
Abstract
The generic term “Gill disease” refers to a wide range of disorders that affect the gills and severely impact salmonid aquaculture systems worldwide. In rainbow trout freshwater aquaculture, various etiological agents causing gill diseases have been described, particularly Flavobacterium and Amoeba species, but research studies suggest a more complex and multifactorial aetiology. Here, a cohort of rainbow trout affected by gill disease is monitored both through standard laboratory techniques and 16S rRNA Next-Generation Sequencing (NGS) analysis during a natural disease outbreak and subsequent antibiotic treatment with Oxytetracycline. NGS results show a clear clustering of the samples between pre- and post-treatment based on the microbial community of the gills. Interestingly, the three main pathogenic bacteria species in rainbow trout (Yersinia ruckeri, Flavobacterium psychrophilum, and Flavobacterium branchiophilum) appear to be weak descriptors of the diversity between pre-treatment and post-treatment groups. In this study, the dynamics of the gill microbiome during the outbreak and subsequent treatment are far more complex than previously reported in the literature, and environmental factors seem of the utmost importance in determining gill disease. These findings present a potential novel perspective on the diagnosis and management of gill diseases, showing the limitations of conventional laboratory methodologies in elucidating the complexity of this disease in rainbow trout. To the authors’ knowledge, this work is the first to describe the microbiome of rainbow trout gills during a natural outbreak and subsequent antibiotic treatment. The results of this study suggest that NGS can play a critical role in the analysis and comprehension of gill pathology. Using NGS in future research is highly recommended to gain deeper insights into such diseases correlating gill’s microbiome with other possible cofactors and establish strong prevention guidelines.
Similar content being viewed by others

Introduction
Aquaculture is poised to play a critical role in providing protein to the ever-growing global population in the future1. Salmonid aquaculture plays a central role within the global seafood industry, accounting for a substantial 18.1% of the total value and 7.4% of the biomass in the worldwide trade of fish and fish products2. Fish diseases represent a significant limitation to salmonid aquaculture, and among them, multifactorial diseases have been increasingly recognized as challenging conditions affecting fish farming3. A complex interplay between the host, various pathogens, and the environment characterizes these disorders, not satisfying classic Koch”s postulates, where one agent is unequivocally associated with a specific illness4.
Gill pathologies in salmonid farming are among the most prevalent and problematic examples of these complex conditions and have a substantial economic impact5. In many cases, their etiology is far from being clarified3, leading to the definition of multifactorial gill disease6. Despite their relatively fragile structure, exposed location, and large surface area making gills susceptible to various pathological conditions, the morphological changes associated with gill diseases are often unspecific and stereotyped, regardless of the causative agent, particularly in chronic cases7,8.
A formidable example of the complexity of gill disorders is represented by bacterial gill disease in freshwater salmonids (BGD). Despite decades of research, BGD pathogenesis and causative agents have remained elusive in field and experimental studies9,10,11,12. The term BGD is derived from its association with rod-shaped, non-invasive bacteria, namely Myxobacteria (Flavobacterium spp.)13. Although Flavobacterium branchiophilum species have consistently appeared linked to gill disease outbreaks14, inducing clinical disease through cohabitation with diseased fish or exposure to Flavobacteria cultures alone proved inconclusive to cause clinical disease15,16. In parallel, environmental and zootechnical factors have been suspected to play a crucial role in the onset of BGD10,11, and research evidenced the influence of stress-inducing (overcrowding or exposure to ammonia and low oxygen) elements17. Within this context, the intricate interplay between biotic and abiotic factors, the role of the gill microbiome, and the onset of BGD remain unclear.
Culture-independent approaches have attempted to clarify the complexity of bacterial populations involved in different forms of gill inflammation, especially in seawater-reared Atlantic salmon18. When analyzing the role of microbiota in multifactorial diseases, the so-called Microbial Dark Matter (MDM), defined as the presence of a vast number of microbial populations that can’t yet be cultivated, represents an essential challenge. MDM is estimated to be about 99% of microbial populations19,20,21. MDM represents an important limiting factor both when cultivation methods are employed19,20,21, and when non-sterile districts, such as gills, are studied. In such cases, a more comprehensive method is required to define the role of the diverse components in the disease onset and progression.
The development of Next-Generation Sequencing (NGS) technology has allowed a better understanding of multifactorial diseases, given its high throughput nature22. Since NGS has become widely available and more affordable, few studies have applied it to study gill diseases, describing the dynamic of the prokaryotic community on Atlantic salmon gills23,24. Unfortunately, similar approaches still need to be improved for freshwater rainbow trout farming, and BGD remains a significant concern in this species.
The present case study describes for the first time an integrated approach to characterize gill microbiome dynamics by high-throughput sequencing of the 16S rRNA gene before and after treatment with antibiotics. To the authors' knowledge, this is the first attempt to monitor and describe the gill prokaryotic microbiome during a natural outbreak and subsequent antibiotic treatment, providing valuable insights and thorough discussion on the roles of the bacterial population in gill disease.
General description of the case study
The case study described here occurred at a freshwater raceway facility in Italy that sources its water from a nearby river, with a water flow of 3,5 m3/s and an annual temperature ranging from 9 ℃ in winter to 15 ℃ in summer. This grow-out facility receives juveniles weighing 10 to 20 g from the hatcheries and grows them until they reach the market size (600 to 800 g). The facility is divided into three sectors, each of them subdivided into several tanks. These tanks measure 70 by 9 m with a volume of 504 m3, and each tank receives a water flow of 600 l/s. The water enters the facility from Sector 1 and leaves the facility from Sector 3. Small fish are introduced in Sector 1 and moved downstream with the grading process, so Sector 1 holds the smallest fish while the biggest ones are in Sector 3. Fish were not vaccinated, and, historically, the most prevalent disease affecting this site was Enteric Red Mouth Syndrome caused by Yersinia ruckeri, suspected during routine necroscopies and confirmed through bacteriological isolation following the method later described. The facility’s basal mortality was < 0.025% of the stock per day during the previous three years.
In late Autumn and Winter 2021, a substantial decrease in rainfall caused a decrease of 1,1 m3 of water in the facility, and in each tank the water flow went down to approximately 400 l/s. Sectors 2 and 3 experienced severe and chronic mortality events, with a mean daily mortality rate of 0.2% of the stock in affected tanks. Affected fish exhibited clinical signs of dyspnea, swimming near the surface and edges of the tanks, as well as open opercula and flared gills, suggesting a potential gill disease. Standard diagnostic procedures were employed at this stage (T0 sampling time-point). The findings suggested the presence of gill pathology, confirmed by histology and bacteriology, and a concurrent Yersinia ruckeri septicemia sensitive to enhanced sulphonamides.
Based on the bacteriological results and the antimicrobial sensitivity testing, it was decided to treat one of the tanks in Sector 2 with enhanced sulphonamides (Neopridimet, Fatro, Ozzano dell’Emilia, Italy), 240 + 48 mg/kg of Body Weight (BW) per day in two administrations for ten days. The treatment failed to produce appreciable results, as the mortality didn’t decrease below 0.2% per day.
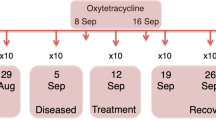
Given the therapeutic failure associated with the use of enhanced sulphonamides, the absence of other isolates in the internal organs able to cause such a mortality event, and the massive gill flavobacteria infection found through histology, it was decided to treat a different but equally affected tank in the same sector with Oxytetracycline at the dose of 120 mg/kg BW administered in a single daily feeding at the rate of 0.5% of BW, for ten days. The antibiotic dose and administration regimen were selected and monitored by the responsible veterinarian, taking into account the severity of the situation, his expertise, and recent studies showing the safety of this drug at the given dose25 and the optimal administration regimen in fish to maximize the drug’s efficacy26. The tank treated with Oxytetracycline had a density of 25 kg of fish/m3 and a water exchange of 400 L s−1, measured with the Bazin equation for a total exchange of 2.8 complete water changes per hour. Oxytetracycline was chosen considering both the massive presence of filamentous bacteria in the gills, highlighted by histology, and the favorable antimicrobial sensitivity testing of the Flavobacterium strains isolated at the first attempt, established by the Minimum Inhibitory Concentration (MIC) test. This treatment reduced mortality from an average of 0.2% (ranging from 0.25 to 0.15%) of tank stock per day to 0.06%. Before and after the treatment with oxytetracycline, the gills of the treated fish were sampled and submitted for bacteriological, histological, and NGS analysis (respectively, T1 and T2 sampling time points). The main phases of the study are represented in Fig. 1.

Sampling and antibiotic treatments timeline.
Results
Water quality and environmental parameters
Throughout all sampling time points, dissolved gases have been within optimal ranges for trout farming (oxygen sat > 80%; residual gasses < 104%; CO2 < 10 ppm), the temperature ranged from 10 ℃ to 12 ℃, and pH was stable at 7.5.
All the environmental analysis results were compatible with fish farming as described in previous literature27,28. These results are available in Tables S1-S5 of the Supplementary materials. The suspended solids values were < 2 mg/L at every time point.
Fish sampling
Wet mounts, necropsy, and histology
At the T0 sampling time-point, the gill wet mounts revealed an evident hyperplastic reaction, and some round vacuolated elements compatible with amoebae were highlighted (Panel 1S-a of the Supplementary Materials). The skin wet mounts highlighted the presence of Gyrodactylus spp. and Icthiophtyrius multifiliis in small amounts (Panel 1S-b, c of the Supplementary Materials). Parasites were identified based on their morphology29.
The necroscopic findings on the gills were an abnormous quantity of mucus, associated with shortening and clubbing of the gill filaments (Panel 1-a), the presence of multifocal to coalescing whitish areas, raised from the surrounding tissue, associated with multifocal to coalescing hemorrhages (Panel 1-b) and, more rarely, oomycetes infestation (Saprolegnia spp.).

Affected fish from Sector 2. (a): The gills show diffuse pallor and hypermucosity, with shortening and rounding of the filaments. (b): The same lesions are also associated with multifocal to coalescing hemorrhages. (c): The most affected filaments show severe alteration of the profile with hyperplasia of the respiratory epithelium, the fusion of the lamellae along the entire filament axis, and diffuse spongiosis, resulting in prominent clubbing of the filaments. (d-e): There are hypertrophy and hyperplasia of the respiratory epithelium causing the thickening and rounding of the lamellae (d) and the presence of numerous clusters of hyperbasophilic filamentous bacteria in the interlamellar spaces, consistent with Flavobacterium spp. (d-e).
At the opening of the coelomic cavity, prominent signs were diffused, marked congestion of all internal organs, and the presence of eggs in the ovaries at different maturation stages. In some specimens, the ovary ruptured with eggs interspersed between the internal organs (this was found on a different degree in the various tanks, up to 100% of analyzed specimens). Other findings included the presence of petechiae on the visceral fat and the swim bladder, brain congestion, splenomegaly, and enteritis with a yellowish mucous content (Panel 1S-d of the Supplementary Materials). Proliferative branchitis aggravated by bacterial septicemia was suspected.
Also, moribund fish from a tank with mortality within normal ranges were sampled as a baseline. Half of the specimens showed gill hyperplasia and hypermucosity, and at the opening of the coelomic cavity the internal signs were milder than the ones of fish from the affected tanks. Splenomegaly and generalized congestion were found in half of the specimens.
At the T1 time point, necropsy findings in moribund specimens were consistent with those observed at the T0 time point. At the T2 time point, the necropsy revealed similar signs, albeit with a milder presentation. In healthy specimens, mild focal gill hyperplasia was evident, while internal organs exhibited no significant abnormalities. At histology, gills from all the groups (affected and controls at T0, healthy and moribund at T1 and T2) presented a common spectrum of lesions that reached a peak of severity in the affected tanks of T0 and moribund fish of T1 and T2.
The main histological findings included marked hyperplasia of the respiratory epithelium, associated with fusion of the lamellae, and moderate to marked goblet cell hyperplasia (Panel 1-c). The hyperplastic reaction not only affected the distal portion of the filaments but frequently extended to their entire length, causing a prominent rounding in their profile (Panel 1-c). In most affected areas, marked spongiosis was identified, coupled with necrosis and apoptosis of the epithelial cells with accumulation of necrotic debris in the inter-filament spaces.
Between the filaments, numerous hyper-basophilic filamentous bacteria were detected in all fish of T0 and T1 groups as cohesive clusters attached to the filaments or dispersed between the inter-filament spaces, consistent with Flavobacterium spp. (Panel 1-d, e). A moderate number of debris admixed with mucous was also noted. The prevalence of filamentous bacteria dropped at T2, where only a few coccobacillary bacteria were noted.
Scarce protozoan elements were inconsistently detected in some sections from T0, T1, and T2 fish, attached to the filaments or encysted within pseudocysts formed by incomplete lamellar fusion. The parasites were 25–30 µm in diameter, and showed a vacuolated cytoplasm, a paracentral micronucleus, and a perinuclear clear halo consistent with freshwater amoebice trophozoites. Other findings included the presence of multifocal telangiectasia, hemorrhages, and thrombi in the lamellar capillaries of the gills, particularly prominent in affected fish from T0 and moribund fish from T1 and T2.
On internal organs evaluation, renal tubules showed a moderate frequency of protein droplet reabsorption at all time-points, consistent with the high number of vitellogenin-phase follicles found in the ovaries, confirming the active reproductive phase reported clinically and at necropsy (Panel 1S-e of the Supplementary Materials).
Occasional mild lymphocytic infiltration of the epicardium and myocardium (Panel 1S-f of the Supplementary Materials) and intraocular hemorrhage were also noted in affected fish at T0 and moribund fish at T1. These findings were consistent with a prolonged diseased status and possible septicemic condition.
Overall, fish groups in all the sampling time-points presented consistently similar fresh mount, necropsy, and histological findings, apart from numerous filamentous bacteria in the gills at histology at T0 and T1 and the presence of few Gyrodactylus spp. on the skin at T1 sampling time-point that was not found at T2.
The final morphological diagnosis for all sample time points was diffuse, severe, chronic hyperplastic branchitis, associated with myriads of intralesional filamentous bacteria in the T0-T1 animals, making the condition consistent with a chronic BGD aggravated by bacterial septicemia (presumed Yersinia ruckeri).
Virology, bacteriology, and antimicrobial sensitivity
Virological analyses were performed only at the T0 time-point. The results of these analyses are shown in Supplementary Table S6. The analyzed sample tested negative for infectious haematopoietic necrosis virus (IHNv), viral haemorrhagic septicaemia virus (VHSv), and salmonid alphavirus (SAV).
At the T0 time-point sampling, microbiological evaluation of the internal organs highlighted Yersinia ruckeri biotype II as the most frequently isolated bacteria; internal organs from negative control specimens tested negative. Flavobacterium spp. were isolated, intermixed with limited polymicrobial growth, from the gills of both affected and control tanks. Among these, Flavobacterium psychrophilum was identified at the species level. Both Kirby-Bauer and MIC tests confirmed the sensitivity of the Yersinia ruckeri isolate to amoxicillin, enrofloxacin, florfenicol, flumequine, oxytetracycline, and enhanced sulphonamides while it tested intermediate to erythromycin. Flavobacterium psychrophilum and Flavobacterium sp. were sensible to all molecules in Kirby-Bauer and MIC testing.
At the T1 time-point, Yersinia ruckeri biotype II was isolated from the internal organs of moribund fish. Gill bacteriology revealed the presence of Flavobacterium spp., Pseudomonas spp., Aeromonas sobria, Yersinia ruckeri biotype II, Lelliottia sp. Microbacterium sp., and Acidovorax sp.
At the T2 time-point (after Oxytetracycline treatment), bacteriological analysis of internal organs of healthy fish was negative, while Yersinia ruckeri biotype II and Pseudomonas spp. were found on the gills; remarkably, bacteria referable to the genus Flavobacterium were not detected. In moribund fish, Yersinia ruckeri biotype II was isolated from the eyes and the internal organs. From the gills of the moribund fish, Yersinia ruckeri biotype II, Aeromonas bestiarum, Aeromonas sobria, Pseudomonas spp., Acinetobacter sp., and Flavobacterium sp. were isolated. At the T2 sampling time-point, Yersinia ruckeri and Flavobacterium sp. were tested for antibiotic resistance with MIC and Kirby Bauer methods. Both strains were sensitive to oxytetracycline: Yersinia ruckeri biotype II showed sensitivity to enhanced sulphonamides, while Flavobacterium sp. was resistant. A summary of the results of the samplings and analyses performed is available in Table S6 of the Supplementary materials.
NGS and bioinformatic analysis
The total number of paired reads used for analysis was 2,279,502 from the eight samples from the “Clinically Healthy” group at T1, 2,665,287 from the eight samples from the “Moribund” group at T1, 1,921,535 from the seven samples from the “Clinically Healthy” group at T2 and 2,823,382 from the eight samples from the “Moribund” group at T2. The total number of reads in the four groups was 9,689,706, with an average of 310,000 paired reads for each sample.
The total number of genera identified was 975. However, 92.7% of the reads were assigned to the 50 most abundant genera.
Samples were clustered using Principal Components Analysis based on bacterial relative abundance (PCA, Fig. 2). One sample belonging to the H-T2 group was an outlier that did not cluster with any other group, so it was removed from further analysis.

Principal component analysis based on gill microbiome composition.
The samples at T1 and T2 are separated on the first principal component, explaining 55% of the total variance. Some separation is also evident in the second component (explaining 15% of the total variance) between ‘’Clinically Healthy ‘’T1 and ‘’Moribund’’ T1, while T2 samples show a similar microbiome profile.
At T1, 18 bacterial genera had significative variation (FDR < 10−5) between healthy and moribund populations. The ten most significant are reported in Table 1. Among these genera, the only one with a prevalence over 1% was the Yersinia genus, with an FDR of 1.96 × 10−14 and representing 4.7% of the total reads in the pretreatment.
Interestingly, the six most represented genera at T1 (Pseudomonas, Psychrobacter, Kaistella, Sphingobacterium, Flavobacterium, and Acinetobacter), accounting for 66.6% of the reads in the sample, didn’t present a significative variation between healthy and moribund groups.
When considering the confrontation on a species level, the analysis identified 19 varying species with FDR < 10−5. Among these, Yersinia ruckeri (representing almost the totality of reads attributed to the Yersinia genus) with an FDR of 2.08 × 10−14 and 3 Flavobacterium species identified by the analysis as F. psycrophilum (FDR 9.55 × 10−24, F1 now on), F. branchiophilum (FDR 5.33 × 10−7, F2 now on) and presumed F. bernardetii (FDR 2.28 × 10−10, F3 now on), were all more prevalent in the moribund group.
At T2, six bacterial genera had significative variation (FDR < 10−5) between healthy and moribund populations (Table 2).
The antibiotic treatment dramatically decreased the diversity of bacterial genera in the T2 samples. Psychrobacter, Kaistella, and Flavobacterium represented 28.5%, 26%, and 13% of the total reads at T2, respectively. Together with Arthrobacter (8.8%), these four genera represented 76.3% of the total reads.
When comparing these two groups on the species level, the analysis identified 11 species with FDR < 10−5. Also in this comparison Yersinia ruckeri, F1, and F2 significantly varied between the ‘’Clinically Healthy’’ and the ‘’Moribund’’ groups (FDR of 1.26 × 10−15, 1.2 × 10−7, and 5.65 × 10−9, respectively). In this case, Y. ruckeri and F1 species were found with greater abundance in the ‘’Clinically Healthy’’ group than in the ‘’Moribund’’ group. F2 maintained the same behavior it had at T1, while F3 didn’t present statistically significant variation among the groups (FDR 1).
125 bacteria genera had FDR < 10−5 in the confrontation between T1 and T2 groups. The ten most significant are reported in Table 3. The genus Yersinia represented, again, a good descriptor of this variation with an FDR of 9.77 × 10−15, but it was only the 59th population in order of importance (lowest to highest FDR). The Flavobacterium genus, also in this confrontation, didn’t have an important variation (FDR of 0.003, ranking > 200th by FDR, with a log2 fold change of − 0.3), although it is the 3rd most represented genera (Fig. 3).

Heatmap and hierarchical clustering based on the fifty most abundant genera ordered by their total abundance from highest (top) to lowest (bottom). The colors of the cells indicate the relative number of reads assigned to a species in every sample, from higher (red) to lower (blue).
In this case, the same four bacterial species considered previously behaved differently: Yersinia ruckeri, although remaining statistically significant (8.56 × 10−13 FDR) between T1 and T2, represented only the 191st species in order of importance.
The three Flavobacterium species previously considered did not show strong variation, with an FDR of 3.6 × 10−3, 0.54, and 0.93 (for F1, F2, and F3, respectively) and ranking > 300th by significance.
30 species of Flavobacterium have been found statistically significant between T1 and T2. Still, these were overall very poorly represented (overall 1.4% of total reads), and the most significant two were in the 30th and 31st positions, each representing less than 0.15% of total reads. The vast majority of the genus Flavobacterium was associated with only 3 species, accounting for 10% of the total reads, without any statistical difference between T1 and T2, mimicking the trend of the entire Flavobacterium genus.
Discussion
To best investigate the role of gill microbiota in a suspected gill disease outbreak in rainbow trout, we employed culture-independent high-throughput analysis on gill tissue before and after administration of antibiotic treatment.
Principal Component Analysis (PCA) based on the composition of microbial genera clearly differentiated three groups that can be indicated as “T1 healthy fish,” “T1 moribund fish,” and “T2 fish.” In the T1 sampling time point, the healthy and moribund populations formed distinct clusters, while in the fish groups at T2, healthy and moribund fish clustered together.
The analysis of the bacterial genera based on NGS showed a very complex scenario identifying more than 400 genera. Overall, three genera (Psychrobacter, Kaistella, and Flavobacterium) represented 54.8% of the bacterial population. Furthermore, the Arthrobacter genus represented 5.3% of the total. Among these four bacterial genera, Psychrobacter, Kaistella, and Arthrobacter significantly varied between samples at T1 and T2 (FDR of 2.49 × 10−11, 3.72 × 10−23, and 1.83 × 10−30). In contrast, the genus Flavobacterium did not show such a strong variation (FDR 0.003). When considering only samples at the T1 sampling time-point, Psychrobacter and Flavobacterium were the only populations with more than 10% of reads. Still, more bacterial genera were well represented with a total of reads > 5%, namely Kaistella, Pseudomonas, Sphingobacterium, and Acinetobacter. Before the treatment, these six bacterial genera represented 66.6% of the reads.
At T2, the population proportions underwent a major change, with a decrease in the Flavobacterium genus and an increase in the Psychrobacter and Kaistella genera. At T2, aside from these three, only the Arthrobacter genus represented more than 5% of total reads. At T2, these four bacteria genera represented 76.3% of the total reads.
125 bacterial genera had a significant variation between T1 and T2 (FDR < 10−5). 11 of these had a variation with FDR < 10−100. These were Acinetobacter, Pseudomonas, Sphingobacterium, Diaphorobacter, Acidovorax, Frigoriflavimonas, Polaromonas, Lampropedia, Epilithonimonas, Janthinobacterium, and Kurthia.
All these bacterial genera have been described as normal environmental inhabitants30, with few of them being reported as pathogenic or opportunistic pathogens to humans (Acinetobacter, Pseudomonas, Sphingobacterium)31,32. Others have been described in fish (Epilithonimonas and Janthinobacterium)33,34.
When comparing healthy and moribund specimens at T1 and T2, much fewer genera changed significantly with FDR < 10−5 (18 in the pretreatment group and 6 in the posttreatment). This was expected, considering the PCA analysis showed a milder variation between the healthy and the moribund specimens belonging to the same group, particularly at the T2 time-point.
Collectively, this data illustrates a significant difference in gill microbiota composition between the T1 and T2 groups, characterized by a notable reduction in population diversity at T2, which correlates with decreased mortality. Some bacterial genera recognized in this study are paramount in fish health35,36,37,38. Still, most of them are typically found in different environments and have not been previously associated with fish-related diseases. These results show that the partial resolution of the mortality event correlated with the variation in environmental populations of bacteria rather than specific pathogens, hinting at the complexity of the disease.
When considering the treatment, the administration of Oxytetracycline significantly decreased mortality rates between T1 and T2. However, the mortality rates remained higher than the facility's average basal mortality. This suggests, in our case, that the development of the disease involved a complex interplay between bacterial populations, hosts, and environmental factors rather than a simple primary infection, as is commonly believed in such cases.
Among the genera identified in our samples, significant focus has been directed towards the Flavobacterium genus, historically linked to Bacterial Gill Disease (BGD)14. The authors expected the antibiotics to reduce the population of Flavobacterium due to its susceptibility. However, the treatment resulted in more substantial changes in other taxa. While the Flavobacterium genus is one of the most prevalent in our study, the primary outcome of the NGS analysis indicates that it remains relatively stable between Healthy and Moribund samples at both T1 and T2 time points. Its False Discovery Rate (FDR) value remains significantly high (FDR 3 × 10−3) even when comparing T1 and T2, in contrast to over 125 genera with significantly lower FDR values. Collectively, these findings enormously diminish the role of Flavobacteria or even exclude their involvement in the mortality event in this case.
Unfortunately, the absence of a genuine, healthy negative control limited our ability to determine the prevalence of this genus in healthy populations. Further research is needed to establish the typical baseline prevalence of the Flavobacterium genus under normal conditions. Another challenge of the present study is the need to improve databases of prokaryotic sequences due to the vast number of yet-undescribed microorganisms20. Identification of bacteria relies on the best match of reads to the taxa archived in public databases. Although rapidly evolving and growing, such databases are still incomplete, which calls for the need to repeat the alignment in the future.
In light of the histological and bacteriological diagnostic results obtained at T1, the role of Flavobacterium spp. may have been initially overestimated. The NGS data has also been examined at a species level, offering additional valuable insights for a more comprehensive understanding of the Flavobacterium genus involvement. At the species level, NGS analysis revealed the presence of Flavobacterium psychrophilum, Flavobacterium branchiophilum, Flavobacterium bernardetii, and Yersinia ruckeri. Flavobacterium branchiophilum (associated with BGD) and Flavobacterium psychrophilum (associated with Bacterial Cold-Water Disease and Rainbow Trout Fry Syndrome) have been isolated in many freshwater environments39. Furthermore, F. psychrophilum may be part of the normal aerobic flora populating salmonids’ skin and gills40. Flavobacterium bernardetii is a novel species recently associated with cutaneous and abdominal hemorrhages in Rainbow trout fry in Turkey, but not associated with gill disease41.
On the other hand, Yersinia ruckeri is recognized as the etiological agent of ERMS. Yersinia ruckeri varied considerably between Healthy and Moribund fish groups and showed high concentration in the moribund fish group at T1. This caused a massive imbalance towards T1 groups, which seems particularly relevant when comparing T1 and T2 samples. The failure of the treatment with enhanced sulphonamides implied that Yersinia was not the primary cause of the mortality. The presence of Yersinia ruckeri on fish’s gills is not surprising as gills can function as the port of entry for this bacterium that secondarily causes septicemia37. Classic histological signs of ERMS on gills include hyperemia, edema, and sloughing37, which were absent here. This bacterium likely aggravated the fragile fish’s health condition, acting as a co-factor of gill disease, probably facilitating the death of the animals.
Although Flavobacterium spp. and Yersinia ruckeri were among the best descriptors of the variation between Healthy and Moribund specimens at T1 (the three Flavobacterium spp. were respectively 11, 445, and 2.5 times more concentrated in the moribund fish while Yersinia was 29 times more concentrated), they weren’t as good in differentiating between T1 and T2 groups.
In this study, certain species of Flavobacterium were found to be highly concentrated in moribund animals exhibiting eventually fatal respiratory distress; this, along with the isolation of a specific bacterial strain through conventional bacteriological methods, may have contributed to the misconception that a single species was responsible for causing BGD in previous research. However, our case study suggests a more intricate network of factors involving numerous other prokaryotic populations and management conditions. Generally speaking, if the latter are suboptimal, they may trigger the onset of gill disease.
Although classical bacteriology was able to isolate a variety of strains generally associated with disease in fish, the bacteria identified proved not to be the sole etiological agents of this gill disease outbreak. The difference between NGS and bacteriology analyses, particularly in the case of Yersinia ruckeri at the T2 time-point, underscores significant limitations associated with the latter. The main drawback of bacteriology is that it relies on bacterial growth in a culture medium; this represents a bias when it comes to quantitative analysis (in our case, it caused a considerable overestimation of the presence of the bacterium since it represented < 0.003% of reads at T2 NGS).
Evident differences among the groups, either at T0 versus T1 versus T2 or Healthy versus Moribund fish groups, were difficult to appreciate through histological analyses due to the generalized chronic condition affecting the gills in all groups. However, similarly to what was also evident in bacteriology, histology did not identify gram-negative rods compatible with Flavobacteria at T2, despite these representing 13% of the NGS reads. These results suggest that histology is a valid tool to detect pathological alterations but must be used in concert with other diagnostics to be fully exploited.
Interestingly, the intermittent presence of amoebic trophozoites within the hyperplastic gills is unlikely to have exerted a significant influence on the current outbreak. Indeed, the role of amoebae in gill disease and as agents of Nodular Gill Disease (NGD) is still controversial, and their presence should be interpreted with caution42,43. NGD has also been described as worsening the BGD condition, speculating that amoebae feed on the bacterial biofilm and act as secondary pathogens, becoming predominant at a later stage in the disease course8. Lastly, an experimental infection of the gills with amoebae, causing NGD in rainbow trout, is still lacking, and Koch’s postulates have not been proven yet. Moreover, the antibiotic treatment’s efficacy and the protozoans’ inconsistent presence in the histological slides strongly suggest that amoebae did not play a significant role in this outbreak.
During the water quality analysis, it was observed that during the outbreak, suspended solids were noticeable to the naked eye and settled at the bottom of the farming tanks. However, the quantitative parameters tested were constantly within normal limits and compatible with fish farming27. The Authors believe that the sampling frequency and the tendency to fast sedimentation could have altered the samples' representativeness in this case.
Finally, it was hypothesized that the change from high-quality raw feed ingredients to low-quality ones due to the COVID-related economic crisis caused a decreased feed digestibility with a consequent increase in suspended solids in the water due to increased feces production27. Moreover, wintertime for Rainbow trout overlaps with the spawning period, which in salmonids is associated with changes including the reduction in the immune competence of the animals, consequently decreasing resistance to pathogens and increasing basal mortality risk44. Without further antibiotic treatment, a complete outbreak resolution was achieved only through a change in the raw feed material, increasing the digestibility of the feed, and restoring the water supply within the facility (due to an increase in rainfall). The reduction in water flow and the removal rate of the system, the rise in suspended solids due to low feed digestibility, and the metabolically active physiological state of the fish possibly played an essential role in facilitating gill disease and increasing mortality.
Conclusions
In conclusion, this case study highlights the complexity of gill disease in rainbow trout species, similar to what is seen for multifactorial gill disease in farmed Atlantic salmon. NGS analysis showed that fish mortality wasn’t due to a single bacterial agent. Microbiota diversity was likely linked to improved fish health, and environmental and zootechnical factors could influence this equation. In the authors’ opinion, classic bacteriological and histological diagnostics should be interpreted cautiously, as they may overestimate the role of some bacteria in gill disease, particularly when considering Flavobacterium species.
In the future, NGS will represent an asset in investigating further complex field cases of supposed multifactorial origin and supporting standard diagnostic procedures addressing or ruling out the actual involvement of the various prokaryotic or eukaryotic populations in the course of the disease. The ability of NGS to produce results partially overcoming the limits imposed by Microbial Dark Matter and classical bacteriology may represent a critical factor in elucidating many complex field situations.
More studies are needed to assess the existence of a basal gill microbiome and correlate the variation of the microbiome with the various triggers possibly involved in the onset and progression of the disease. Further on-site investigations are necessary to fully understand the relationship between bacteria, environmental factors, and husbandry conditions. The research should focus on the interaction between prokaryotic and eukaryotic biomes, management conditions, total suspended solids, and water quality parameters.
Material and methods
Ethical statement
The tissue sampling used in this study was conducted at the fish farm by the attending veterinarians as part of a thorough diagnostic iter and following routine farm practices. No procedure was conducted on live fish. All the methods and results applied and reported in the study are the results of this diagnostic iter, which integrated high-throughput technology with classical investigation methods during a disease outbreak.
As per Directive 2010/63/EU of the European Parliament and of the Council of September 22, 2010, regarding the protection of animals used for scientific purposes, the Italian legislation (D. Lgs. n. 26/2014) does not require approval from ethics committees for the use of samples submitted or taken for diagnostic purposes.
Water quality and environmental parameters
Water samples were collected to assess water quality throughout the study. Total Gas Pressure, Oxygen content, and temperature were tested through the use of OxyGuard® TGP Probe (OxyGuard International A/S; Farum, Denmark) (https://www.oxyguard.dk/en/probes/tgp-probe/). Oxygen and temperature were also continuously monitored through the site’s monitoring system. CO2 was measured with OxyGuard® CO2 analyzer (OxyGuard International A/S; Farum, Denmark) (https://www.oxyguard.dk/en/product-overview-hand-held-portable/co2-analyzer/), and pH was measured with pH-meter HannaHI98128 (Hannah Instruments; Woonsocket, RI, USA) (https://hannacan.com/hi98128-phep-ph-tester).
Water samples were taken at 5 points throughout the facility and immediately transported to a lab for analysis. The 5 points were:
-
1 at the water entrance in the facility.
-
2, 3, and 4 in the middle of the facility.
-
5 at the end of the facility before settling tanks.
All these locations were sampled at 4 different time points:
-
7.30 AM before the feeding starts.
-
Noon, after the last feed.
-
2.00 PM, 2 h after feeding.
-
4.00 PM, 4 h after feeding.
The parameters tested in every location in all the time points were NH4, NO2, NO3, and PO4.
Active chlorine and fecal coliforms were tested only at the water inlet as indicators of human pollution in waters. Total suspended solids were tested only at point 4 immediately after tanks with a significant mortality event. All water analyses followed the protocols described in the APAT-ISRA-CNR manual45.
Fish sampling
Samples were taken at three time-points for diagnostic procedures named T0 (before enhanced sulphonamide treatment), T1 (before starting the oxytetracycline treatment), and T2 (after eight days of treatment with oxytetracycline).
All fish were humanely euthanized with tricaine methanesulfonate overdose (Tricaine, Pharmaq®).
At the T0 time-point, samples for wet mounts, necropsy, and histology (4 fish) and for virology, bacteriology, and antimicrobial sensitivity testing (6 fish) were taken from moribund fish from 5 affected tanks of Sector 2 and one non-affected tank as a negative control sampled from sector 1 with mortality lower than 0.01% per day on the stock.
At the time-point T1, before oxytetracycline treatment, fish were collected by another affected tank from Sector 2 (different from the ones sampled during T0) and subdivided into two subgroups based on the feeding behavior, which was considered an indicator of the progression of the disease; fish were therefore categorized as ‘’clinically healthy’’, still feeding and healthful from a clinical perspective, and ‘’moribund’’ fish groups, showing anorexia and lethargy, swimming near tank’s borders. At this time-point, moribund diseased fish were sampled for wet mounts, necropsies, histology (4 fish), and bacteriology (6 fish). Additionally, gill samples from 16 fish (8 from the ‘’clinically healthy’’ group and 8 from the ‘’moribund’’ group) were taken for 16S NGS analysis.
After oxytetracycline treatment, a second batch of samples was taken from the same tank at the time-point T2. The samples included both healthy and moribund specimens for wet mounts, necropsies, histology (4 fish in each group), bacteriology (6 fish in each group), and 16S NGS analysis, as already described for the T1 time-point. Details of sampling and results of necropsies, wet mounts, bacteriology, and virology are summarized in Table S6 of the Supplementary material.
Wet mounts, necropsy, and histology
Wet mounts were performed by scraping the skin with a glass coverslips and squeezing the coverslip to the histological slide. The gills were analyzed by cutting the filaments’ base, laying them down on the histological glass with a drop of physiological solution, and pressing them down with the coverslips. Standard necropsies were performed in two steps29: macroscopic evaluation of the gills and the integument, opening of the coelomic cavity and macroscopic observation of the internal organs. Histological sampling was, in all cases, performed on-site, collecting and fixing fish tissues in 10% neutral buffered formalin (NBF). Gills and organ pools (liver, head and trunk kidney, spleen, heart, pancreas, intestine, and ovary) were fixed in NBF for 48 h. After fixation, the pools were trimmed in separate biocassettes, dehydrated in ethanol series, and embedded in paraffin. Tissue sections were cut at 4 µm with a microtome and stained with Hematoxylin and Eosin (H&E).
Virology
Portions of the head kidney, spleen, heart, and brain were collected from fish at the T0 time-point and pooled for biomolecular analysis targeting infectious haematopoietic necrosis virus (IHNv), viral haemorrhagic septicaemia virus (VHSv), and salmonid alphavirus (SAV).
The samples were manually grounded with sterile quartz sand. Minimum Essential Medium (MEM) was added to obtain a 1:3 w/v dilution. The homogenized sample was centrifuged at 8000 g for 2 min, and the supernatant was collected. Total nucleic acids were extracted from 250 μl of supernatant using QIAsymphony DSP Virus/Pathogen Midi Kit (Qiagen) and the automated QIAsymphony SP (Qiagen) system. Isolation of the nucleic acids was performed following the manufacturer’s recommendations. Real-time RT-PCR was employed to detect IHNv, VHSv, and SAV46,47,48.
Bacteriology and antimicrobial sensitivity test
Bacteriological examination of the spleen, kidney, and brain specimens for all sampling time points was performed on Columbia Blood Agar (Biolife, Milano—Italy), and gill mucus was cultured on both Columbia Blood Agar and Enriched Anacker and Ordal agar49.
The Columbia Blood Agar plates were incubated at 22 ± 2 ℃ for 24–48 h, while the Enriched Anacker and Ordal Agar plates were incubated at 15 ± 2 ℃ for 48–96 h49.
Single colonies were isolated based on their macroscopic appearance; bacterial cell morphology, oxidase, catalase, and KOH tests were employed to characterize the isolated strains49.
Initial bacterial screening and identification have been done through the MALDI-TOF MS assay50 (Bruker Daltonic, Bremen, Germany). Thanks to the high reliability of this method in identifying those pathogens, Yersinia ruckeri and Flavobacterium psychrophilum identifications were considered final with MALDI-TOF MS51,52. For other important genera associated with pathology in rainbow trout (in this case, the Aeromonas genera), the identification to the species level has been deepened through 16S rRNA gene amplification and sequencing53 and gyrB amplification and sequencing54.
The characterization of other genera not associated with major diseases in rainbow trout was stopped at the genus level. For Yersinia ruckeri, the biotype has been attributed through the evaluation of the motility by cultivating the colonies overnight in semisolid TSA at 22 ± 2 ℃55. Yersinia ruckeri is defined as biotype 1 if it has motility and biotype 2 if it is immobile36.
A summary of the identification methods employed for the various infectious agents found by bacteriology and virology is shown in Table S7 of the Supplementary Materials.
Antimicrobial sensitivity testing was performed using both the Kirby-Bauer method and the MIC test56. The tested antibiotics were Amoxicillin, Enrofloxacin, Florfenicol, Flumequine, Oxytetracycline, Enhanced Sulphonamides, and Erythromycin.
NGS and bioinformatic analysis
All frozen gill samples from T1 and T2 (32 samples) were transported in dry ice to the IGAtech lab (Udine, Italy), where metagenomic analyses were performed. Fish undergoing metagenomic analyses were divided in four groups: T1 ‘’Clinically Healthy’’ Fish (H-T1), T1 ‘’Moribund’’ Fish (M-T1), T2 ‘’Clinically Healthy’’ Fish (H-T2), and T2 ‘’Moribund’’ Fish (M-T2). Samples were thawed, and a gentle scrape on the surface of the gills was done to collect as much mucous as possible and limit the contamination of the sample from the host’s DNA24,57. Serum and stool protocol of Mag-Bind®Universal Pathogen 96 Kit (Omega Bio-tek, Norcross, GA) were used for DNA extraction with minor modification of the initial step: mucous was resuspended in 600 µl SLX-Mlus buffer and briefly vortexed 10 s at max speed to homogenize samples. Subsequently, 300 µl of lysate was collected and vortexed at maximum speed for 5 min to lyse samples. The final elution was done with 30 µl of 10 mM Tris HCl.
Libraries were prepared by following Illumina 16S Metagenomic Sequencing Library Preparation protocol in two amplification steps: an initial PCR amplification using locus-specific PCR primers with overhangs 16S-341F 5′- TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGCCTACGGGNGGCWGCAG -3′ and 16S-805R 5′- GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGGACTACNVGGGTATCTAATCC -3′, and a subsequent amplification that integrates relevant flow-cell binding domains and unique indices (NexteraXT Index Kit, FC‐131‐1001/FC‐131‐1002). PNA clamping was applied during the first amplification step to block host mitochondrial 16S sequences amplification following the manufacturer’s protocol (PNA Bio Inc, Newbury Park, CA). Libraries were sequenced on a NovaSeq 6000 instrument (Illumina, San Diego, CA) in paired-end 250-bp mode.
Reads were classified using Kraken2 with default parameters58 on the 16 s curated RefSeq database (https://www.ncbi.nlm.nih.gov/refseq/targetedloci/16S_process/) at the species and genus level. Kraken2 associates short genomic substrings (k-mers) with the lowest common ancestor (LCA) taxa58, it is memory intensive, ultrafast, and has been previously used on several complex matrices59. All subsequent analyses were performed at the taxonomic level of species and genus.
Abundance estimation was refined using bracken (“Bracken: estimating species abundance in metagenomics data”60) with the following parameters.
-
The threshold was set to 10 reads. The threshold is the minimum number of reads attributed by kraken2 to a taxa to perform taxa abundance estimation with bracken2. This step was used to remove extremely rare taxa from the analysis.
-
Read length was set to 250. This corresponds to the length of the reads after the removal of adapters performed by the sequencing facility.
Beta diversity and differential abundance were assessed using DESeq261.
Differential abundance results for the comparisons M-T1 VS H-T1 and M-T2 VS H-pT2 were filtered to remove false positives due to rare taxa. Results were considered only for species satisfying the following criteria:
-
The number of reads assigned to a taxa in the two groups must be at least 500.
-
At least one read must be assigned to the taxon in at least five samples (representing the 50% + 1 of a single group).
Results of the differential abundance were sorted by false discovery rate (FDR). Taxa with FDR < 10−5 were prioritized in the presentation and discussion of the results.
Data availability
The datasets generated and analysed during the current study are available in the SRA repository under accession number PRJNA1096935, https://dataview.ncbi.nlm.nih.gov/object/PRJNA1096935?reviewer=9n8lvmb09ts8d1635k9oq1qkbh. Scripts and functions used in this work are available at https://github.com/fabiomarroni/BGD.
References
Boyd, C. E., McNevin, A. A. & Davis, R. P. The contribution of fisheries and aquaculture to the global protein supply. Food Sec. 14, 805–827 (2022).
Food and Agriculture Organization (FAO) The State of World Fisheries and Aquaculture 2018—Meeting the Sustainable Development Goals; Food and Agriculture Organization of the United Nations: Rome, Italy (2018). https://www.fao.org/documents/card/en?details=I9540EN.
Sommerset, I. et al. Norwegian Fish Health Report 2022, Norwegian Veterinary Institute Report, series #5a/2023, published by the Norwegian Veterinary Institute in 2023. (2022) https://www.vetinst.no/rapporter-og-publikasjoner/rapporter/2023/norwegian-fish-health-report-2022.
Walker, L., Levine, H. & Jucker, M. Koch’s postulates and infectious proteins. Acta Neuropathol. 112, 1–4 (2006).
Scottish Government. Marine Scotland Science Scotland’s 10 year Farmed Fish Health Framework (The Scottish Government, Edinburgh, 2018).
Boerlage, A. S. et al. Epidemiology of marine gill diseases in Atlantic salmon (Salmo salar) aquaculture: a review. Rev. Aquac. 12, 2140–2159 (2020).
Bruno, D. W., Noguera, P. A. & Poppe, T. T. A Colour Atlas of Salmonid Diseases 2nd edn. (Springer, 2013).
Speare, D. J. & Ferguson, H. W. Gills and Pseudobranchs Systemic Pathology of Fish, 2nd, 51 (Scotian Press, 2012).
MacPhee, D. D. et al. Influence of feeding on the development of bacterial gill disease in rainbow trout Oncorhynchus mykiss. Dis. Aquat. Organ 21, 163–170 (1995).
Bullock, G. L. et al. Observations on the occurrence of bacterial gill disease and amoeba gill infestation in rainbow trout cultured in a water recirculation system. J. Aquat. Anim. Health. 6, 310–317 (1994).
Good, C. M. et al. A prospective matched nested case–control study of bacterial gill disease outbreaks in Ontario, Canada government salmonid hatcheries. Prev. Vet. Med. 95, 152 (2010).
Good, C. et al. Flavobacterium branchiophilum and F. succinicans associated with bacterial gill disease in rainbow trout Oncorhynchus mykiss (Walbaum) in water recirculation aquaculture systems. J. Fish Dis. 38, 409–413 (2015).
Ordal, E. J. & Rucker, R. R. Pathogenic myxobacteria. Proc. So.c. Exp Biol. Med. 56, 15–18 (1944).
Wakabayashi, H., Huh, G. J. & Kimura, N. Flavobacterium branchiophila sp. nov., a causative agent of bacterial gill disease of freshwater fishes. Int. J. Syst. Bacteriol. 39, 213–216 (1989).
Bullock, G. L. Studies on selected myxobacteria pathogenic for fishes and on bacterial gill disease in hatchery-reared salmonids 30 (US Fish Wildl. Serv. Tech. Pap, Washington, 1972).
Wakabayashi, H., Egusa, S. & Fryer, J. L. Characteristics of filamentous bacteria isolated from a gill disease of salmonids. Can. J. Fish Aquat. Sci. 37, 1499–1504 (1980).
Rucker, R. R., Johnson, H. E. & Kaydas, G. M. An interim report on gill disease. Prog. Fish. Cult. 14, 10–14 (1952).
Steinum, T. et al. An RT PCR-DGGE survey of gill-associated bacteria in Norwegian seawater-reared Atlantic salmon suffering proliferative gill inflammation. Aquaculture 293, 172–179 (2009).
Solden, L., Lloyd, K. & Wrighton, K. The bright side of microbial dark matter: Lessons learned from the uncultivated majority. Curr. Opin. Microbiol. 31, 217–226 (2016).
Jiao, J. Y. et al. Microbial dark matter coming to light: challenges and opportunities. Natl. Sci. Rev. 8, nwaa280 (2020).
Hutson, K. S. et al. Assigning cause for emerging diseases of aquatic organisms. Trends Microbiol. 31, 681–691 (2023).
Slatko, B. E., Gardner, A. F. & Ausubel, F. M. Overview of next-generation sequencing technologies. Curr. Protoc. Mol. Biol. 122, e59 (2018).
Slinger, J., Adams, M. B. & Wynne, J. W. Bacteriomic profiling of branchial lesions induced by Neoparamoeba perurans challenge reveals commensal dysbiosis and an association with Tenacibaculum dicentrarchi in AGD-affected Atlantic salmon (Salmo salar L.). Microorganisms 8, 1189 (2020).
Birlanga, V. B. et al. Dynamic gill and mucus microbiomes during a gill disease episode in farmed Atlantic salmon. Sci. Rep. 12, 16719 (2022).
Leal, J. F., Santos, E. B. H. & Esteves, V. I. Oxytetracycline in intensive aquaculture: Water quality during and after its administration, environmental fate, toxicity and bacterial resistance. Rev. Aquacult. 11, 1176–1194 (2019).
Rigos, G. et al. Best therapeutic practices for the use of antibacterial agents in finfish aquaculture: A particular view on European seabass (Dicentrarchus labrax) and gilthead seabream (Sparus aurata) in Mediterranean aquaculture. Rev. Aquacult. 13, 1285–1323 (2021).
Schumann, M. & Brinker, A. Understanding and managing suspended solids in intensive salmonid aquaculture: A review. Rev. Aquacult. 12, 2109–2139 (2020).
Roberts, R. J. The Aquatic Environment. In Fish Pathology (ed. Roberts, R. J.) 1–16 (Wiley-Blackwell, 2012).
Noga, E. J. Fish Disease: Diagnosis and Treatment 2nd edn. (Wiley-Blackwell, 2010).
Parte, A. C. et al. List of Prokaryotic names with standing in nomenclature (LPSN) moves to the DSMZ. Int. J. Syst. Evol. Micr. 70, 5607–5612 (2020).
Doughari, H. J. et al. The ecology, biology and pathogenesis of Acinetobacter spp. An overview. Microbes Environ. 26, 101–112 (2011).
Barahona, F. & Slim, J. Sphingobacterium multivorum: case report and literature review. New Microbes New Infect. 7, 33–36 (2015).
Ayala, M. et al. Draft genome sequence of Epilithonimonas sp. FP211-J200, isolated from an outbreak episode on a Rainbow trout (Oncorhynchus mykiss). Farm. Genome Announc. 5, e00819-17 (2017).
Oh, W. T. et al. Janthinobacterium lividum as an emerging pathogenic bacterium affecting Rainbow trout (Oncorhynchus mykiss) fisheries in Korea. Pathogens 8, 146 (2019).
Vendrell, D. et al. Lactococcus garvieae in fish: A review. Comp. Immunol. Microbiol. Infect. Dis. 29, 177–198 (2006).
Tomás, J. M. The main Aeromonas pathogenic factors. ISRN. 2012, 256261 (2012).
Kumar, G. et al. Yersinia ruckeri, the causative agent of enteric redmouth disease in fish. Vet. Res. 46, 103 (2015).
Fernández-Bravo, A. & Figueras, M. J. An update on the genus Aeromonas: Taxonomy, epidemiology, and pathogenicity. Microorganisms 8, 129 (2020).
Loch, T. P. & Faisal, M. Flavobacterium spp. In Fish Viruses and Bacteri: Pathobiology and Protection (eds Woo, P. T. K. & Cipriano, R. C.) 211–232 (CABI International, 2017).
Nematollahi, A. et al. Flavobacterium psychrophilum infections in salmonid fish. J. Fish Dis. 26, 563–574 (2003).
Saticioglu, I. B. et al. Flavobacterium bernardetii sp. nov., a possible emerging pathogen of farmed rainbow trout (Oncorhynchus mykiss) in cold water. Aquaculture 540, 736717 (2021).
Vannetti, S. M. et al. Amoeba species colonizing the gills of rainbow trout (Oncorhynchus mykiss) in Swiss aquaculture. J. Fish Dis. 46, 987–999 (2023).
Brocca, G. A. et al. Identification of new amoebae strains in rainbow trout (Oncorhynchus mykiss, Walbaum) farms affected by nodular gill disease (NGD) in Northeastern Italy. J. Fish Dis. 00, e13933 (2024).
Campbell, J. H., Dixon, B. & Whitehouse, L. M. The intersection of stress, sex and immunity in fishes. Immunogenetics 73, 111–129 (2021).
Belli, M. et al. Sezione 4000 - Inorganici non metallici, Sezione 5000 – Organici. In: Metodi analitici per le acque. APAT, Rapporti 29/2003 (2003).
Overturf, K., LaPatra, S. & Powell, M. Real-time PCR for the detection and quantitative analysis of IHNV in salmonids. J. Fish Dis. 24, 325–333 (2001).
Jonstrup, S. P. et al. Development and validation of a novel Taqman-based real-time RT-PCR assay suitable for demonstrating freedom from viral haemorrhagic septicaemia virus. J. Fish Dis. 36, 9–23 (2013).
Hodneland, K. & Endresen, C. Sensitive and specific detection of Salmonid alphavirus using real-time PCR (TaqMan). J. Virol. Methods 131, 184–192 (2006).
Buller, N. B. Bacteriological culture techniques: microscopy culture and identification. In: Bacteria and fungi from fish and other aquatic animals: A practical identification manual. (2nd ed.) 425–427 (CABI International, 2014).
Li, D. et al. MALDI-TOF Mass Spectrometry in Clinical Analysis and Research. ACS Meas. Sci. Au. 2, 385–404 (2022).
Fernández-Álvarez, C., Torres-Corral, Y. & Santos, Y. Use of ribosomal proteins as biomarkers for identification of Flavobacterium psychrophilum by MALDI-TOF mass spectrometry. J. Proteom. 170, 59–69 (2018).
Ojasanya, R. A. et al. Development and validation of main spectral profile for rapid identification of Yersinia ruckeri isolated from Atlantic salmon using matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. Front. Vet. Sci. 9, 1031373 (2022).
Davidovich, N. et al. An outbreak of crayfish rickettsiosis caused by Coxiella cheraxi in redclaw crayfish (Cherax quadricarinatus) imported to Israel from Australia. Transbound. Emerg. Dis. 69, 204–212 (2022).
Gonçalves Pessoa, R. B. et al. The genus aeromonas: A general approach. Microb. Pathog. 130, 81–94 (2019).
Riborg, A., Colquhoun, D. J. & Gulla, S. Biotyping reveals loss of motility in two distinct Yersinia ruckeri lineages exclusive to Norwegian aquaculture. J. Fish Dis. 45, 641–653 (2022).
CLSI. Performance Standards for Antimicrobial Susceptibility Testing of Bacteria Isolated From Aquatic Animals. 3rd ed. CLSI supplement VET04. Wayne, PA: Clinical and Laboratory Standards Institute, (2020).
Clinton, M. et al. Sampling the fish gill microbiome: a comparison of tissue biopsies and swabs. BMC Microbiol. 21, 313 (2021).
Wood, D. E., Lu, J. & Langmead, B. Improved metagenomic analysis with Kraken 2. Genome Biol. 20, 257 (2019).
Cattonaro, F. et al. Do you cov me Effect of coverage reduction on metagenome shotgun sequencing studies. F1000Res. 7, 1767 (2018).
Lu, J. et al. Bracken: estimating species abundance in metagenomics data. PeerJ. Comput. Sci. 3, e104 (2017).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Acknowledgements
The authors thank Cortinovis Luana, Marsella Andrea, Pretto Tobia, and Toffan Anna from the “Istituto Zooprofilattico Sperimentale delle Venezie, Legnaro (PD), Italy” for the bacteriological and virological analyses and the histological pictures employed in Panel 1; Paola Beraldo from the “Università degli Studi di Udine” and David Groman from the “AVC Diagnostic Services, University of Prince Edward Island” for the help with the wet mounts and histological pictures.
Author information
Authors and Affiliations
Contributions
SZ: Conceptualization, investigation, funding acquisition, writing—original draft, writing—review and editing; MO: sample processing, writing—original draft, writing—review and editing; GB: writing—original draft, writing—review and editing, histological analysis; CC: Investigation, sample processing, funding acquisition; SR: Methodology and NGS analysis; FM: Bioinformatic and statistical analyses; RV, LM: histological analysis, writing—review and editing; MG: Funding acquisition, supervision, writing—review and editing.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zamparo, S., Orioles, M., Brocca, G. et al. Novel insights on microbiome dynamics during a gill disease outbreak in farmed rainbow trout (Oncorhynchus mykiss). Sci Rep 14, 17791 (2024). https://doi.org/10.1038/s41598-024-68287-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-68287-w
- Springer Nature Limited