (Fig. 9A) for both groups. In addition, proteins altered by MeOH were involved in protein export from the nucleus, whereas DMSO-altered proteins participated in ubiquitin protein ligase binding, nucleocytoplasmic transport, and nuclear pore organization. The molecular function annotation confirmed a strong association with binding and catalytic activity for both groups (Fig. 9A). IPA further revealed a significant association between cell viability, glycolysis and gluconeogenesis and the levels of sperm leakage proteins in both the MeOH and DMSO groups. The top ten IPA biological processes related to leakage proteins are shown in Table 1.

Gene ontology (GO) analysis of biological process (BP), cellular component (CC) and molecular function (MF) for sperm leakage proteins (downregulated proteins in spermatozoa and upregulated proteins in the EM (A) and upregulated proteins in spermatozoa (B)) after cryopreservation with MeOH and DMSO.
The sperm leakage proteins, regardless of the cryoprotectant used, were localized to extracellular vesicles, the proteasome complex, mitochondria, nuclear pores, sperm flagella and the ZP receptor complex (Fig. 9A). DMSO-altered proteins also localized to the cAMP-dependent protein kinase complex. All functional analyses are shown in Supplementary Table S7.
GO analysis revealed that the upregulated sperm proteins, regardless of the cryoprotectant used, were involved in biological processes such as microtubule-based movement, nucleoside triphosphate biosynthetic processes and cilium organization. In the MeOH group, proteins involved in energy derivation by the oxidation of organic compounds, left/right pattern formation, transmembrane transport and ventricular system development were identified. Conversely, upregulated proteins in spermatozoa following cryopreservation with DMSO were enriched in cellular respiration, regulation of brood size, spermatid development and the carboxylic acid metabolic process. GO molecular function annotations indicated that most proteins were associated with microtubule motor activity and metabolism in both groups (Fig. 9B). Localization analysis revealed that the upregulated sperm proteins in the DMSO group were primarily located in cilia and mitochondria, whereas in the MeOH group, they were localized to the axoneme, motile cilia, the microtubule cytoskeleton and the inner mitochondrial membrane protein complex. Functional analysis of the upregulated proteins in cryopreserved spermatozoa treated with MeOH or DMSO is shown in Supplementary Table S8.
Functional annotation of sperm leakage proteins uniquely altered by cryopreservation with MeOH and DMSO
A comparison of sperm leakage proteins following cryopreservation with MeOH and DMSO revealed 36 proteins uniquely altered by MeOH and 39 proteins uniquely altered by DMSO, with a shared group of 340 proteins commonly affected by both cryoprotectants (Fig. 10).

Venn diagram illustrating the common and distinct sperm leakage proteins following cryopreservation with MeOH and DMSO, along with functional annotation of proteins uniquely altered by each cryoprotectant. Functional analysis was performed using ShinyGO (v0.80; http://bioinformatics.sdstate.edu/go/).
ShinyGO analysis revealed that sperm leakage proteins unique to MeOH were predominantly involved in small molecule metabolic processes, organic acid catabolic processes, fatty acid catabolic processes, cytoskeleton organization and microtubule-based processes. In contrast, DMSO-specific alterations involved mRNA transport and fertilization processes, among others. Functional categorization revealed that the MeOH-specific proteins were largely related to oxidoreductase activity, hydrolase activity acting on acid anhydrides and magnesium ion binding, while the DMSO-specific proteins included structural constituents of the nuclear pore, arylsulfatase activity, calmodulin-dependent protein kinase activity, and ATP-dependent protein folding chaperones. Localization analysis revealed the mitochondrial matrix and microtubules for MeOH-specific proteins, whereas DMSO-specific proteins were primarily found in the BBSome, secretory granules, primary lysosomes and nuclear pores (Fig. 10).
The protein‒protein interaction networks identified via the STRING database illustrated distinct clusters related to metabolic processes and the microtubule cytoskeleton for MeOH (Fig. 11A) and transport processes, fertilization, and kinase activity for DMSO (Fig. 11B). Functional analysis of sperm leakage proteins uniquely altered by cryopreservation in MeOH or DMSO is shown in Supplementary Table S9.

Protein‒protein interaction networks for sperm leakage proteins uniquely altered by cryopreservation with MeOH and DMSO. The nodes correspond to the proteins, and the edges represent the interactions (thick lines indicate a high score > 0.9; thin lines indicate a medium score > 0.7). Model statistics and an explanation of edge colors are presented on the left.
Validation of proteomic results by western blotting
To confirm the proteomic findings, seven proteins (LDHA, CKB, ENO3, ALDOA, H2A.Z, FH and ACR) were selected for validation by western blotting in spermatozoa and EM. In details, in spermatozoa, LDHA and H2A.Z decreased in abundance following cryopreservation with MeOH and DMSO, while CKB abundance decreased only in the MeOH group (Fig. 12A). Although no significant differences in protein levels were found for ENO3, ALDOA or FH, the changes in protein expression agreed with the 2D-DIGE results. On the other hand, the abundance of all proteins increased in the EM after cryopreservation in the MeOH and DMSO groups (Fig. 12B). In fresh and cryopreserved spermatozoa, anti-acrosin antibodies identified two protein bands: a predominant band at 38 kDa and a less pronounced band at 24 kDa (see Fig. 13A). Cryopreservation did not alter the intensity of these bands (Fig. 13B). On the other hand, in fresh EM samples, one predominant band of 73 kDa was recognized using anti-acrosin antibodies. After cryopreservation, 3–5 additional bands with molecular weights ranging from 53 to 24 kDa were detected, which varied among individuals (Fig. 13A). A decrease in the 73 kDa band corresponding in our opinion to proacrosin was observed following cryopreservation with DMSO and MeOH. In contrast, an increase in bands of lower molecular mass (38, 33, 24 kDa) was observed after cryopreservation (Fig. 13C). The band intensities at 73 and 38 kDa differed between the DMSO and MeOH groups.

Immunoblotting validation of l-lactate dehydrogenase A chain (LDHA), creatine kinase B-type (CKB), enolase 3 (ENO3), fructose-bisphosphate aldolase A (ALDOA), histone (H2A.Z) and mitochondrial fumarate hydratase (FH) and acrosin (ACR) in fresh and cryopreserved spermatozoa (A) and the extracellular medium (B). The full-length blots are presented in Supplementary Figs. S7 and S8. The intensity of the protein bands on the TGX Stain-Free gels was analyzed using Image Lab 6 software (Bio-Rad).

Validation of acrosin (ACR) immunoblotting between fresh and cryopreserved spermatozoa with DMSO and MeOH. Western blot profile (A), graph of the relative intensity of acrosin bands of different molecular masses in spermatozoa (B) and extracellular medium (C). The results are expressed as the means ± SDs. Different letters indicate statistical significance at a p value ≤ 0.05, n = 7 per group. The full-length blots are presented in Supplementary Figs. S7 and S8.
Discussion
This study represents the first comprehensive examination of the alterations in the proteome of Siberian sturgeon spermatozoa resulting from cryopreservation, delineating the distinct effects of DMSO and MeOH cryoprotectants on sperm fertilization. Our findings contribute to a deeper understanding of the molecular mechanisms leading to a decrease in sperm quality following cryopreservation, particularly by identifying significant alterations in proteins essential for flagellar motility, energy metabolism, nuclear pore organization and fertilization processes. Notably, we identified proteins distinctly affected by DMSO and MeOH, underscoring the cryoprotectant-specific mechanisms potentially responsible for the observed differences in sperm fertilization ability (Fig. 14).

Consistent with previous studies on sturgeons, we observed a decrease in sperm motility parameters (MOT, VCL, ALH, and PROG) and viability following cryopreservation9,19, which was further corroborated by increased ROS production, a known factor in semen quality degradation31. Using flow cytometry, we observed a significant increase in the percentage of dead spermatozoa with reacted acrosomes following cryopreservation, reaching 22–24%, without notable differences between the cryoprotectants used. This increase correlated with elevated percentages of nonviable sperm. The proportion of viable spermatozoa with reacted acrosomes, while remaining low (0.3% in fresh samples), exhibited notable changes due to cryopreservation (approximately 1%), irrespective of the cryoprotectant used. In contrast, Lahnsteiner et al.32 reported no change in acrosome-reacted sperm after cryopreservation in sterlets, ranging from 15 to 19% for both fresh and cryopreserved semen with MeOH and DMSO, as measured under a phase contrast microscope through acrosomal filament determination. On the other hand, Psenicka et al.9 reported that DMSO specifically induced acrosome damage (12%) in sterlet semen, whereas MeOH maintained acrosome integrity comparable to that of fresh semen (5–8%), as determined by microscopic observation with SBTI-Alexa488 staining. These disparities underscore the variability in cryopreservation outcomes, likely due to variations in species and methods used for assessing acrosome status. Our results indicated that despite similar motility parameters, membrane fluidity, and acrosome status between the two cryoprotectants and greater viability in the DMSO group, we observed a marked decrease in the fertilization success of semen cryopreserved with DMSO compared to MeOH, which aligns with the findings of a previous study on sterlet and paddlefish32,33,34 and was explained by acrosomal reaction activation after cryopreservation. Our results suggest that standard sperm quality parameters may not adequately predict fertilization potential after cryopreservation, highlighting the significant role of molecular factors, such as protein composition, in determining fertilization success.
We utilized two complementary methods, gel-based and MS-based methods, to maximize the number of proteins altered due to cryopreservation. This combined approach identified more proteins than did LC‒MS or 2D-DIGE alone. The complementary nature of 2D-DIGE and LC-based methods has been shown in other proteomics studies35. The observed low overlap of differential proteins between the methods could result from their inherent strengths and analytical bases36. 2D-DIGE separates intact proteins based on pI and Mw, allowing visualization of isoforms and posttranslational modifications (PTMs). This method excels in distinguishing subtle protein variations but overall has lower sensitivity and a limited dynamic range. MS-based methods separate peptides based on hydrophobicity, thus detecting low-abundance proteins more effectively. However, MS-based techniques may struggle to differentiate between isoforms and modifications without additional targeted analyses. These fundamental differences in separation principles and detection capabilities have led to the identification of distinct sets of differentially expressed proteins by each approach.
Our results revealed that, unlike MeOH, DMSO selectively altered 39 sturgeon sperm proteins during cryopreservation. This unique subset of proteins, which diverges from the 340 proteins altered by both cryoprotectants, highlights the distinct impact of DMSO on sperm function and can be vital for understanding the loss of sperm fertilization ability when DMSO was used. Remarkably, a substantial majority of these proteins (33 proteins) were involved in fertilization processes, especially in the acrosome reaction, binding of sperm to the zona pellucida, nuclear pore organization, and the flagellum/centrosome structure. Most importantly, DMSO-induced changes shed new light on proteins important for sperm fertilization ability. This knowledge is new for sturgeons and is potentially important for better understanding how cryoprotectants influence sperm functionality and fertilization success.
Cryopreservation with DMSO led to alterations in acrosin (ACR), acrosin binding protein (ACRBP) and other proteins essential for the acrosome reaction (Arf-GAP domain and FG repeat-containing protein 1 (AGFG1), Rab GDP dissociation inhibitor beta (GDI2) and lysosomal Pro-X carboxypeptidase (PRCP)), which highlights the unique effect of DMSO on this crucial fertilization step. Unique to sturgeons and lampreys, the acrosome reaction involves the formation of actin-based filaments which occurs within the micropylar canal37. ACR, a serine protease found in the sturgeon acrosome, is associated with the acrosome reaction38,39 and may be facilitated by ACRBP, which regulates its activity. The presence of the acrosin/proacrosin system was identified previously in Siberian sturgeon spermatozoa40, and alterations of this system in seminal plasma following cryopreservation were detected in the present study. AGFG1 is involved in the fusion of proacrosomal vesicles and facilitates the structural rearrangements necessary for enzyme release during the acrosome reaction41, while GDI2 plays a key role in activating Rab proteins essential for this process42. Moreover, proteins such as vacuolar protein sorting-associated protein 26A (VPS26A), which is involved in vesicular trafficking, likely influence the acrosome reaction43, suggesting that extracellular vesicles participate in the acrosome reaction, aligning with their general role in the maturation of fish spermatozoa. Moreover, alterations in signaling proteins such as calcium/calmodulin-dependent protein kinase type IV (CAMK4), cAMP-dependent protein kinase catalytic subunit alpha (PRKACA), cAMP-dependent protein kinase type II-beta regulatory subunit (PRKAR2B), and LOC117435672, similar to calcium-binding and tyrosine phosphorylation-regulated proteins, highlight the effect of DMSO on sperm motility and the acrosome reaction. Taken together, our results provide the first evidence supporting the role of the acrosome reaction in sturgeon fertilization.
The uniqueness of sturgeons is further exemplified by their eggs being in possession of numerous micropyles at the animal pole, necessitating potentially more intricate sperm‒egg interactions than those in teleost species with a single micropyle. The identified changes in proteins crucial for sperm-egg recognition and binding uniquely altered by DMSO, such as zona pellucida-binding protein 2 (ZPBP2), ARSA, a vital enzyme in sturgeon sperm44, serine protease 55 (PRSS55), a chymotrypsin-like serine protease, and sialidase-1 (NEU1), which are responsible for the desialylation of sperm surface glycoproteins, underscore their role in the modification of the sperm surface to enable proper acrosome reactions and subsequent attachment to the zona pellucida45. In addition, PRSS55 was identified as a sperm-borne marker crucial for oocyte fertilization and early embryo development in mammals46. Notably, the functions of zona pellucida-binding proteins have been established in mammals, but no information on the role of these proteins in sturgeons is available. Nevertheless, our results clearly indicate the presence of these proteins in sturgeon sperm with a possible role in sperm-oocyte interactions. Further studies are needed to unravel their specific role in fertilization.
Cryopreservation with DMSO distinctively altered nuclear pore complexes (NPCs) in spermatozoa, especially affecting nucleoporins such as the nuclear pore complex proteins Nup54 (NUP54), Nup155 (NUP155), and Nup 85 (NUP85), which are key for NPC integrity and function. Nucleoporins are involved in nucleocytoplasmic transport and are crucial for cell functions such as proliferation, DNA repair, and cell cycle regulation, with significant roles in spermatogenesis, including aiding chromatin compaction, flagellar biogenesis, nuclear elongation and epigenetic regulation affecting embryonic development47,48,49. NUP54, a central channel protein, is crucial for nucleocytoplasmic transport. Mirroring the role of NUP54 in oocyte pronucleus formation50, NUP54 in spermatozoa could aid chromatin decondensation and nuclear envelope reassembly, which are vital for zygote formation. Alterations in NUP54 have been associated with increased cell death, mitotic disruption, and prolonged cell cycle phases, which exacerbate chromosomal aberrations in mammals51. NUP155 is crucial for nuclear envelope integrity, and its depletion causes embryonic lethality, underscoring its role in embryogenesis52. The importance of NUP85 in NPC assembly suggests that changes in this protein could impact genomic stability and transcriptional regulation, which are essential for embryo development53. While DMSO and MeOH affect various nucleoporins, the alteration of these nucleoporins by DMSO may impair fertilization outcomes by impacting genomic stability, nucleocytoplasmic transport, gamete fusion, and transcriptional activities, which are crucial for successful fertilization and early embryo development.
DMSO-induced alterations in flagellar and metabolic proteins significantly affected sperm functionality, potentially decreasing fertility. The BBSome complex, which includes tetratricopeptide repeat protein 8 (TTC8), Bardet-Biedl syndrome 1 protein (BBS1), and Bardet-Biedl syndrome 7 protein (BBS7), plays significant role in intraciliary transport, cell membrane protein trafficking and mitochondrial activity54,55. Alterations to these proteins cause flagellar defects and reduced motility56,57. IFT25, which is involved in intraflagellar transport, is vital for flagellar assembly58,59, while the kinesin-like protein KIFC3 (KIFC3), a motor protein, has been implicated in cellular transport and mitotic spindle assembly60. These proteins are also categorized as centrosome sperm proteins61. In addition to its roles in spermatogenic cell division and sperm formation, the centrosome is essential for regulating sperm tail beating and organizing zygote centrosomes after fertilization62,63. This finding underscores the novel insight that fertilization failure in DMSO-cryopreserved spermatozoa may also stem from disturbances to centrosome function. Our study also revealed a decrease in dynein axonemal intermediate chain 2 (DNAI2) and thioredoxin domain-containing protein 6 (NME9), which are crucial components for flagellar movement and axonemal organization, respectively, suggesting compromised flagellar integrity and potentially decreased fertility64,65. The significance of sperm tail morphology for fertilization capacity, particularly during the early postfertilization stages, has been emphasized66. Additionally, reductions in phosphorylase b kinase regulatory subunit alpha, skeletal muscle isoform (PHKA1) and ASRGL1, which are necessary for glycogen and asparagine metabolism, respectively, may affect energy production, impacting motility. These alterations could explain differences in motility parameters, such as ALH and BCF, between sperm cryopreserved with MeOH and those cryopreserved with DMSO. These parameters, while not related to major movement characteristics, such as the percentage of moving spermatozoa or their velocity, may reflect specific alterations to flagellar morphology. Overall, our study highlights a set of proteins that play a specific role in flagellar structure maintenance, centrosome function and metabolic processes, affecting both sperm movement patterns and fertilization ability, particularly in early embryo development. Further research is needed to explore the roles of these proteins in fish fertilization.
Among the DMSO-treated proteins, stress response proteins were altered. NHL repeat-containing protein 3 (NHLRC3) is critical for the ubiquitination process, and its reduction may disrupt the regulatory mechanisms of sperm essential for protein quality control. Proteasome subunit alpha type-6 (PSMA6), which supports protein degradation and turnover, is essential for cellular functions such as cell cycle regulation and DNA repair. The ubiquitin‒proteasome system (UPS) is instrumental in the targeted degradation of proteins, including the removal of misfolded proteins, thus ensuring cellular homeostasis. In spermatozoa, the UPS plays a role in fertilization, particularly in the degradation of sperm mitochondrial components67. Heat shock proteins (HSPs), such as HSPA8 and HSPE1, are vital for protecting spermatozoa against stress-induced damage. They act as molecular chaperones, ensuring proper protein folding and assembly, which is crucial for maintaining the structural and functional integrity of sperm68. FTH1, which is involved in scavenging free radicals, is crucial for shielding spermatozoa from oxidative damage. The alterations observed in these proteins due to DMSO cryopreservation suggest a compromised ubiquitin‒proteasome system and heat shock protein response; this may affect the proper folding of sperm proteins and the degradation of mitochondria following fertilization, leading to embryo death.
We identified several DMSO-specific alterations in sperm proteins whose roles in sperm functionality are not well understood. These proteins are involved in various biological processes, including nucleotide production, protein synthesis and immune response, suggesting a multifaceted impact on sperm functionality and fertility. Cytoplasmic C-1-tetrahydrofolate synthase (MTHFD1) has emerged as a key player in folate metabolism and is essential for nucleotide production and homocysteine methylation. Elevated homocysteine levels lead to recurrent miscarriage and compromised embryo quality69,70,71. Characterized by its leucine-rich repeat motifs, LRRC37A was predominantly expressed in the testis72. Leucine-rich repeat motifs, which are involved in protein‒protein interactions, suggest that LRRC37A may play a role in spermatogenesis or other testicular functions, although its precise role has not been fully elucidated. Although the specific function of fibrillin-1 (FBN1) in spermatozoa is unknown, it is known to be critical during embryonic development, and mutations or dysfunction of this protein can lead to significant developmental disorders73. Furthermore, elongation factor 1-gamma (EEF1G) is involved in the elongation step of protein synthesis, which is fundamental for cell growth, division, and function. We also observed alterations in butyrophilin-like protein 2 (BTNL2) and CD276 antigen (CD276), proteins involved in immune regulation74,75,76. In summary, our findings underscore the intricate roles of DMSO-specific protein alterations in sperm functionality and fertility, highlighting the necessity for further research to unravel the complexities of their contributions to fertilization ability.
MeOH specifically influenced proteins associated with mitochondrial functionality, including those involved in energy production, as well as flagellar structure. Key mitochondrial enzymes, such as mitochondrial succinate-semialdehyde dehydrogenase (ALDH5A1), ethylmalonyl-CoA decarboxylase (ECHDC1), mitochondrial glutaryl-CoA dehydrogenase (GCDH), acyl-coenzyme A diphosphatase NUDT19 (NUDT19), mitochondrial methylcrotonoyl-CoA carboxylase beta chain (MCCC2), and HADH, which are involved in carboxylic and fatty acid catabolic processes, are crucial for ATP synthesis and maintaining controlled ROS levels, thereby ensuring cellular energy and reducing oxidative stress77. Additionally, alteration in mitochondrial citramalyl-CoA lyase, (CLYBL), mitochondrial ribosome-releasing factor 2 (GFM2) and bis(5'-nucleosyl)-tetraphosphatase [asymmetrical] (NUDT2) suggest potential disruption in mitochondrial integrity and function. Metabolic and regulatory proteins, including lysophosphatidic acid phosphatase type 6 (ACP6), inositol monophosphatase 1 (IMPA1), and SPRY domain-containing protein 4 (SPRYD4), which are involved in lipid metabolism, myo-inositol synthesis, osmoregulation and signaling pathways, respectively, also show significant changes78. Notable changes in proteins related to the stress response, such as oxidoreductase activities (glutaredoxin-1 (GLRX), delta-1-pyrroline-5-carboxylate synthase (ALDH18A1), mitochondrial adrenodoxin (FDX1), protein disulfide-isomerase (P4HB)), the ubiquitin‒proteasome system (polyubiquitin-B (UBB) and 26S proteasome regulatory subunit 6A (PSMC3)), heat shock 70 kDa protein 4 L (HSPA4) and glutathione synthetase (GSS)), further suggest an elevated risk of oxidative damage, exacerbating susceptibility to oxidative stress, which is a common consequence of cryopreservation79,80. Alterations in axonemal proteins, including cilia- and flagella-associated protein 20 (CFAP20), CFAP96, microtubule-associated protein RP/EB family member 1 (MAPRE1), SPACA9, VIM, TUBB4, LEXM, and outer dense fiber protein 2 (ODF2), as well as the actin dynamics regulators WD repeat-containing protein 1 (WDR1) and tropomyosin alpha-3 chain (TPM3), may be indicative of decreased sperm motility due to compromised structural integrity and reduced tail flexibility81. Additionally, MeOH-induced modifications in the sperm axonemal maintenance protein CFAP97D1, Rho GDP-dissociation inhibitor 2 (ARHGDIB), and the SNARE protein vesicle-trafficking protein SEC22b (SEC22B), which is critical for vesicle trafficking, likely contribute to nuanced reductions in fertilization capability. Alterations in chromosomal structure-related proteins such as histone H2B (H2BC26) and the nuclear migration protein NudC (NUDC) suggest disturbances in the nucleosome structure of the chromosomal fiber, which may impact genetic integrity and expression. Collectively, these findings underscore the profound impact of MeOH on sperm functionality, particularly highlighting the decreased sperm motility and potential reductions in fertilization capacity following freeze‒thaw cycles. Nevertheless, these changes in the proteome seem to be sublethal, as they did not produce a dramatic decrease in sperm fertilization ability compared to that of DMSO.
Our findings align with the literature on mammalian and fish species, demonstrating that sperm cryopreservation induces significant proteomic changes, impacting proteins essential for flagellar structure maintenance, membrane fluidity, sperm motility, energy production, antioxidative defense mechanisms and fertilization ability10,11,12,13,14,15,16,17,18,82,83,84,85,86. We identified major alterations in proteins involved in metabolic pathways (DBI, 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 1 (PFKFB1), PYGM, GAPDHS, ENO3, leukotriene A-4 hydrolase (LTA4H), AASS, PDE7A, and CKMT1A), suggesting a potential correlation with decreased semen quality after cryopreservation. Additionally, our findings revealed disruptions in oxidative and immune stress defenses (PDIA3, peroxiredoxin-4 (PRDX4), S-(hydroxymethyl)glutathione dehydrogenase (ADH5), HSPD1, ASS1) and protein degradation pathways (26S proteasome non-ATPase regulatory subunits 1, 6 and 7 (PSMD1, PSMD6, PSMD7), E3 ubiquitin-protein ligase UBR4 (UBR4)) that are critical for sperm motility, suggesting vulnerabilities in cellular integrity and fertilization processes. Additionally, as the most affected proteins, we identified nuclear proteins such as histones (H2A.Z) and Ran GTPase-activating protein 1 (RANGAP1), which belong to the nuclear complex family, and those involved in reproductive processes, including binding to the zona pellucida (IDH2, T-complex protein 1 subunit epsilon and subunit zeta (CCT5, CCT6A), CABYR, and ALDOA), which suggest disturbances in chromatin integrity, nucleocytoplasmic transport and fertilization ability. We observed that some proteins showed more significant alterations in response to different cryoprotectants. Moreover, this study revealed species-specific impacts, with only a few of the most altered proteins being commonly affected across sturgeons and carp (PDIA3)26, rainbow trout (CCT6A and GAPDHS)16, and sterlets (ENO3)17,18, underscoring the nuanced effects of cryopreservation across species.
Interestingly, our study identified proteins with significantly increased abundance in sperm, despite sperm cells being transcriptionally and translationally inactive. This finding aligns with observations from previous proteomic studies examining the impact of cryopreservation on sperm proteomes in aquatic and mammalian species26,82. These prior studies suggest that the observed increase could be the result of stress-induced responses, potentially leading to selective protein exclusion from the membrane or the induction of PTMs, such as phosphorylation. This explanation was supported in our study by the identification of both up- and downregulated proteins that exhibited shifts in their pI, altering their positions on 2D-DIGE gels (Supplementary Fig. S3). Moreover, structural alterations to proteins have been posited as another potential mechanism for the observed increase in protein levels following cryopreservation11. However, further research could help to fully understand this issue using different methodological approaches.
The underlying mechanisms through which DMSO affects the fertilization capabilities of sturgeon remain largely unclear; however, recent studies have highlighted its substantial impact on cellular processes. The use of DMSO in cryopreservation has been associated with substantial modifications in mitochondrial function, including increased ROS levels and reduced ATP production in spermatozoa87. Additionally, the broader cytotoxic effects of DMSO, as evidenced by extensive DNA methylation disruptions in human cells88, may explain the adverse effects observed in sturgeon spermatozoa. The proteomic alterations in sturgeon sperm following cryopreservation with DMSO likely reflect secondary damage rather than direct effects of the cryoprotectant, suggesting deeper implications of cellular disturbances linked to the mechanisms of DMSO action. Interestingly, while DMSO decreased fertilization efficacy in our studies, contrasting findings in other species, such as the Indian red jungle fowl, have shown enhanced fertilization potential when sperm were cryopreserved with 8% DMSO compared to glycerol89. This variation underscores the species-specific responses to cryoprotectants.
Conclusions
In summary, we unravel several significant changes in the sperm proteome specific to sturgeon in response to cryopreservation. Among the DMSO-specific proteins, those involved in the acrosome reaction, zona pellucida binding, flagellar structure and nuclear pore organization could explain why spermatozoa cryopreserved with DMSO had low hatching rate. These changes were not as pronounced when semen was cryopreserved with MeOH. The unique structure of sturgeon sperm and its specialized fertilization process, underscore the importance of tailored cryopreservation methods. Our research supports the earlier recommendation that methanol is the most effective cryoprotectant for sturgeon semen. This work also lays the groundwork for future research aimed at refining cryopreservation techniques, which is crucial for the preservation and successful reproduction of this ecologically and economically valuable species.
Data availability
The mass spectrometry proteomics data have been deposited in the ProteomeXchange Consortium via the PRIDE90 partner repository with the dataset identifiers PXD052170 and https://doi.org/https://doi.org/10.6019/PXD052170. Additional data that support the findings of this study are available from the corresponding author (M.A.D.) upon reasonable request.
References
Ruban, G. & Mugue, N. Acipenser baerii. The IUCN red list of threatened species. https://doi.org/10.2305/IUCN.UK.2022-1.RLTS.T244A156718817.en (2022).
Pšenička, M. & Ciereszko, A. Sperm and Spermatozoa Characteristics in the Siberian Sturgeon 307–326 (Springer, 2018).
Alavi, S. M. H. et al. Sperm biology and control of reproduction in sturgeon: (II) sperm morphology, acrosome reaction, motility and cryopreservation. Rev. Fish. Biol. Fish. 22, 861–886 (2012).
Kolyada, M. N., Osipova, V. P. & Berberova, N. T. Use of cryoprotectors and antioxidants in sturgeon semen cryopreservation. Cryobiology 111, 30–39 (2023).
Leung, L. K. P. Principles of Biological Cryopreservation 231–244 (Cambridge University Press, 1991).
Bui, T. V. L., Ross, I. L., Jakob, G. & Hankamer, B. Impact of procedural steps and cryopreservation agents in the cryopreservation of chlorophyte microalgae. PLoS One 8, e78668 (2013).
Glogowski, J. et al. Fertilization rate of Siberian sturgeon (Acipenser baeri, Brandt) milt cryopreserved with methanol. Aquaculture 211, 367–373 (2002).
Ciereszko, A., Dabrowski, K., Froschauer, J. & Wolfe, T. D. Cryopreservation of Semen from Lake Sturgeon. T. Am. Fish. Soc. 135, 232–240 (2006).
Psenicka, M. et al. Acrosome staining and motility characteristics of sterlet spermatozoa after cryopreservation with use of methanol and DMSO. Cryobiology 56, 251–253 (2008).
Chen, X. et al. Identification of differentially expressed proteins in fresh and frozen-thawed boar spermatozoa by iTRAQ-coupled 2D LC-MS/MS. Reproduction 147, 321–330 (2014).
Bogle, O. A. et al. Identification of protein changes in human spermatozoa throughout the cryopreservation process. Andrology 5, 10–22 (2017).
Perez-Patiño, C. et al. Cryopreservation differentially alters the proteome of epididymal and ejaculated pig spermatozoa. Int. J. Mol. Sci. 20, 1791 (2019).
Li, P. et al. Ice-age endurance: The effects of cryopreservation on proteins of sperm of common carp, Cyprinus carpio L.. Theriogenology 74, 413–423 (2010).
Zilli, L. et al. Effect of cryopreservation on sea bass sperm proteins. Biol. Reprod. 72, 1262–1267 (2005).
Dietrich, M. A. et al. Proteomic analysis of extracellular medium of cryopreserved carp (Cyprinus carpio L.) semen. Comp. Biochem. Phys. D 15, 49–57 (2015).
Nynca, J., Arnold, G. J., Fröhlich, T. & Ciereszko, A. Cryopreservation-induced alterations in protein composition of rainbow trout semen. Proteomics 15, 2643–2654 (2015).
Xin, M. et al. Impact of cryopreservation on sterlet, Acipenser ruthenus sperm motility and proteome. Anim. Reprod. Sci. 192, 280–289 (2018).
Horokhovatskyi, Y. et al. Cryopreservation effects on a viable sperm sterlet (Acipenser ruthenus) subpopulation obtained by a Percoll density gradient method. PLoS One 13, 0202514 (2018).
Judycka, S., Szczepkowski, M., Ciereszko, A. & Dietrich, G. J. New extender for cryopreservation of Siberian sturgeon (Acipenser baerii) semen. Cryobiology 70, 184–189 (2015).
Conte, F. S., Doroshov, S. I., Lutes, P. B. & Strange, E. M. Hatchery Manual for the White Sturgeon Acipenser transmontanus Richardson: With Application to Other North American Acipenseridae Vol. 88 (University of California, 1988).
Szczepkowski, M. & Kolman, R. A simple method for collecting sturgeon eggs using a catheter. Arch. Pol. Fish. 19, 123–128 (2011).
Jähnichen, H. et al. Motility and fertilizing capability of cryopreserved Acipenser ruthenus L. sperm. J. Appl. Ichthyol. 15, 204–206 (1999).
Guthrie, H. D. & Welch, G. R. Effects of hypothermic liquid storage and cryopreservation on basal and induced plasma membrane phospholipid disorder and acrosome exocytosis in boar spermatozoa. Reprod. Fertil. Dev. 17, 467–477 (2005).
Park, C. & Chapman, F. A. An extender solution for the short term storage of sturgeon semen. N. Am. J. Aquacult. 67, 52–57 (2005).
Mostek, A. et al. Identification of oxidatively modified proteins due to cryopreservation of carp semen. J. Anim. Sci. 96, 1453–1465 (2018).
Dietrich, M. A. & Ciereszko, A. Proteomic characterization of fresh spermatozoa and supernatant after cryopreservation in relation to freezability of carp (Cyprinus carpio L) semen. PLoS One 13, 0192972 (2018).
Nynca, J. et al. Triploidization of rainbow trout affects proteins related to ovary development and reproductive activity. Aquaculture 565, 739145 (2023).
Dietrich, M. A., Dietrich, G. J., Mostek, A. & Ciereszko, A. Motility of carp spermatozoa is associated with profound changes in the sperm proteome. J. Proteomics 138, 124–135 (2016).
Kodzik, N., Ciereszko, A., Szczepkowska, B., Malinowska, A. & Dietrich, M. A. Comparative proteomic analysis of the ovarian fluid and eggs of Siberian sturgeon. BMC Genom. 25, 451 (2024).
Posch, A., Kohn, J., Oh, K., Hammond, M. & Liu, N. V3 stain-free workflow for a practical, convenient, and reliable total protein loading control in western blotting. J. Vis. Exp. 82, e50948 (2013).
Shaliutina, A., Hulak, M., Gazo, I., Linhartova, P. & Linhart, O. Effect of short-term storage on quality parameters, DNA integrity, and oxidative stress in Russian (Acipenser gueldenstaedtii) and Siberian (Acipenser baerii) sturgeon sperm. Anim. Reprod. Sci. 139, 127–135 (2013).
Lahnsteiner, F., Berger, B., Horvath, A. & Urbanyi, B. Studies on the semen biology and sperm cryopreservation in the starlet, Acipenser ruthenus L.. Aquac. Res. 35, 519–528 (2004).
Linhart, O. et al. Effect of cryoprotectants and male on motility parameters and fertilization rate in paddlefish (Polyodon spathula) frozen-thawed spermatozoa. J. Appl. Ichthyol. 22, 389–94 (2007).
Boryshpolets, S. et al. Cryopreservation of sterlet (Acipenser ruthenus) spermatozoa using different cryoprotectants. J. Appl. Ichthyol. 27, 1147–1149 (2011).
Jankowska, U. et al. Proteome analysis of PC12 cells reveals alterations in translation regulation and actin signaling induced by clozapine. Neurochem. Res. 46, 2097–2111 (2021).
Jungblut, P. R., Holzhütter, H. G., Apweiler, R. & Schlüter, H. The speciation of the proteome. Chem. Cent. J. 2, 1–10 (2008).
Cherr, G. N. & Clark, J. R. W. H. Fine structure of the envelope and micropyles in the eggs of the white sturgeon, Acipenser transmontanus Richardson. Dev. Growth Differ. 24, 341–52 (1982).
Ciereszko, A., Dabrowski, K. & Ochkur, S. I. Characterization of acrosin-like activity of lake sturgeon (Acipenser fulvescens) spermatozoa. Mol. Reprod. Dev. 45, 72–77 (1996).
Ciereszko, A., Dabrowski, K., Mims, S. D. & Glogowski, J. Characteristics of sperm acrosin-like activity of paddlefish (Polyodon spathula Walbaum). Comp. Biochem. Physiol. B Biochem. Mol. Biol. 125, 197–203 (2000).
Słowińska, M. A. et al. Effect of season on proteases and serine protease inhibitors of Siberian sturgeon (Acipenser baerii Brandt, 1869) semen. J. Appl. Ichthyol. 31, 125–131 (2015).
Kang-Decker, N., Mantchev, G. T., Juneja, S. C., McNiven, M. A. & van Deursen, J. M. Lack of acrosome formation in Hrb-deficient mice. Science 294, 1531–1533 (2001).
Bae, J. W. et al. Ras-related proteins (Rab) are key proteins related to male fertility following a unique activation mechanism. Reprod. Biol. 19, 356–362 (2019).
Da Ros, M., Hirvonen, N., Olotu, O., Toppari, J. & Kotaja, N. Retromer vesicles interact with RNA granules in haploid male germ cells. Mol. Cell. Endocrinol. 401, 73–83 (2015).
Sarosiek, B., Ciereszko, A., Kolman, R. & Glogowski, J. Characteristics of arylsulfatase in Russian sturgeon (Acipenser gueldenstaedti) semen. Comp. Biochem. Physiol. B. Biochem. Mol. Biol. 139, 571–579 (2004).
Shang, X. et al. Serine protease PRSS55 is crucial for male mouse fertility via affecting sperm migration and sperm-egg binding. Cell. Mol. Life Sci. 75, 4371–4384 (2018).
Vallet-Buisan, M., Mecca, R., Jones, C., Coward, K. & Yeste, M. Contribution of semen to early embryo development: Fertilization and beyond. Hum. Reprod. Update 29, 395–433 (2023).
Guglielmi, V., Sakuma, S. & D’Angelo, M. A. Nuclear pore complexes in development and tissue homeostasis. Development 147, dev183442 (2020).
Major, A. T., Whiley, P. A. & Loveland, K. L. Expression of nucleocytoplasmic transport machinery: Clues to regulation of spermatogenic development. Biochim. Biophys. Acta 1813, 1668–1688 (2011).
Arafah, K. et al. Defect in the nuclear pore membrane glycoprotein 210-like gene is associated with extreme uncondensed sperm nuclear chromatin and male infertility: A case report. Hum. Reprod. 36, 693–701 (2021).
Payne, C., Rawe, V., Ramalho-Santos, J., Simerly, C. & Schatten, G. Preferentially localized dynein and perinuclear dynactin associate with nuclear pore complex proteins to mediate genomic union during mammalian fertilization. J. Cell Sci. 116, 4727–4738 (2003).
Rodriguez-Berriguete, G. et al. Nucleoporin 54 contributes to homologous recombination repair and post-replicative DNA integrity. Nucleic Acids Res. 46, 7731–7746 (2018).
Franz, C. et al. Nup155 regulates nuclear envelope and nuclear pore complex formation in nematodes and vertebrates. EMBO J. 24, 3519–3531 (2005).
Gonzalez-Estevez, A., Verrico, A., Orniacki, C., Reina-San-Martin, B. & Doye, V. Integrity of the short arm of the nuclear pore Y-complex is required for mouse embryonic stem cell growth and differentiation. J. Cell Sci. 134, jcs258340 (2021).
Jin, H. & Nachury, M. V. The BBSome. Curr. Biol. 19, 472–473 (2009).
Guo, D. F. et al. The BBSome regulates mitochondria dynamics and function. Mol. Metab. 67, 101654 (2023).
Prasai, A. et al. The BBSome assembly is spatially controlled by BBS1 and BBS4 in human cells. J. Biol. Chem. 295, 14279–14290 (2020).
Tian, X., Zhao, H. & Zhou, J. Organization, functions, and mechanisms of the BBSome in development, ciliopathies, and beyond. Elife 12, e87623 (2023).
Taschner, M., Bhogaraju, S., Vetter, M., Morawetz, M. & Lorentzen, E. Biochemical mapping of interactions within the intraflagellar transport (IFT) B core complex: IFT52 binds directly to four other IFT-B subunits. J. Biol. Chem. 286, 26344–26352 (2011).
Keady, B. T. et al. IFT25 links the signal-dependent movement of Hedgehog components to intraflagellar transport. Dev. Cell 22, 940–951 (2012).
Hu, J. R. et al. Gene expression pattern of KIFC3 during spermatogenesis of the skink Eumeces chinensis. Gene 556, 206–212 (2015).
Amargant, F. et al. The human sperm basal body is a complex centrosome important for embryo preimplantation development. Mol. Hum. Reprod. 27, gaab062 (2021).
Firat-Karalar, E. N., Sante, J., Elliott, S. & Stearns, T. Proteomic analysis of mammalian sperm cells identifies new components of the centrosome. J. Cell Sci. 127, 4128–4133 (2014).
Aljiboury, A. & Hehnly, H. The centrosome- diverse functions in fertilization and development across species. J. Cell Sci. 136, jcs261387 (2023).
Walton, T., Wu, H. & Brown, A. Structure of a microtubule-bound axonemal dynein. Nat. Commun. 12, 477 (2021).
Koscinski, I. et al. Reproduction function in male patients with Bardet Biedl syndrome. J. Clin. Endocrinol. Metab. 105, 4417–4429 (2020).
Kai, Y., Iseki, H., Matsui, T. & Yamashita, N. Morphological analysis of the human sperm tail during the early post-fertilization stage. Reproduction 167, e230290 (2024).
Song, E. J., Yim, S. H., Kim, E., Kim, N. S. & Lee, K. J. Human Fas-associated factor 1, interacting with ubiquitinated proteins and valosin-containing protein, is involved in the ubiquitin-proteasome pathway. Mol. Cell. Biol. 25, 2511–2524 (2005).
Meccariello, R., Chianese, R., Ciaramella, V., Fasano, S. & Pierantoni, R. Molecular chaperones, cochaperones, and ubiquitination/deubiquitination system: Involvement in the production of high quality spermatozoa. Biomed. Res. Int. 2014, 561426 (2014).
Benchaib, M. et al. Quantitation by image analysis of global DNA methylation in human spermatozoa and its prognostic value in in vitro fertilization: A preliminary study. Fertil. Steril. 80, 947–953 (2003).
Benchaib, M. et al. Influence of global sperm DNA methylation on IVF results. Hum. Reprod. 20, 768–773 (2005).
Cao, J. et al. DJ-1 suppresses ferroptosis through preserving the activity of S-adenosyl homocysteine hydrolase. Nat. Commun. 11, 1251 (2020).
Giannuzzi, G. et al. Evolutionary dynamism of the primate LRRC37 gene family. Genome Res. 23, 46–59 (2013).
Quondamatteo, F. et al. Fibrillin-1 and fibrillin-2 in human embryonic and early fetal development. Matrix Biol. 21, 637–646 (2002).
Cristiano, L. EEF1G (eukaryotic translation elongation factor 1 gamma). Atlas Genet. Cytogenet. Oncol. Hematol. 24, 58–68 (2020).
Arnett, H. A. et al. BTNL2, a butyrophilin/B7-like molecule, is a negative costimulatory molecule modulated in intestinal inflammation. J. Immunol. 178, 1523–1533 (2007).
Sun, M. et al. Characterization of mouse and human B7–H3 genes. J. Immunol. 168, 6294–6297 (2002).
Amaral, A., Lourenço, B., Marques, M. & Ramalho-Santos, J. Mitochondria functionality and sperm quality. Reproduction 146, 163–174 (2013).
Samanta, L., Sharma, R., Cui, Z. & Agarwal, A. Proteomic analysis reveals dysregulated cell signaling in ejaculated spermatozoa from infertile men. Asian J. Androl. 21, 121–130 (2019).
Mańkowska, A., Gilun, P., Zasiadczyk, Ł, Sobiech, P. & Fraser, L. Expression of TXNRD1, HSPA4L and ATP1B1 genes associated with the freezability of boar sperm. Int. J. Mol. Sci. 23, 9320 (2022).
Ribas-Maynou, J. & Yeste, M. Oxidative stress in male infertility: Causes, effects in assisted reproductive techniques, and protective support of antioxidants. Biology (Basel) 9, 77 (2020).
Inaba, K. & Mizuno, K. Sperm dysfunction and ciliopathy. Reprod. Med. Biol. 15, 77–94 (2015).
Zilli, L. et al. Comparative proteome analysis of cryopreserved flagella and head plasma membrane proteins from sea bream spermatozoa: Effect of antifreeze proteins. PLoS One 9, e99992 (2014).
Wang, S. et al. Proteomic characteristics of human sperm cryopreservation. Proteomics 14, 298–310 (2014).
Westfalewicz, B., Dietrich, M. A. & Ciereszko, A. Impact of cryopreservation on bull (Bos taurus) semen proteome. J. Anim. Sci. 93, 5240–5253 (2015).
Arunkumar, R. et al. The cryopreservation process induces alterations in proteins associated with bull sperm quality: The equilibration process could be a probable critical control point. Front. Endocrinol. (Lausanne) 13, 1064956 (2022).
He, Y. et al. Differential proteome association study of freeze-thaw damage in ram sperm. Cryobiology 72, 60–68 (2016).
Ding, Y., Liu, S., Liu, J., Jin, S. & Wang, J. Cryopreservation with DMSO affects the DNA integrity, apoptosis, cell cycle and function of human bone mesenchymal stem cells. Cryobiology 114, 104847 (2024).
Verheijen, M. et al. DMSO induces drastic changes in human cellular processes and epigenetic landscape in vitro. Sci. Rep. 9, 4641 (2019).
Rakha, B. A. et al. Use of dimethylsulfoxide for semen cryopreservation in Indian red jungle fowl (Gallus gallus murghi). Theriogenology 122, 61–67 (2018).
Perez-Riverol, Y. et al. The PRIDE database resources in 2022: A Hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res. 50, 543–552 (2022).
Acknowledgements
We extend our sincere thanks to Halina Karol for excellent technical assistance, Ewa Liszewska for oxidative stress analysis and Joanna Wiśniewska for microscopy analysis.
Funding
This work was supported by grants from the National Science Centre to research project 2019/35/B/NZ9/03501. The equipment used for proteomic analysis was sponsored in part by the Centre for Preclinical Research and Technology (CePT), a project cosponsored by the European Regional Development Fund and Innovative Economy of the National Cohesion Strategy of Poland.
Author information
Authors and Affiliations
Contributions
N.K.: Formal analysis, data curation, visualization, methodology, writing-original draft, writing-review & editing. A.C.: Conceptualization, writing-original draft, writing-review & editing. S.J.: Formal analysis and data curation. M.S.: Methodology. B.S.: Resources. B.Ś.: Formal analysis and data curation. M.A.D.: Conceptualization, methodology, writing-original draft, writing-review & editing, supervision, and funding acquisition. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Kodzik, N., Ciereszko, A., Judycka, S. et al. Cryoprotectant-specific alterations in the proteome of Siberian sturgeon spermatozoa induced by cryopreservation. Sci Rep 14, 17707 (2024). https://doi.org/10.1038/s41598-024-68395-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-68395-7
- Springer Nature Limited