
Abstract
Obesity is characterized by dysregulated adipogenesis that leads to increased number and/or size of adipocytes. Understanding the molecular mechanisms governing adipogenesis is therefore key to designing therapeutic interventions against obesity. In our study, we analyzed 3’-end sequencing data that we generated from human preadipocytes and adipocytes, as well as previously published RNA-seq datasets, to elucidate mechanisms of regulation via long non-coding RNA (lncRNA), alternative splicing (AS) and alternative polyadenylation (APA). We discovered lncRNAs that have not been previously characterized but may be key regulators of white adipogenesis. We also detected 100 AS events and, using motif enrichment analysis, identified RNA binding proteins (RBPs) that could mediate exon skipping—the most prevalent AS event. In addition, we show that usage of alternative poly(A) sites in introns or 3’-UTRs of key adipogenesis genes leads to isoform diversity, which can have significant biological consequences on differentiation efficiency. We also identified RBPs that may modulate APA and defined how 3’-UTR APA can regulate gene expression through gain or loss of specific microRNA binding sites. Taken together, our bioinformatics-based analysis reveals potential therapeutic avenues for obesity through manipulation of lncRNA levels and the profile of mRNA isoforms via alternative splicing and polyadenylation.
Similar content being viewed by others

Introduction
Obesity is a major global public health burden. It is characterized by excessive accumulation of fat mass in white adipose tissue, which occurs due to increased adipocyte volume, increased number of cells, or both1,2. Adipocytes develop from mesenchymal stem cells (MSCs), which become committed preadipocytes and then fully differentiated adipocytes. There are two main types of adipocytes—white and brown adipocytes. White adipose tissue (WAT) functions in fat storage and is characterized by adipocytes containing large unilocular lipid droplets. It is an active endocrine organ that regulates insulin sensitivity, lipid metabolism and satiety. It is distributed throughout the body in subcutaneous regions and surrounds visceral organs. The main function of brown adipose tissue (BAT) is thermogenesis. It is composed of multiloculated adipocytes that contain large numbers of mitochondria. Its location is restricted to the paravertebral, supraclavicular and periadrenal regions3. Our focus is on white adipogenesis since obesity is caused by WAT outgrowth4.
Murine cells lines, such as 3T3-L1, 3T3-F422A and OP9, are commonly used in adipogenesis research1, with the most popular model being 3T3-L1 cells. However, these murine cells cannot completely recapitulate the process in human cells due to several differences. For example, 3T3-L1 and 3T3-F442A cell lines express the adipocyte-secreted factor leptin at much lower levels than primary adipocytes. Recently, an increasing number of studies are using human Adipose-Derived Stem Cells (ASCs) and human preadipocytes to better capture characteristics of adipose tissue when addressing questions of adipose-related biology1. However, in depth sequencing analysis of these human cells is limited but is critical to dissecting the molecular regulatory mechanisms that characterize human adipogenesis. In our study, we examine gene expression regulation during differentiation of human primary preadipocytes into white adipocytes by using high-depth sequencing libraries and advanced bioinformatic tools, and in this way, expand our understanding of the role of long non-coding RNAs (lncRNAs), alternative splicing, and alternative polyadenylation (APA).
LncRNAs are small RNA transcripts that are longer than 200 nucleotides, generally have a 5’-cap and 3’-polyA tail, and do not code functional proteins. They regulate various processes and some play important roles in adipogenesis based on studies using cell lines and primary cells5,6. Since lncRNAs have low conservation between human and mouse7, studies using primary human cells are of greater relevance to translational research. Studies using primary human adipocytes have characterized individual lncRNAs identified from microarray or RNA-sequencing8,9 or studied expression in cells from obese and lean individuals10. One study in 2015 examined global changes in lncRNA expression during differentiation of human adipose-derived stromal cells11. However, the improvement of sequencing strategies may now allow the identification of novel lncRNAs with important roles in adipogenesis that may have been missed in the earlier studies. In our study, we sought to use global profiling to identify such lncRNAs and provide a resource for comparative analysis with future datasets.
APA during adipogenesis has been less extensively studied. APA is a pre-mRNA processing mechanism that generates different mRNA isoforms based on usage of alternative polyA (pA) sites. APA often results in isoforms with the same coding sequence but different 3’-UTR lengths, with downstream effects on mRNA stability, translatability, localization, and RNA-seeded protein/protein interactions, while intronic APA can alter both the coding sequence and 3’-UTR sequences12,13,14,15. Use of pA sites located in introns affects the coding sequence, leading to transcript degradation or expression of different protein isoforms12,13,15. Previously, analysis of 3’-end reads showed an overall lengthening during 3T3-L1 differentiation16. Also, in 3T3-L1 cells, the shorter transcript of heme oxygenase 1 (HO1) was demonstrated to have a stronger inhibitory effect on differentiation of preadipocytes17. In 2013, a different group used RNA sequencing to investigate APA in the 3’-UTR and found that differentiated human adipocytes have a modest trend towards longer 3’-UTRs compared to undifferentiated adipocytes18. However, a comprehensive analysis of APA at a genome-wide level with standard RNA-seq data is difficult as few reads are localized in the 3’-UTR. A goal of our study was to use 3’-focused sequencing data to accurately and quantitatively characterize alternative pA site usage and location and identify specific white adipogenesis-related genes undergoing such regulation.
Alternative splicing is another pre-mRNA processing mechanism that diversifies the transcriptome and regulates gene expression19. It is carried out by the spliceosome and various RNA-binding proteins (RBPs) that activate or repress splicing of regulated exons by binding to enhancer or silencer elements, respectively, in the pre-mRNA19. Spliced isoforms and splicing factors relevant to adipogenesis have for the most part been identified in 3T3-L1 cells and mouse animal models19. Some studies have characterized specific spliced isoforms in human adipogenesis20,21, but to our knowledge, only one paper addressed global alternative splicing during the process by inducing mesenchymal stem cells from bone marrow to differentiate into adipocytes22. In the current study, we sought to take advantage of a recent dataset23 on differentiation of stem cells derived from subcutaneous adipose tissue and analyze global alternative splicing with a commonly used tool, rMATS24.
By utilizing multiple sequencing datasets and newly available bioinformatics tools, combined with a thorough investigation into available experimental evidence, we have identified potentially new mechanisms of regulation of white adipocyte differentiation at the level of lncRNAs, alternative splicing and APA.
Methods
RNA samples and sequencing
Commercially available total RNA of human preadipocytes (Day 0) and their respective differentiated adipocytes (Day 14) were purchased from Zen-Bio Inc (Durham, NC, USA). The preadipocytes were isolated from human subcutaneous adipose tissue, plated and then differentiated into mature adipocytes for 14 days using the company’s Adipocyte Differentiation Medium (catalog number DM-2), yielding cells that were rounded with large lipid droplets apparent in the cytoplasm (Supplementary Fig. S1). Zen-Bio has also confirmed that the resulting mature adipocytes expressed the fatty acid binding protein aP2/FABP4, responded to lipolytic agents, and secreted leptin and adiponectin. We also tested expression of markers by reverse transcription of the RNA samples to cDNA using using NEB LunaScript RT SuperMix kit (New England Biolabs, M3010L) followed by qPCR using NEB Luna Universal qPCR Master Mix (New England Biolabs, M3003L) and primers listed in Supplementary Table S1. Expression of adipogenesis markers were normalized to that of RPL13A and the qPCR results were quantified using the ddCt method. Details of the donors of the adipose tissue samples are provided in Supplementary Table S2. These samples were sent to Admera Health LLC (NJ, USA) for 3’-end sequencing using the Quant-Seq FWD library kit. The generated cDNA libraries were sequenced as 150 bp paired-end reads with read depth of 60 million reads on an Illumina [NovaSeq X Plus](Illumina, California, USA) instrument.
Differential gene expression analysis
The raw sequence reads of the samples were processed to remove adapters using Cutadapt25 (v2.8) and the quality of these trimmed reads passed using FastQC26 (v0.11.8) and MultiQC27 (v1.7.0). The FastQC reports are uploaded to GEO database (GSE250525). R1 reads were aligned using STAR28 (2.6.1d) to the Human hg38 genome. Aligned reads were then quantified using featurecounts29 (v1.6.3) and log2-fold changes were calculated with DESeq230(1.40.2) using default parameters and the UCSC annotation file (created on 2015-08-14). A fold change of 1 and an adjusted p-value of less than 0.05 were applied to select differentially expressed genes. The enrichment of Gene Ontology Biological Processes (GOBP) and Kyoto Encyclopedia of Genes and Genomes (KEGG) Pathways for upregulated and downregulated genes were carried out using the DAVID Functional Annotation tools31 and the default settings, with background genes restricted to those detected in our analysis.
To obtain a comprehensive list of lncRNAs, we obtained a complete list of gene symbols from GeneCards32, along with their categories, from https://www.genecards.org/cgi-bin/cardlisttxt.pl. We then filtered for the “RNA Gene” category and then further filtered these “RNA Genes” by removing RNAs such as piRNAs and miRNAs, that are not lncRNAs. Using this list as reference, we extracted data for differentially expressed lncRNAs from the differential gene expression data of the DESeq2 analysis above.
For analysis of differential gene expression of lncRNAs during brown adipogenesis, we used the publicly available GEO dataset GSE16838733 which contained three replicates each of brown preadipocytes (Day 0) and differentiated adipocytes (Day 14) from the human immortalized brown adipocyte Paz6 cell line. Paired-end reads of samples (GSM5137833, GSM5137834, GSM5137835, GSM5137836, GSM5137837, GSM5137838) were aligned to the hg38 genome, quantified, and the log2-fold changes were calculated as above. Differentially expressed lncRNAs were selected based on adjusted p-value < 0.05 and log2 fold change > 1 or <− 1.
Alternative polyadenylation analysis
APA analysis was performed using a polyA_DB-based APA analysis. Adapter and poly(T) sequences from QuantSeq FWD raw read 2 data were trimmed. Trimmed reads were then aligned to a human (hg38) polyA_DB34 reference (− 100nt, polyA_DB-annotated-PolyA Site (PAS), + 25 nt) using STAR-2.7.7a. The last aligned position of each mapped read was compared to polyA_DB-annotated-PASs, allowing ± 24 nt flexibility. Matched reads were considered pA site-supporting (PASS) reads, which were used for further APA analysis. UCSC genome browser plots shown in this paper are based on PASS reads. For 3’-UTR APA analysis, two pA sites with the highest usage in the 3’-UTR were compared, and for intronic APA analysis, all intronic isoforms were combined and compared to all isoforms using pA sites in the terminal exon. A RED value (Relative Expression Difference) was calculated as difference in log2-ratio of isoform abundance (isoforms from use of distal pA sites vs those from proximal pA site) between undifferentiated and differentiated samples. A “shortening” or “lengthening” of gene was called when RED <− log2(1.2) or > log2(1.2), respectively, and BH (Benjamini and Hochberg)-adjusted P-value < 0.05 (Fisher’s exact test).
The enrichment of GOBP and KEGG Pathways for shortened and lengthened genes were carried out using the DAVID Functional Annotation tools31 and the default settings, with background genes restricted to those detected in our analysis.
The presence of miRNA-binding sites was identified using the “Custom prediction tool” in the mirDB35,36 database. MiRDB includes target prediction scores between 50 and 100. For scores below 60, there is low confidence in the prediction, and it is recommended to have additional supporting evidence. Therefore, miRNA-binding predictions with “target scores” less than 60 were excluded from our analysis. We also examined whether binding sites of these miRNAs are enriched in the 3’-UTRs of genes undergoing 3’-UTR APA relative to the 3’-UTRs of all genes regardless of their APA status. To do this, we used miRDB to determine the number of genome-wide targets for the miRNAs of interest (miRNA targets with target score less than 60 were filtered out) and calculated the ratio of these miRNA targets to all genes expressed in the system. We then compared that to the ratio of miRNA-target genes undergoing APA and all genes that undergo APA in our analysis. Statistical analysis was done using a two-tailed Poisson test.
The presence of RBP-binding sites was identified through RBPmap37,38 (v1.2), using the hg38 database assembly, all 223 human/mouse motifs included in RBPmap, the stringency level set to high, and default settings for other parameters. For analysis of RBP-binding, we overlapped results from RBPmap38 with CLIP-seq data from the POSTAR339 database to identify RBP-binding to 200 bp upstream and downstream of proximal, distal and intronic polyadenylation sites.
Alternative splicing analysis
We used the GEO dataset GSE17602023 for alternative splicing analysis. In this dataset, adipose tissue-derived stem cells (ASCs) were induced to differentiate in vitro. We analyzed data from undifferentiated (Day 0) and differentiated (Day 9) samples derived from abdominal subcutaneous adipose tissue (GSM5352869, GSM5352872, GSM5352874, GSM5352877, GSM5352879, GSM5352882). FASTQ files from two runs of each sample were merged, followed by alignment of the paired-end reads to human hg38 genome using STAR28 (v2.6.1d). The BAM (alignment) files generated were used as input to the rMATS24 (v4.1.2) program, using all default parameters and UCSC annotation file (created on 2015-08-14), to detect splicing events between undifferentiated and differentiated adipocyte samples. The “JCEC” output files containing both junction and exon counts were used to detect significant differentially spliced events. To increase confidence in the rMATS calls, counts of each sample in both conditions were only considered if greater than 25. Differentially spliced events were called if dPsi [IncLevelDifference, or psi (percent-spliced-in) values between undifferentiated and differentiated samples] is greater than 0.05 or less than -0.05, and if the FDR value is less than 0.05. The enrichment of GOBP and KEGG Pathways for differentially spliced genes were carried out using the DAVID Functional Annotation tools31 and the default settings, with background genes restricted to those detected in our analysis. Sashimi plots for specific genes were generated using rmats2sashimiplot (v3.0.0) (https://github.com/Xinglab/rmats2sashimiplot).
The splicing output files from rMATS were input into “Motif Map” analysis of rMAPS240,41 (http://rmaps.cecsresearch.org/MTool/) using default parameters. This analysis involves scanning for occurrences of binding motifs of known RBPs in the intronic sequences 250 bp upstream or downstream of the target exon or flanking exons, and the first 50 bp of the 5’- or 3’-end of exonic sequences of included, skipped and non-regulated or background exons. A motif score (density) within a 50 bp sliding window is calculated, along with a P-value given in comparison between regulated and control exons40,41. Motif maps generated by rMAPS2 were edited in Inkscape (www.inkscape.org).
Image generation
Volcano plots and bar graphs were generated using Rstudio (2023.09.0) with the packages dplyr (v1.1.4)42, ggplot2 (v3.4.3)43, Enhanced volcano (v1.18.0)44 and smplot2 (v0.1.0)45. Pie charts and bar graphs were generated using GraphPad Prism version 10.0.2 for Windows, GraphPad Software, Boston, Massachusetts USA, www.graphpad.com.
Results
Long non-coding RNAs LINC00312, LINC00607 and TYMSOS may be key regulators of white adipogenesis
We sequenced polyadenylated RNAs from three different sets of preadipocytes isolated from human subcutaneous adipose tissue and compared these 3’-end datasets to those from white adipocytes derived from these precursors. Differential gene expression analysis of the sequencing data shows upregulation of previously established adipogenesis markers FABP4, ADIPOQ and PPARG46 (Supplementary Fig. S2A), and this was confirmed by RT-qPCR analysis of the same RNA samples used for sequencing (Supplementary Fig. S3). This analysis supports the efficient differentiation of our samples. GOBP and KEGG enrichment analysis of upregulated genes shows significantly enriched terms related to adipogenesis, such as fatty acid metabolism, PPAR signaling, and lipid metabolic process, whereas that of downregulated genes show enriched terms related to cell cycle and cell adhesion (Supplementary Figs. S2B and S2C).
LncRNAs have been reported to have deregulated expression in obese individuals, and some are known to play a role in the differentiation of both brown and white adipocytes5. Analysis of our dataset detected upregulation and downregulation of 158 and 77 lncRNAs, respectively, during differentiation into white adipocytes (Fig. 1A and Supplementary Table S3). Sixteen of the 158 upregulated lncRNAs were previously reported, based on integrative co-expression analysis, as enriched in adipocytes, and 5 of the downregulated 77 lncRNAs were enriched in adipose progenitor cells47. The others have not been identified before as enriched in any particular tissue. Among the top 5 downregulated and top 5 upregulated lncRNAs (Fig. 1A), only ADIPOQ-AS1 is known to function in adipogenesis through the regulation of murine adiponectin48. The large change in the expression of the nine novel lncRNAs warrants further investigation into their role in human adipogenesis. Most of these lncRNAs have deregulated expression in cancer, affecting properties like proliferation, migration, and invasion of cancer cells through their target miRNA/gene axis49,50,51,52,53,54,55. Considering that cell cycle and proliferation genes are downregulated during early adipogenesis56,57, these lncRNAs may promote adipogenesis by targeting cell proliferation.

Multiple lncRNAs are differentially expressed during white adipogenesis. (A) Volcano plot depicts differential gene expression of lncRNAs at Day 0 vs Day 14 of differentiation. Red represents upregulated genes and blue represents downregulated genes that have p-adjusted values less than 0.05 and log2-fold change of more than or equal to 1, or less than or equal to − 1, respectively. The top 5 upregulated and top 5 downregulated lncRNAs are highlighted. (B) Venn diagram shows overlap analysis of lncRNAs differentially expressed during white and brown adipogenesis. (C) Possible mechanisms of lncRNAs LINC00312, LINC00607 and TYMSOS in the regulation of adipogenesis.
To identify lncRNAs differentially expressed in the same or opposite manner during white and brown adipogenesis, we carried out differential expression analysis of a previously published human brown adipogenesis dataset33. Overlap analysis with our white adipogenesis dataset (Fig. 1B) showed upregulation of 13 and downregulation of 8 lncRNAs during differentiation of both brown and white adipocytes (Table 1). Additional lncRNAs were up- or down-regulated specifically in brown or white adipocytes.
Among the lncRNAs upregulated or downregulated during both brown and white adipogenesis, only LIPE-AS1 and LINC01119 have been shown to have a functional role and are differentially expressed during either murine or human adipogenesis58,59. LncRNAs FOXD2-AS1, LINC00968, LINC01116 and SENCR regulate glioma stem cell60, osteogenic61, neuronal62 and endothelial63 differentiation, respectively, and may play similar roles in adipogenic differentiation via regulation of target genes and miRNAs. The other lncRNAs (LINC01085, LINC01003, LINC00663, LINC01140, RAMP2-AS1, NIFK-AS1, LINC00673, FRMD6-AS1, SLC8A1-AS1) have been associated with cancer, playing diverse roles in cancer proliferation and migration, cell cycle and signaling pathways64,65,66,67,68,69,70,71,72,73.
No lncRNAs were upregulated during white adipogenesis but downregulated during brown adipogenesis. Three lncRNAs, LINC00312, LINC00607 and TYMSOS, were upregulated during brown adipogenesis but downregulated during white adipogenesis. None of these have been characterized in adipogenesis, but we hypothesized their regulatory mechanisms based on literature review (Fig. 1C).
LINC00312 may regulate adipogenesis by two potential mechanisms.
-
(1)
LINC00312 directly binds to and negatively regulates expression of miR-9 in breast cancer74. Also, miR-9 is upregulated during 3T3-L1 adipogenesis, but surprisingly, its overexpression inhibits 3T3-L1 differentiation75. LINC00312 potentially regulates adipogenesis by regulating availability of miR-9.
-
(2)
LINC00312 directly binds to and negatively regulates expression of miR-21 in colorectal cancer cells76. miR-21 is reported to positively regulate adipogenesis and is upregulated in the adipose tissue of obese individuals77. miR-21 in 3T3-L1 cells decreased expression of adipogenic markers and increased expression of genes involved in thermogenesis and browning78. Therefore, miR-21 is a possible link between LINC00312 and adipogenesis.
LINC00607 may also regulate adipogenesis through miR-607. LINC00607 acts as a sponge of miR-607 in osteosarcoma79. Mir-607 was shown to downregulate EGFR in triple-negative breast cancer cells80—a gene required for white adipogenic differentiation81.
TYMSOS may regulate adipogenesis via three mechanisms.
-
(1)
TYMSOS may affect adipogenesis via PI3K-AKT signaling, which is important for the differentiation and browning of white preadipocytes82. TYMSOS was shown to activate the PI3K/AKT signaling pathway in thyroid cancer cells83.
-
(2)
TYMSOS sponges miR-4739 in gastric cancer cells84, and miR-4739 was shown to promote adipogenic differentiation of hBMSC (human bone marrow stromal cells)85.
-
(3)
TYMSOS was also shown to act as a sponge for miR-214-3p in NSCLSC (non–small cell lung cancer) cells86 and miR-214-3p was shown to promote adipogenesis of 3T3-L1 cells87.
In summary, our analysis suggests that LINC00312, LINC00607 and TYMSOS may regulate adipogenesis by acting as sponges to limit the availability of specific miRNAs.
Alternative polyadenylation could regulate gene expression during adipogenesis
APA is a co-transcriptional process that can generate multiple isoforms through differential usage of alternative pA sites in the 3’-UTR as well as the coding region14. We analyzed our 3’-end RNA-seq data of preadipocytes (Day 0) and adipocytes (Day 14) to identify differential usage of pA sites during adipogenesis. Our analysis of 4211 genes for significant 3’-UTR APA identified 70 shortened genes and 78 lengthened genes (Fig. 2A). Genes undergoing 3’-UTR APA did not show a correlation of shortening or lengthening with gene expression (Fig. 2B). GOBP and KEGG enrichment analysis (Supplementary Fig, S4) showed that genes undergoing 3’-UTR shortening are involved in multiple pathways and processes, among which are adipogenesis-related terms such as “fatty acid metabolism” and “PPAR signaling pathway”. The lengthened genes encompassed more general terms, such as “protein modification” and “collagen fibril organization”.

Alternative polyadenylation in the 3’-UTR region is regulated during adipogenesis. (A) Volcano plot depicts shortened (red) and lengthened (blue) genes (p-adjusted value < 0.05, RED values > = 0.26 (lengthening) or < = − 0.26 (shortening). The clear circles are the 4063 genes that fell below these thresholds. The P-adjusted value and RED value thresholds are shown by dashed lines. (B) Scatter plot shows both RED and Log2-Fold expression change of shortened and lengthened genes. Genes with p-adjusted value < 0.05 and log2-Fold change > 1 (upregulated) are shown by triangles pointed upward and those with p-adjusted value < 0.05 and log2-Fold change < − 1 (downregulated) are shown by triangles pointed downward. Genes unchanged in expression are shown by black circles. The color of triangle outline represents shortened (red) and lengthened (blue) genes. The correlation between log2-fold changes and RED values is represented by the green dashed line of best fit, with the Spearman’s correlation value and associated p-value shown in the top right of the graph. (C) UCSC genome browser plots depict shortened and lengthened genes. The genes are oriented as 5’ (left) to 3’ (right). The tracks show gene, annotated polyA-sites (blue indicates RNAs are transcribed from the minus whereas red are transcribed from the plus strand), and peaks (or reads) of merged triplicates of undifferentiated (black) and differentiated (purple) cells. Ranges of normalized coverage signals (reads per million) are given on the right. The gene direction is represented by the green arrow and the exact proximal (P) and distal (D) pA site positions are shown.
Several key genes in adipogenesis undergo 3’-UTR APA during differentiation (Fig. 2C). Shortened genes SCD and ACSL5, which are involved in fatty acid metabolism88,89, are upregulated, whereas CDK16, a gene involved in cell cycle with known roles in differentiation90, is unchanged in expression. Lengthened genes WWTR1 and E2F1 play important roles in adipogenesis91,92 and are upregulated and downregulated, respectively, in gene expression. HADHB, another lengthened gene, is involved in fatty acid oxidation93 and is upregulated during adipogenesis. This suggests that adipogenesis is directly affected by APA-mediated regulation of genes.
To assess the gain or loss of miRNA sites due to changes in 3’-UTR length of genes in Fig. 2C, we used miRDB35 to investigate the presence of miRNA-binding sites in the 3’-UTR region between the differentially used pA sites. We then checked if any of the predicted miRNAs were known to play a role in adipogenesis based on literature review77,94,95, and if they are differentially expressed during adipogenesis96 (Table 2). Nine miRNAs known to be important for adipogenesis and multiple miRNAs that are differentially expressed had binding sites in the genes undergoing APA. However, among the nine miRNAs important for adipogenesis, none were significantly enriched for APA genes relative to all targets detected in the system (Supplementary Table S4).
Based on Table 2 and literature review, we propose below how APA can modulate miRNA-mediated regulation of adipogenesis:
-
1.
The shortening of SCD leads to loss of binding sites for miR-181a-5p, miR-16-1-3p, and miR-125b-2-3p. Both miR-181a-5p and miR-16-1-3p promote 3T3-L1 differentiation97,98 whereas miR-125b-2 inhibits lipogenesis and negatively regulates expression of SCD in mice fed with high-fat diet99. Therefore, the shortening of SCD and consequent avoidance of miRNA-mediated regulation, may contribute to increased expression of SCD and its correct timing, during adipogenesis.
-
2.
Shortening of CDK16 leads to loss of binding sites for miR-23a-5p which is a negative regulator of adipogenesis100, and for miR-23b-5p, which inhibits the thermogenic program of brown adipocytes101.
-
3.
Shortened gene ACSL5 lost binding sites for miRNAs known to be differentially expressed during adipogenesis.
-
4.
HADHB lengthening led to gain of binding sites for miR-30a-3p, which triggers anti-inflammatory responses in adipose tissue and increases insulin sensitivity in diet-induced obesity mice77. Overexpression of miR-30a can also activate the beige fat transcriptional program102. Extended 3’-UTRs can act as sponges to trap miRNAs and prevent their action at other mRNAs12. Therefore, lengthening of HADHB may not subject it to miR-30a-mediated regulation since HADHB is upregulated during adipogenesis, but it could competitively bind to miR-30a-3p to prevent browning of differentiated white adipocytes.
-
5.
Lengthening of WWTR1 leads to gain of binding sites for miR-9-3p, miR-17-3p and miR-10b-3p. Among these miRNAs, miR-17-3p103 may promote adipogenesis, while the others negatively regulate adipogenesis75,104. WWTR1 negatively regulates adipogenesis in mice by down regulating PPARγ activity91,105 and its expression is upregulated in our dataset. Therefore, the presence of binding sites of these miRNAs in the lengthened 3’-UTR suggests that it may be subjected to miRNA-mediated regulation that affects its protein levels. Alternatively, the extended WWTR1 sequence could competitively bind to miRNAs that inhibit adipogenesis.
-
6.
Lengthened gene E2F1 did not gain any miRNA-binding sites of adipogenesis-related or differentially expressed miRNAs.
We also assessed which RBPs may regulate APA of genes shown in Fig. 2C. To do so, we checked for RNA-binding motifs by RBPmap38 and evidence of binding from CLIP-Seq data in POSTAR339 in the regions 200 bp upstream and downstream of proximal and distal pA sites. Table 3 lists RBPs satisfying both criteria that are known to function in adipogenesis19,106,107,108, as well as other RBPs that are not reported to have roles in adipogenesis.
Among the RBPs known to function in adipogenesis, IGF2BP2 was predicted to bind around the proximal pA site of CDK16, the distal pA site of HADHB, and around both proximal and distal pA sites of SCD and WWTR1. IGF2BP1 motifs were found around the proximal sites of SCD and HADHB, and SRSF1 bound around both the distal and proximal pA sites of E2F1 and only the distal site of CDK16. These RBPs have not yet been identified as polyadenylation regulators. While the other RBPs have not been shown to play a role in adipogenesis, they have been shown to regulate APA in other systems109 and may therefore regulate adipogenesis through APA. For example, both U2AF2 and SRSF7 regulate 3’-UTR length109, and in our adipogenesis dataset, U2AF2 bound around proximal and distal pA sites of multiple genes while SRSF7 bound only around the proximal pA site of the shortened CDK16 gene.
In addition to 3’-UTR APA, multiple genes, out of a total of 8449 genes, were identified as undergoing intronic APA. Shortening (increased intronic pA site usage) was found for 441 genes and lengthening (decreased intronic pA site usage) for 816 genes (Fig. 3A). Genes undergoing intronic APA showed a strong correlation with expression (read counts), whereby genes with suppressed usage of intronic pA sites (increased RED values) were upregulated, while those with increased usage (decreased RED values) were downregulated (Fig. 3B). GOBP and KEGG enrichment analysis shows that most genes with increased intronic APA are related to cell cycle and DNA replication, while those with suppressed intronic APA are enriched in categories involved in lipid and fatty acid metabolism and insulin signaling, consistent with the increased need of adipocytes for these gene products (Supplementary Fig. S4).

Alternative polyadenylation in introns is regulated during adipogenesis (A) Volcano plot depicts shortened (red) and lengthened (blue) genes (p-adjusted value < 0.05, RED values >= 0.26 (lengthening) or <= − 0.26 (shortening). The clear circles are genes that undergo no APA. The P-adjusted value and RED value thresholds are shown by dashed lines. (B) Scatter plot shows both RED and Log2-Fold expression change of shortened and lengthened genes. Genes with p-adjusted value < 0.05 and log2-Fold change > 1 (upregulated) are shown by triangles pointed upward and those with p-adjusted value < 0.05 and log2-Fold change < − 1 (downregulated) are shown by triangles pointed downward. Genes unchanged in expression are shown by black circles. The color of triangle outline represents shortened (red) and lengthened (blue) genes. The correlation between log2-fold changes and RED values is represented by the green dotted line of best fit, with the Spearman’s correlation value and associated p-value shown in the top right of graph. (C) UCSC genome browser plots depict shortened and lengthened genes. The genes are oriented as 5’ (left) to 3’ (right). The tracks show gene, annotated pA-sites (blue indicates RNAs are transcribed from the minus whereas red are transcribed from the plus strand), and peaks (or reads) of merged triplicates of undifferentiated (black) and differentiated (purple) cells. Ranges of normalized coverage signals (reads per million) are given on the right. The gene direction is represented by the green arrow and the exact intronic pA site (INT) position is shown.
Examples of genes with changes in intronic APA are shown in Fig. 3C. Usage of intronic pA sites increases in the NCAPG2, ORC1 and CDCA2 transcripts during adipogenesis. NCAPG2 and CDCA2 are involved in cell cycle regulation110,111 and ORC1 in DNA replication112, and all are downregulated in expression during adipogenesis. Intronic pA site usage is suppressed in NDUFB3, MLYCD, and PIK3R1. NDUFB3 has been implicated in brown adipogenesis113, while MLYCD is needed for fatty acid biosynthesis114 and PIK3R1 in the PI3K/AKT pathway is important for insulin signaling in adipocytes115. Both MLYCD and PIK3R1 increase in expression during adipogenesis. NDUFB3 expression is unchanged but the proportion of transcripts ending in the intron decreases.
We analyzed the presence of RBP motifs and binding as described earlier by considering the sequence 200 bp upstream and 200 bp downstream of the target intronic pA site of genes in Fig. 3C (Table 3). For most intronic pA sites, we did not detect binding of an RBP. However, among the RBPs known to function in adipogenesis, SRSF1 bound around intronic pA sites of NCAPG2 and CDCA2. The binding sites of RBPs HNRNPA1 and HNRNPC were found around the intronic pA sites of CDCA2, NCAPG2, NDUFB3, and PIK3R1 and both RBPs are known to function in APA116,117.
Alternative splicing regulates key genes in adipogenesis
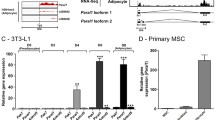
By changing which exons are included in the final mRNA, alternative splicing can regulate gene expression by generating more than one mRNA isoform per gene, hence contributing to transcriptomic and proteomic diversity. It happens during adipogenesis, and obese and lean individuals have different alternative splicing profiles106. To analyze global changes in alternative splicing during adipogenesis, we used the program rMATS24 on a recently published RNA-seq dataset of the differentiation of human primary preadipocytes23. We identified 100 alternative splicing events, with the most common types of splicing being “skipped exon” (SE) events (Fig. 4A,B). One gene can undergo multiple events of the same event type as well as multiple event types.

Analysis of the global splicing profile reveals important genes being regulated by splicing during adipogenesis (A) Volcano plots of distribution of significant splicing events (FDR less than 0.05 and dPsi value greater than or less than 0.05 or − 0.05, respectively) in each type of splicing event. Red denotes splicing events where psi value is greater than 0.05 and the target exon/region is included, whereas blue denotes splicing events where dPsi value is less than − 0.05 and the target exon/region is skipped. For the graphical representation of splicing events, blank rectangles represent constitutive exons and shaded exons or introns represent alternatively spliced regions. (B) Percentages of splicing events distributed across the 5 types of splicing events. (C) Sashimi plots depict splicing of genes during adipogenesis. Each plot is labelled with the splicing event type and gene symbol above. Genomic coordinates of the exons in question are shown on top of plot. Tracks show junction counts and the psi value with top (purple) being the undifferentiated sample and the bottom (red) being differentiated. At the bottom, exons are shown as black boxes and introns as lines with arrows showing directions of genes.
To understand the roles the alternatively spliced genes might play in adipogenesis, we carried out GOBP and KEGG enrichment analyses (Supplementary Fig. S5). Although the enriched terms are not directly associated with adipogenesis, the spliced genes are involved in various processes important for adipogenesis such as “positive regulation of GTPase activity” and “ECM-receptor interaction”118,119. We analyzed some of the spliced genes in greater detail (Fig. 4C):
-
(1)
Skipping of exon 6 of COL6A3: COL6A3 encodes the alpha-3 chain of type VI collagen, which is highly enriched in adipose tissue. The expression of COL6A3 expression in abdominal subcutaneous adipose tissue positively correlated with BMI and total body fat mass120. Exon 6 is skipped less frequently in preadipocytes compared to adipocytes. Studies have shown that the inclusion of exon 6 of COL6A3 is correlated with upregulation in cancer121,122. This suggests that the skipping of exon 6 may be responsible for the downregulation that we observe in adipocytes.
-
(2)
Alternative 5’ splice site usage in MAP4K4: In mice, MAP4K4 negatively regulates adipogenesis and insulin sensitivity123. Also, during brown adipogenesis in mice, alternatively spliced MAP4K4 isoforms had different effects on the phosphorylation of JNK, which in turn was correlated with the differentiation and metabolic signature of brown adipocytes124. MAP4K4 was not studied in human adipogenesis, but its silencing in myotubes was shown to prevent TNF-α-mediated insulin resistance and increase glucose uptake125. In our analysis, the expression of MAP4K4 decreases during adipogenesis, and alternative 5’- splice site usage in the target exon of MAP4K4 may regulate adipogenesis through changes in MAP4K4 expression and/or through signaling via JNK phosphorylation.
-
(3)
Alternative 3’ splice site usage in FN1: FN1 is an extracellular matrix (ECM) protein with roles in cell adhesion, migration, and fibrosis. FN1 is also known to undergo alternative splicing that results in proteins with different tissue localization126. It is dysregulated in obese adipose tissue127, and we also observed that its expression is downregulated during adipogenesis.
-
(4)
Mutually exclusive exons of RBPJ: RBPJ has been labelled as “mutually exclusive exons” event by rMATS. It is involved in both transcriptional activation and repression of genes and is a downstream effector of the Notch signaling pathway128. Though RBPJ is not directly associated with adipogenesis, Notch signaling is a known regulator of the process129,130. Therefore, alternatively spliced transcripts of RBPJ may regulate adipogenesis through Notch signaling.
-
(5)
Intron retention of SRSF1: SRSF1 is a splicing factor that negatively regulates adipogenesis19, but there have been no studies on the expression of its variants in the adipocyte system. Intron retention in the 3’-UTR region of SRSF1 is higher in differentiated cells in our analysis. With intron retention, the transcript was shown to be more stable and produce more protein131,132. Since SRSF1 is a negative regulator of adipogenesis, splicing out of the intron may be required for undifferentiated cells to undergo differentiation, but after differentiation, SRSF1 may have other functions in mature adipocytes.
To identify splicing factors that could contribute to global alternative splicing changes, we used rMAPS240,41 (RNA map analysis and plotting server). The program rMAPS2 carries out motif enrichment analysis (based on known consensus RBP motifs from the literature) on various regions of alternatively spliced exons—specifically, intronic sequences 250 bp upstream or downstream of target and flanking exons, as well as the first 50 bp of the 5’- or 3’-end of the exons40. Since SE events had the highest number of regulated exons, we limited our analysis to enrichment of RBP motifs in SE events. Among the RBPs identified by rMAPS2, we observed nine RBPs that are already known to influence adipogenesis19,106,107,108,133. Predictions of where these known RBPs could bind (along with the motifs) to regulate global changes in splicing during adipogenesis are shown in Fig. 5 and Supplementary Fig. S6 and summarized in Fig. 5D. For example, SRSF1 could mainly promote exon skipping by binding in the intronic sequence upstream of the target exon. Sam68 (KHDRBS1), on the other hand, could lead to exon inclusion if it binds just downstream of the 3’-end of the upstream flanking exon, but can also cause a skipped exon event if it binds just downstream of 3’-end of the target exon. IGF2BP2 could stimulate inclusion of exons by binding in the intronic sequence downstream of the target exon or promote skipping if it binds upstream of the target exon (Fig. 5B).

Specific splicing factors may regulate global “skipped exon” events. (A) Specific motif sequences used to search for binding sites in the vicinity of the skipped exon for the splicing regulators shown in panels (B) and (C) and key to the binding profiles of RBPs traced in the graphs. The solid lines are motif scores (Y axis, left side), calculated as the overall percentage of nucleotides covered by the motif in a 50 bp window, with red denoting the motif density score for exons with increased inclusion (38 events), blue for exons with increased skipping (22 events), and black for background (non-regulated) exons (70 events). The dotted lines are − log10 (P-value) (Y-axis, right side) based on comparison of motif scores between regulated exons against background exons. Only -log10 (P-value) greater than 1.3 are considered. The green exon is the target exon (that is skipped or included) and the flanking exons are shown in grey. (B) Maps of the RBPs SRSF1, Sam68 (KHDRBS1), and IGF2BP2, which are known to play a role in adipogenesis. (C) Maps of novel RBPs ESRP1, LIN28A, CPEB4, PABPC1, KHDRBS2, and HNRNPA1 that could alter the global alternative splicing profile during adipogenesis. (D) Summary of binding profiles of the RBPs, with proteins promoting inclusion indicated as red circles and those promoting skipping as blue circles.
We also identified six other RBPs that are not known to have roles in adipogenesis but could potentially be major regulators of alternative splicing (Fig. 5C,D). Both ESRP1 and PABPC1 could cause skipping of exons. LIN28A and KHPRBS2 could mainly promote inclusion of exons by binding to intronic regions downstream and upstream of the target exon, respectively. CPEB4 could lead to skipping or inclusion of the target exon by binding upstream or downstream of the exon, respectively. HNRNPA1 is predicted to mainly bind to regions downstream of the target exon and promote inclusion and would lead to skipping only if it binds to a region within 125 bp upstream of the downstream flanking exon.
Discussion
Obesity is an increasing global health problem. It is therefore important to understand the basic mechanisms which regulate human adipogenesis because its dysregulation can lead to obesity and obesity-related disorders3. In this study, we have used multiple sequencing datasets to identify potentially important regulators of human adipogenesis at the level of lncRNAs, alternative splicing and alternative polyadenylation.
Long non-coding RNAs
Using the sequencing dataset that we generated from human primary preadipocytes, we first explored lncRNA-mediated regulation. We reasoned that the most important lncRNAs were likely to be those with high change in expression during white adipogenesis, and similarities or differences in expression when compared to their changes during brown adipogenesis. The changes in lncRNAs levels may drive adipogenesis or facilitate function of the differentiated adipocytes. It will be important to characterize their role in adipogenic systems as they may be crucial in maintaining the differentiated state or in determining susceptibility to develop obesity or related disorders and may therefore be important therapeutic targets. Additionally, adiposity has been associated with certain types of cancer4, and because some of the lncRNAs highlighted in our study have been associated with cancer, they may affect the susceptibility to developing cancer. LncRNAs that undergo a similar change in expression in both white and brown adipogenesis may function in the commitment of mesenchymal stem cells into the adipocyte lineage or the maintenance of the differentiated cell state. The lncRNAs that change in opposite directions during white and brown adipogenesis are especially interesting, since browning of white adipose tissue has received increased attention as a therapeutic approach to obesity and its related disorders134,135. Therefore, the lncRNAs LINC00312, LINC00607 and TYMSOS, that increase during brown adipogenesis but decrease during white adipogenesis, could be important therapeutic candidates.
Our analysis of lncRNAs is limited by lack of time-course data. For example, we did not see a change in expression of HOTAIR between preadipocytes at Day 0 versus mature adipocytes at Day 14, but this finding is consistent with the previous report that HOTAIR increases during early adipogenesis and decreases at later stages23. There are also depot-specific46 as well as sex-specific136 differences in adipose tissue gene expression. For example, HOTAIR was shown to vary in expression in gluteal versus abdominal subcutaneous tissue137. Since our samples are not depot-specific or sex-specific and only examined the terminal differentiated state, our experimental design does not allow discrimination of these reported differences.
Alternative polyadenylation
APA during adipogenesis has not been extensively investigated. The two studies on global APA changes were published ten years ago and mainly focused on identifying a trend to longer 3′ UTRs during adipogenesis16,18, and the one examining human cells used total RNA-seq data and did not characterize intronic APA. Our study is the first to generate 3’-end sequencing data of human preadipocytes and adipocytes, which provides much more accurate and quantitative characterization of pA site usage. We observed only a slight trend towards 3’-UTR lengthening during human adipogenesis, consistent with the modest tendency towards longer 3’-UTRs that was previously reported18, but unlike the more notable trend seen during mouse 3T3-L1 adipogenesis16. This divergence may be due to differences in models used to study the process, as well as differences in experimental design and analysis. Interestingly, increased or decreased use of intronic pA sites was much more prevalent in our data set than changes in the 3’-UTR at the gene′s end. Suppression of an intronic pA site was most common, and a prime example is PIK3R1, a gene that regulates adipogenesis and insulin sensitivity115. This APA event is expected to yield more transcripts extending to the end of the gene and being translated into full-length protein. The strong enrichment in this group of genes involved in lipid and fatty acid metabolism and insulin signaling would be consistent with adipocytes needing more of these types of proteins.
Changes in 3’-UTR length could subject genes to different degrees of miRNA-mediated regulation. We found that several miRNAs with known functions in adipogenesis had potential binding sites in 3’-UTR sequences of the shortened or lengthened transcripts that we examined, suggesting miRNA-mediated regulation of these genes. We also identified several other miRNAs that are differentially expressed during adipogenesis and may play important, yet undefined roles in this transition. While none of the miRNAs that we tested were enriched in the 3’-UTRs of genes undergoing APA compared to all 3’-UTRs expressed in adipogenesis, it is still possible that APA regulates the miRNA action on the predicted targets.
To identify RBPs that may function in both 3’-UTR APA and intronic APA, we used motif enrichment analysis and global protein/RNA crosslinking data to identify binding sites in the regions 200 bp upstream and downstream of pA sites. We found that RBPs with known roles in adipogenesis bind around alternate pA sites of seven of the twelve genes that we examined, and also identified several other RBPs that could be regulating ten of these twelve genes. Interestingly, IGF2BP2, SRSF1, and HNRNPA1 are also possible regulators of skipped exon events during adipogenesis, and thus might be regulating both alternative splicing and APA.
For ORC1 and MLYCD that are undergoing intronic APA, no RBPs were detected to bind around the intronic pA site. Besides RBPs, other factors such as transcriptional dynamics and expression levels of subunits of the cleavage and polyadenylation complex can regulate APA14, and may function in regulating intronic APA of these genes during adipogenesis. Regulation may also occur by suppressing recognition of the splice sites of the flanking exons15. It is important to note that the CLIP-seq evidence for our RBP-binding predictions was based on experiments with non-adipogenic cell lines, such as HEK293, which may not be expressing all of the APA regulators present during adipogenesis, such as ones that would act on the ORC1 and MLYCD intronic pA sites. Further experiments are needed to demonstrate a direct function of the various RBPs on regulating APA during adipogenesis.
Alternative splicing
Spliced isoforms have been shown to play roles in adipogenesis19,20,21, and our analysis of a publicly available RNA-seq dataset revealed 100 alternative splicing events occurring during human adipogenesis. Like our study, the global analysis of alternative splicing in human adipogenesis published by Yi et al.22, also identified exon skipping as the most prevalent type of alternative splicing. However, they analyzed cells derived from bone marrow whereas the cells in our study originated from subcutaneous adipose tissue. The use of primary human preadipocytes, the stringent thresholds that we have used, and the application of a widely used and reliable alternative splicing analysis tool, rMATS24 makes our analysis a valuable resource for future studies to dissect the importance of alternative splicing to adipogenesis.
Our study uncovered possible mechanisms of adipogenesis regulation by specific splicing events. Of note is splicing of MAP4K4 transcripts, which encode a potential therapeutic target that is alternatively spliced during brown adipogenesis in mice124. Alternative splicing of RBPJ, a gene involved in the Notch signaling pathway128, is interesting as this pathway is associated with adipocyte dedifferentiation, a therapeutic potential in the field of obesity138. SRSF1, which shows increased intron retention in mature adipocytes, is an important regulator of alternative splicing in adipogenesis19, and in our analysis, it is a potential mediator of both exon skipping and APA. Additionally, by motif enrichment analysis, we demonstrated how RBPs previously characterized in adipogenesis can potentially regulate alternative splicing of multiple genes during differentiation, thus extending their roles in adipogenesis beyond the few targets examined in previous studies. We have also identified novel RBPs that could regulate the global alternative splicing profile during adipogenesis but are as yet uncharacterized.
To the best of our knowledge, our study presents the first comprehensive analysis of different modes of gene regulation during human adipogenesis. We generated 3’-sequencing datasets from matched preadipocyte and adipocyte RNA samples, in addition to using publicly available datasets for analysis with specialized tools. With these resources, we identified multiple potential mechanisms of regulation that can be addressed in future research. In addition to illustrating how lncRNAs, alternative polyadenylation and alternative splicing may regulate adipogenesis, we also identified novel lncRNA, miRNA, and RBP regulators that may play a crucial role in fine-tuning adipocyte differentiation. Further research into these potential regulators of mRNA and protein output during adipogenesis will aid in designing therapeutics to combat obesity and its related disorders.
Data availability
The raw sequencing reads are deposited and made publicly available on the gene expression omnibus database (GEO; GSE250525).
References
Ruiz-Ojeda, F. J., Rupérez, A. I., Gomez-Llorente, C., Gil, A. & Aguilera, C. M. Cell models and their application for studying adipogenic differentiation in relation to obesity: A review. Int. J. Mol. Sci. 17, 1040 (2016).
Lin, X. & Li, H. Obesity: Epidemiology, pathophysiology, and therapeutics. Front Endocrinol. (Lausanne) 12, 706978 (2021).
Cristancho, A. G. & Lazar, M. A. Forming functional fat: A growing understanding of adipocyte differentiation. Nat. Rev. Mol. Cell Biol. 12, 722–734 (2011).
Evseeva, M. N., Balashova, M. S., Kulebyakin, K. Y. & Rubtsov, Y. P. Adipocyte biology from the perspective of in vivo research: Review of key transcription factors. Int. J. Mol. Sci. 23, 322 (2021).
Zhang, P. et al. LncRNA-mediated adipogenesis in different adipocytes. Int. J. Mol. Sci. 23, 7488 (2022).
Sufianov, A. et al. The role of long non-coding RNAs in the development of adipose cells. Noncoding RNA Res. 8, 255–262 (2023).
Ding, C. et al. De novo reconstruction of human adipose transcriptome reveals conserved lncRNAs as regulators of brown adipogenesis. Nat. Commun. 9, 1–14 (2018).
Xiao, T. et al. Long noncoding RNA ADINR regulates adipogenesis by transcriptionally activating C/EBPα. Stem Cell Rep. 16, 1006 (2021).
Fan, L., Xu, H., Li, D., Li, H. & Lu, D. A novel long noncoding RNA, AC092834.1, regulates the adipogenic differentiation of human adipose-derived mesenchymal stem cells via the DKK1/Wnt/β-catenin signaling pathway. Biochem. Biophys. Res. Commun. 525, 747–754 (2020).
Rey, F. et al. Transcriptional characterization of subcutaneous adipose tissue in obesity affected women highlights metabolic dysfunction and implications for lncRNAs. Genomics 113, 3919–3934 (2021).
Luan, A. et al. RNA sequencing for identification of differentially expressed non-coding transcripts during adipogenic differentiation of adipose-derived stromal cells. Plast. Reconstr. Surg. 136, 752 (2015).
Zhang, Q. & Tian, B. The emerging theme of 3’UTR mRNA isoform regulation in reprogramming of cell metabolism. Biochem. Soc. Trans. 51, 1111–1119 (2023).
Kamieniarz-Gdula, K. & Proudfoot, N. J. Transcriptional control by premature termination: A forgotten mechanism. Trends Genet. 35, 553 (2019).
Gallicchio, L., Olivares, G. H., Berry, C. W. & Fuller, M. T. Regulation and function of alternative polyadenylation in development and differentiation. RNA Biol. 20, 908–925 (2023).
Mitschka, S. & Mayr, C. Context-specific regulation and function of mRNA alternative polyadenylation. Nat. Rev. Mol. Cell Biol. 23, 779–796 (2022).
Hoque, M. et al. Analysis of alternative cleavage and polyadenylation by 3′ region extraction and deep sequencing. Nat. Methods 10, 133–139 (2013).
Cui, J. et al. Shortening of HO1 3′UTRs by alternative polyadenylation suppresses adipogenesis in 3T3-L1. J. Agric. Food Chem.s 69, 8049 (2021).
Spangenberg, L., Correa, A., Dallagiovanna, B. & Naya, H. Role of alternative polyadenylation during adipogenic differentiation: An in silico approach. PLoS ONE 8, e75578 (2013).
Naing, Y. T. & Sun, L. The role of splicing factors in adipogenesis and thermogenesis. Mol. Cells 46, 268 (2023).
Carter, G. et al. Dysregulated alternative splicing pattern of PKCδ during differentiation of human preadipocytes represents distinct differences between lean and obese adipocytes. ISRN Obes. 2013, 1–9 (2013).
Aprile, M. et al. PPARγΔ5, a naturally occurring dominant-negative splice isoform, impairs PPARγ function and adipocyte differentiation. Cell Rep. 25, 1577-1592.e6 (2018).
Yi, X., Yang, Y., Wu, P., Xu, X. & Li, W. Alternative splicing events during adipogenesis from hMSCs. J. Cell Physiol. 235, 304–316 (2020).
Potolitsyna, E., Hazell Pickering, S., Germier, T., Collas, P. & Briand, N. Long non-coding RNA HOTAIR regulates cytoskeleton remodeling and lipid storage capacity during adipogenesis. Sci. Rep. 12, 10157 (2022).
Shen, S. et al. rMATS: Robust and flexible detection of differential alternative splicing from replicate RNA-Seq data. Proc. Natl. Acad. Sci. U. S. A. 111, E5593–E5601 (2014).
Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 17, 10–12 (2011).
Andrews, S. FastQC: A quality control tool for high throughput sequence data [Online]. http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (2010).
Ewels, P., Magnusson, M., Lundin, S. & Käller, M. MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics 32, 3047–3048 (2016).
Dobin, A. et al. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Liao, Y., Smyth, G. K. & Shi, W. FeatureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Sherman, B. T. et al. DAVID: A web server for functional enrichment analysis and functional annotation of gene lists (2021 update). Nucleic Acids Res. 50, W216–W221 (2022).
Safran, M. et al. The GeneCards suite. Pract. Guide Life Sci. Databases 27, 56. https://doi.org/10.1007/978-981-16-5812-9_2/FIGURES/8 (2022).
Castellá, M. et al. Adipose tissue plasticity in pheochromocytoma patients suggests a role of the splicing machinery in human adipose browning. iScience 26, 106847 (2023).
Wang, R., Nambiar, R., Zheng, D. & Tian, B. PolyA_DB 3 catalogs cleavage and polyadenylation sites identified by deep sequencing in multiple genomes. Nucleic Acids Res. 46, D315–D319 (2018).
Chen, Y. & Wang, X. miRDB: An online database for prediction of functional microRNA targets. Nucleic Acids Res. 48, 127–131 (2019).
Liu, W. & Wang, X. Prediction of functional microRNA targets by integrative modeling of microRNA binding and target expression data. Genome Biol. 20, 1–10 (2019).
Paz, I., Argoetti, A., Cohen, N., Even, N. & Mandel-Gutfreund, Y. RBPmap: A tool for mapping and predicting the binding sites of rna-binding proteins considering the motif environment. Methods Mol. Biol. 2404, 53–65 (2022).
Paz, I., Kosti, I., Ares, M., Cline, M. & Mandel-Gutfreund, Y. RBPmap: A web server for mapping binding sites of RNA-binding proteins. Nucleic Acids Res. 42, W361–W367 (2014).
Zhao, W. et al. POSTAR3: An updated platform for exploring post-transcriptional regulation coordinated by RNA-binding proteins. Nucleic Acids Res. 50, D287–D294 (2022).
Park, J. W., Jung, S., Rouchka, E. C., Tseng, Y.-T. & Xing, Y. rMAPS: RNA map analysis and plotting server for alternative exon regulation. Nucleic Acids Res. 44, 333–338 (2016).
Hwang, J. Y. et al. rMAPS2: An update of the RNA map analysis and plotting server for alternative splicing regulation. Nucleic Acids Res. 48, W300–W306 (2020).
Wickham, H., François, R., Henry, L., Müller, K. & Vaughan, D. Dplyr: A Grammar of data manipulation. https://dplyr.tidyverse.org (2023).
Hadley Wickham. Ggplot2: Elegant graphics for data analysis. https://ggplot2.tidyverse.org (2016).
Blighe, K., Rana, S. & Lewis, M. EnhancedVolcano: Publication-ready volcano plots with enhanced colouring and labeling. https://bioconductor.org/packages/EnhancedVolcano (2023) https://doi.org/10.18129/B9.bioc.EnhancedVolcano.
Min, S. H. & Zhou, J. Smplot: An R package for easy and elegant data visualization. Front Genet. 12, 802894 (2021).
Mathur, N. et al. Human visceral and subcutaneous adipose stem and progenitor cells retain depot-specific adipogenic properties during obesity. Front Cell Dev. Biol. 10, 983899 (2022).
Norreen-Thorsen, M. et al. A human adipose tissue cell-type transcriptome atlas. Cell Rep. 40, 111046 (2022).
Cai, R. et al. Adiponectin AS lncRNA inhibits adipogenesis by transferring from nucleus to cytoplasm and attenuating Adiponectin mRNA translation. Biochim. et Biophys. Acta (BBA) Mol. Cell Biol. Lipids 1863, 420–432 (2018).
Ou, Y. et al. Targeting antisense lncRNA PRKAG2-AS1, as a therapeutic target, suppresses malignant behaviors of hepatocellular carcinoma cells. Front Med. (Lausanne) 8, 649279 (2021).
Zhang, F. et al. LINC00673 silencing inhibits cell migration and invasion by suppressing PI3K/AKT signaling in glioma. Neuroreport 29, 718–722 (2018).
Ji, Z. et al. C-Myc-activated long non-coding RNA LINC01050 promotes gastric cancer growth and metastasis by sponging miR-7161-3p to regulate SPZ1 expression. J. Exp. Clin. Cancer Res. 40, 351 (2021).
Cen, D., Huang, H., Yang, L., Guo, K. & Zhang, J. Long noncoding RNA STXBP5-AS1 inhibits cell proliferation, migration, and invasion through inhibiting the PI3K/AKT signaling pathway in gastric cancer cells. Onco Targets Ther. 12, 1929–1936 (2019).
Shao, S., Wang, C., Wang, S., Zhang, H. & Zhang, Y. LncRNA STXBP5-AS1 suppressed cervical cancer progression via targeting miR-96-5p/PTEN axis. Biomed. Pharmacother. 117, 109082 (2019).
Zhang, W. et al. LINC01088 inhibits tumorigenesis of ovarian epithelial cells by targeting miR-24-1-5p. Sci. Rep. 8, 1–10 (2018).
Zhang, Q. et al. LINC01060 knockdown inhibits osteosarcoma cell malignant behaviors in vitro and tumor growth and metastasis in vivo through the PI3K/Akt signaling. Cancer Biol. Ther. 24, 2198904 (2023).
Marquez, M. P. et al. The role of cellular proliferation in adipogenic differentiation of human adipose tissue-derived mesenchymal stem cells. Stem Cells Dev. 26, 1578 (2017).
Marcon, B. H. et al. Cell cycle genes are downregulated after adipogenic triggering in human adipose tissue-derived stem cells by regulation of mRNA abundance. Sci. Rep. 9, 1–10 (2019).
Thunen, A., La Placa, D., Zhang, Z. & Shively, J. E. Role of lncRNA LIPE-AS1 in adipogenesis. Adipocyte 11, 11 (2022).
Chen, K., Xie, S. & Jin, W. Crucial lncRNAs associated with adipocyte differentiation from human adiposederived stem cells based on co-expression and ceRNA network analyses. PeerJ 2019, e7544 (2019).
Wang, Y., Cheng, Y., Yang, Q., Kuang, L. & Liu, G. Overexpression of FOXD2-AS1 enhances proliferation and impairs differentiation of glioma stem cells by activating the NOTCH pathway via TAF-1. J. Cell Mol. Med. 26, 2620 (2022).
Liao, C., Zhou, Y., Li, M., Xia, Y. & Peng, W. LINC00968 promotes osteogenic differentiation in vitro and bone formation in vivo via regulation of miR-3658/RUNX2. Differentiation 116, 1–8 (2020).
Douka, K. et al. Cytoplasmic long noncoding rnas are differentially regulated and translated during human neuronal differentiation. RNA 27, 1082–1101 (2021).
Boulberdaa, M. et al. A role for the long noncoding RNA SENCR in commitment and function of endothelial cells. Mol. Therapy 24, 978 (2016).
Pan, M. et al. The effect and mechanism of LINC00663 on the biological behavior of glioma. Neurochem. Res. 46, 1737–1746 (2021).
Fei, Y., Li, Y. & Chen, F. LncRNA-IQCH-AS1 sensitizes thyroid cancer cells to doxorubicin via modulating the miR-196a-5p/PPP2R1B signalling pathway. J. Chemother 35, 250–258 (2023).
Song, H. et al. A novel biomarker NIFK-AS1 promotes hepatocellular carcinoma cell cycle progression through interaction with SRSF10. J. Gastrointest. Oncol. 13, 1927–1941 (2022).
Xin, Y. et al. SLC8A1 antisense RNA 1 suppresses papillary thyroid cancer malignant progression via the FUS RNA binding protein (FUS)/NUMB like endocytic adaptor protein (Numbl) axis. Bioengineered 13, 12572–12582 (2022).
Zhang, J. et al. Docetaxel resistance-derived LINC01085 contributes to the immunotherapy of hormone-independent prostate cancer by activating the STING/MAVS signaling pathway. Cancer Lett. 545, 215829 (2022).
Zhu, X. et al. m6A-mediated upregulation of LINC01003 regulates cell migration by targeting the CAV1/FAK signaling pathway in glioma. Biol. Direct 18, 1–13 (2023).
Li, L., Gan, Y. P. & Peng, H. RAMP2-AS1 inhibits CXCL11 expression to suppress malignant phenotype of breast cancer by recruiting DNMT1 and DNMT3B. Exp. Cell Res. 416, 113139 (2022).
Huang, S. K. et al. Overexpression of LINC00673 promotes the proliferation of cervical cancer cells. Front Oncol. 11, 669739 (2021).
Sun, W. et al. LncRNA FRMD6-AS1 promotes hepatocellular carcinoma cell migration and stemness by regulating SENP1/HIF-1α axis. Pathol. Res. Pract. 243, 154377 (2023).
Peng, G., Yan, J., Shi, P. & Li, H. LINC01140 hinders the development of breast cancer through targeting miR-200b-3p to downregulate DMD. Cell Transplant. 32, 1–14 (2023).
Chen, Y. et al. Long non-coding RNA LINC00312 regulates breast cancer progression through the miR-9/CDH1 axis. Mol. Med. Rep. 21, 1296–1303 (2020).
Xu, B. et al. Overexpression of microRNA-9 inhibits 3T3-L1 cell adipogenesis by targeting PNPLA3 via activation of AMPK. Gene 730, 144260 (2020).
Li, G., Wang, C., Wang, Y., Xu, B. & Zhang, W. LINC00312 represses proliferation and metastasis of colorectal cancer cells by regulation of miR-21. J. Cell Mol. Med. 22, 5565 (2018).
Kurylowicz, A. MicroRNAs in human adipose tissue physiology and dysfunction. Cells 10, 3342 (2021).
Lhamyani, S. et al. MiR-21 mimic blocks obesity in mice: A novel therapeutic option. Mol. Therapy Nucleic Acid 26, 401–416 (2021).
Zheng, Y., Chen, Z., Zhou, Z., Xu, X. & Yang, H. Silencing of long non-coding RNA LINC00607 prevents tumor proliferation of osteosarcoma by acting as a sponge of miR-607 to downregulate E2F6. Front Oncol. 10, 584452 (2021).
Yang, T. et al. Cantharidin induces apoptosis of human triple negative breast cancer cells through mir-607-mediated downregulation of EGFR. J. Transl. Med. 21, 597 (2023).
Latorre, J. et al. The relevance of EGFR, ErbB receptors and neuregulins in human adipocytes and adipose tissue in obesity. Biomed. Pharmacother. 156, 113972 (2022).
Pan, J., Kothan, S., Moe Moe, A. T. & Huang, K. Dysfunction of insulin-AKT-UCP1 signalling inhibits transdifferentiation of human and mouse white preadipocytes into brown-like adipocytes. Adipocyte 11, 213 (2022).
Jia, J. et al. LncRNA TYMSOS promotes epithelial-mesenchymal transition and metastasis in thyroid carcinoma through regulating MARCKSL1 and activating the PI3K/Akt signaling pathway. Crit. Rev.&Trade Eukaryot. Gene Expr. 33, 1–14 (2023).
Gu, Y. et al. TYMSOS drives the proliferation, migration, and invasion of gastric cancer cells by regulating ZNF703 via sponging miR-4739. Cell Biol. Int. 45, 1710–1719 (2021).
Elsafadi, M. et al. MicroRNA-4739 regulates osteogenic and adipocytic differentiation of immortalized human bone marrow stromal cells via targeting LRP3. Stem Cell Res. 20, 94–104 (2017).
Yuan, Y. et al. FOXM1/lncRNA TYMSOS/miR-214-3p–mediated high expression of NCAPG correlates with poor prognosis and cell proliferation in non–small cell lung carcinoma. Front Mol. Biosci. 8, 785767 (2022).
Xi, F. X. et al. MicroRNA-214-3p targeting Ctnnb1 promotes 3T3-L1 preadipocyte differentiation by interfering with the Wnt/β-Catenin signaling pathway. Int. J. Mol. Sci. 20, 1816 (2019).
Flowers, M. T. & Ntambi, J. M. Role of stearoyl-coenzyme A desaturase in regulating lipid metabolism. Curr. Opin. Lipidol. 19, 248–256 (2008).
Luo, Q. et al. Role of ACSL5 in fatty acid metabolism. Heliyon 9, e13316 (2023).
Wang, X. et al. The roles, molecular interactions, and therapeutic value of CDK16 in human cancers. Biomed. Pharmacother. 164, 114929 (2023).
El Ouarrat, D. et al. TAZ is a negative regulator of PPARγ activity in adipocytes and TAZ deletion improves insulin sensitivity and glucose tolerance. Cell Metab. 31, 162 (2020).
Chen, J. et al. E2F1 regulates adipocyte differentiation and adipogenesis by activating ICAT. Cells 9, 1024 (2020).
Sekine, Y. et al. HADHB, a fatty acid beta-oxidation enzyme, is a potential prognostic predictor in malignant lymphoma. Pathology 54, 286–293 (2022).
Ru, W. et al. Non-coding RNAs and adipogenesis. Int. J. Mol. Sci. 24, 9978 (2023).
Zhang, X., Price, N. L. & Fernández-Hernando, C. Non-coding RNAs in lipid metabolism. Vascul. Pharmacol. 114, 93–102 (2019).
Ortega, F. J. et al. MiRNA expression profile of human subcutaneous adipose and during adipocyte differentiation. PLoS ONE 5, e9022 (2010).
Ouyang, D. et al. MiR-181a-5p regulates 3T3-L1 cell adipogenesis by targeting Smad7 and Tcf7l2. Acta Biochim. Biophys. Sin. 48, 1034–1041 (2016).
Xu, J., Zhang, L., Shu, G. & Wang, B. microRNA-16–5p promotes 3T3-L1 adipocyte differentiation through regulating EPT1. Biochem. Biophys. Res. Commun. 514, 1251–1256 (2019).
Wei, L. M. et al. MiR-125b-2 knockout increases high-fat diet-induced fat accumulation and insulin resistance. Sci. Rep. 10, 1–10 (2020).
Shen, L. et al. MicroRNA-23a regulates 3T3-L1 adipocyte differentiation. Gene 575, 761–764 (2016).
You, L. et al. The role of microRNA-23b-5p in regulating brown adipogenesis and thermogenic program. Endocr. Connect 9, 457 (2020).
Saha, P. K. et al. Browning and beiging of adipose tissue: its role in the regulation of energy homeostasis and as a potential target for alleviating metabolic diseases: miR-30a targets gene networks that promote browning of human and mouse adipocytes. Am. J. Physiol. Endocrinol. Metab. 319, E667 (2020).
Wang, Q. et al. miR-17-92 cluster accelerates adipocyte differentiation by negatively regulating tumor-suppressor Rb2/p130. Proc. Natl. Acad. Sci. U. S. A. 105, 2889–2894 (2008).
Li, H. et al. MiRNA-10b reciprocally stimulates osteogenesis and inhibits adipogenesis partly through the TGF-β/SMAD2 signaling pathway. Aging Dis. 9, 1058 (2018).
Hong, J. H. et al. TAZ, a transcriptional modulator of mesenchymal stem cell differentiation. Science 309, 1074–1078 (2005).
Chao, Y. et al. Regulatory roles and mechanisms of alternative RNA splicing in adipogenesis and human metabolic health. Cell Biosci. 11, 1–16 (2021).
Zhang, P. et al. RNA-Binding proteins in the regulation of adipogenesis and adipose function. Cells 11, 2357 (2022).
Chen, J. et al. Integrative analyses of mRNA expression profile reveal the involvement of IGF2BP1 in chicken adipogenesis. Int. J. Mol. Sci. 20, 2923 (2019).
Tian, B. & Manley, J. L. Alternative polyadenylation of mRNA precursors. Nat. Rev. Mol. Cell Biol. 18, 18–30 (2016).
Zhan, P. et al. NCAPG2 promotes tumour proliferation by regulating G2/M phase and associates with poor prognosis in lung adenocarcinoma. J. Cell Mol. Med. 21, 665 (2017).
Tao, Q. et al. The roles of the cell division cycle-associated gene family in hepatocellular carcinoma. J. Gastrointest. Oncol. 12, 781–794 (2021).
DePamphilis, M. L. The ‘ORC cycle’: A novel pathway for regulating eukaryotic DNA replication. Gene 310, 1–15 (2003).
Kim, J. et al. Regulation of brown and white adipocyte transcriptome by the transcriptional coactivator NT-PGC-1α. PLoS ONE 11, 0159990 (2016).
Zhou, L. et al. Fatty acid oxidation mediated by malonyl-CoA decarboxylase represses renal cell carcinoma progression. Cancer Res. 83, 3920–3939 (2023).
Tsay, A. & Wang, J. C. The role of PIK3R1 in metabolic function and insulin sensitivity. Int. J. Mol. Sci. 24, 12665 (2023).
Jia, Q. et al. HNRNPA1-mediated 3’ UTR length changes of HN1 contributes to cancer- and senescence-associated phenotypes. Aging 11, 4407–4437 (2019).
Gruber, A. J. et al. A comprehensive analysis of 3’ end sequencing data sets reveals novel polyadenylation signals and the repressive role of heterogeneous ribonucleoprotein C on cleavage and polyadenylation. Genome Res. 26, 1145–1159 (2016).
Ruiz-Ojeda, F. J., Méndez-Gutiérrez, A., Aguilera, C. M. & Plaza-Díaz, J. Extracellular matrix remodeling of adipose tissue in obesity and metabolic diseases. Int. J. Mol. Sci. 20, 4888 (2019).
Hasegawa, K. et al. Regulation of de novo lipid synthesis by the small GTPase Rac1 in the adipogenic differentiation of progenitor cells from mouse white adipose tissue. Int. J. Mol. Sci. 24, 4608 (2023).
Pasarica, M. et al. Adipose tissue collagen VI in obesity. J. Clin. Endocrinol. Metab 94, 5155 (2009).
Thorsen, K. et al. Alternative splicing in colon, bladder, and prostate cancer identified by exon array analysis. Mol. Cell Proteomics 7, 1214–1224 (2008).
Arafat, H. et al. Tumor-specific expression and alternative splicing of the COL6A3 gene in pancreatic cancer. Surgery 150, 306–315 (2011).
Tang, X. et al. An RNA interference-based screen identifies MAP4K4/NIK as a negative regulator of PPARγ, adipogenesis, and insulin-responsive hexose transport. Proc. Natl. Acad. Sci. U. S. A. 103, 2087 (2006).
Peng, H. Y. et al. RBM4A-SRSF3-MAP4K4 splicing cascade constitutes a molecular mechanism for regulating brown adipogenesis. Int. J. Mol. Sci. 19, 2646 (2018).
Bouzakri, K. & Zierath, J. R. MAP4K4 gene silencing in human skeletal muscle prevents tumor necrosis factor-alpha-induced insulin resistance. J. Biol. Chem. 282, 7783–7789 (2007).
Spada, S., Tocci, A., Di Modugno, F. & Nisticò, P. Fibronectin as a multiregulatory molecule crucial in tumor matrisome: From structural and functional features to clinical practice in oncology. J. Exp. Clin. Cancer Res. 40, 1–14 (2021).
Berger, E. et al. Pathways commonly dysregulated in mouse and human obese adipose tissue: FAT/CD36 modulates differentiation and lipogenesis. Adipocyte 4, 161 (2015).
Lake, R. J., Tsai, P.-F., Choi, I., Won, K.-J. & Fan, H.-Y. RBPJ, the major transcriptional effector of notch signaling, remains associated with chromatin throughout mitosis, suggesting a role in mitotic bookmarking. PLoS Genet 10, 1004204 (2014).
Shan, T., Liu, J., Wu, W., Xu, Z. & Wang, Y. Roles of notch signaling in adipocyte progenitor cells and mature adipocytes. J. Cell. Physiol. 232, 1258–1261 (2017).
Liu, M. C., Logan, H. & Newman, J. J. Distinct roles for Notch1 and Notch3 in human adipose-derived stem/stromal cell adipogenesis. Mol. Biol. Rep. 47, 8439–8450 (2020).
Jacob, A. G. & Smith, C. W. J. Intron retention as a component of regulated gene expression programs. Hum. Genet. 136, 1043 (2017).
Valacca, C. et al. Sam68 regulates EMT through alternative splicing–activated nonsense-mediated mRNA decay of the SF2/ASF proto-oncogene. J. Cell Biol. 191, 87 (2010).
Lin, J. C. Impacts of alternative splicing events on the differentiation of adipocytes. Int. J. Mol. Sci. 16, 22169 (2015).
Schirinzi, V., Poli, C., Berteotti, C. & Leone, A. Browning of adipocytes: A potential therapeutic approach to obesity. Nutrients 15, 2229 (2023).
Roth, C. L., Molica, F., Kwak, B. R., Alvarez-Llamas, G. & Martin-Lorenzo, M. Browning of white adipose tissue as a therapeutic tool in the fight against atherosclerosis. Metabolites 11, 319 (2021).
Anderson, W. D. et al. Sex differences in human adipose tissue gene expression and genetic regulation involve adipogenesis. Genome Res. 30, 1379–1392 (2020).
Divoux, A. et al. Identification of a novel lncRNA in gluteal adipose tissue and evidence for its positive effect on preadipocyte differentiation. Obesity 22, 1781–1785 (2014).
Liu, S. et al. Potential key factors involved in regulating adipocyte dedifferentiation. J. Cell Physiol. 237, 1639–1647 (2022).
Acknowledgements
We would like to acknowledge Tufts Research Technology team for advice and guidance, especially with using the Tufts University High-Performance Computing (HPC) Cluster for our analysis. We also thank all members of the Moore laboratory for their comments on the manuscript. This work was supported by Grants from the National Institutes of Health, USA (R01 GM101010-01A1 and R01AI152337 awarded to CM).
Author information
Authors and Affiliations
Contributions
CM and SMM conceived and designed the work; SMM and LW acquired and analyzed the data, and all authors interpreted the data. SMM and CM drafted the manuscript and SMM, CM, JG, LW and BT revised and edited it.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Mostafa, S.M., Wang, L., Tian, B. et al. Transcriptomic analysis reveals regulation of adipogenesis via long non-coding RNA, alternative splicing, and alternative polyadenylation. Sci Rep 14, 16964 (2024). https://doi.org/10.1038/s41598-024-67648-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-67648-9
- Springer Nature Limited