
Abstract
Numerous studies have indicated that Morocco’s indigenous sheep breeds are genetically homogenous, posing a risk to their survival in the challenging harsh climate conditions where they predominantly inhabit. To understand the genetic behind genetic homogeneity through the lens of runs of homozygosity (ROH), we analyzed the whole genome sequences of five indigenous sheep breeds (Beni Guil, Ouled Djellal, D’man, Sardi, Timahdite and Admixed).The results from principal component, admixture, Fst, and neighbour joining tree analyses consistently showed a homogenous genetic structure. This structure was characterized by an average length of 1.83 Mb for runs of homozygosity (ROH) segments, with a limited number of long ROH segments (24–48 Mb and > 48 Mb). The most common ROH segments were those ranging from 1–6 Mb. The most significant regions of homozygosity (ROH Islands) were mostly observed in two chromosomes, namely Chr1 and Chr5. Specifically, ROH Islands were exclusively discovered in the Ouled Djellal breed on Chr1, whereas Chr5 exhibited ROH Islands in all breeds. The analysis of ROH Island and iHS technique was employed to detect signatures of selection on Chr1 and Chr5. The results indicate that Chr5 had a high level of homogeneity, with the same genes being discovered across all breeds. In contrast, Chr1 displays some genetic variances between breeds. Genes identified on Chr5 included SLC39A1, IL23A, CAST, IL5, IL13, and IL4 which are responsible for immune response while genes identified on Chr1 include SOD1, SLAMF9, RTP4, CLDN1, and PRKAA2. ROH segment profile and effective population sizes patterns suggests that the genetic uniformity of studied breeds is the outcome of events that transpired between 250 and 300 generations ago. This research not only contributes to the understanding of ROH distribution across breeds but helps design and implement native sheep breeding and conservation strategies in Morocco. Future research, incorporating a broader sample size and utilizing the pangenome for reference, is recommended to further elucidate these breeds’ genomic landscapes and adaptive mechanisms.
Similar content being viewed by others
Introduction
The escalating threat of climate change poses a substantial risk to biodiversity, manifesting in the alarming rate of species extinction. This phenomenon severely impacts global food security as the planet grapples with increasing temperatures and an increasing human population. The impending reality is stark: by 2030, global temperatures are projected to rise by 2 degrees Celsius, with a subsequent increment of 0.15 degrees Celsius every decade1. Such shifts in climate are expected to exert profound heat stress on livestock, significantly undermining agricultural productivity. The situation is further exacerbated by climate-induced health hazards that compromise animal immunity, necessitating urgent and sustainable interventions to preserve livestock productivity against the backdrop of climate change2,3.
Among small ruminants, sheep emerge as pivotal contributors to global animal protein sources. Their domestication in the Fertile Crescent around 11,000 years ago ushered in a series of evolutionary adaptations, enabling their proliferation across diverse geographical landscapes4,5,6. This evolutionary journey, shaped by both natural and selective breeding, has culminated in the emergence of distinct sheep breeds, each exhibiting unique phenotypic traits indicative of their adaptive capacities. Historically, sheep populations in North Africa have been in existence for seven millennia now, and they constitute a highly diverse sheep populations that have been bred under conventional agricultural practices for many years. Based on the historical records of African sheep, it is likely that the first sheep migrated from the center of domestication to Africa, possibly in response to droughts and other climate-related calamities7. Subsequently, there were many instances of dispersal, including a movement towards the North to Libya about 6700 BP, a movement towards the south to the middle Nile Valley, and a movement towards the west to the central Sahara around 6000 BP. The most recent dispersal episode occurred around 3700 years ago, towards West Africa according to MacDonald8. The evolution of sheep breeds in North Africa developed through many stages, resulting in the creation of sheep genetic resources that have historically adapted to local conditions. Nevertheless, as a result of ambiguous circumstances, the breeds have recently undergone intermixing and genetic homogeneity, resulting in the emergence of crossbred populations.Recent statistics highlight that, global sheep population is at over 1.28 billion, with Africa and Morocco contributing 10.3% and 1.75%, respectively9,10. The sheep demographic in Morocco predominantly features indigenous breeds, characterized by their resilience to the arid conditions prevalent in desert regions11,12. This resilience underscores the importance of understanding and harnessing their genetic adaptability, especially in the face of climatic adversities.
The advent of advanced genomic and bioinformatics tools has significantly enriched our comprehension of the genetic landscape, offering insights into the adaptability and diversity of animal genetic resources, particularly within Morocco11,13,14,15,16. While recent studies into the genetic diversity of Moroccan sheep breeds have revealed a general trend of between-breed genetic homogeneity, they concurrently highlight a substantial within-breed genetic diversity13,17,18. Moreover, the exploration of genomic signatures indicative of local adaptation has revealed unique genetic regions pivotal for environmental resilience, thereby accentuating the remarkable adaptive traits of these indigenous breeds11.
Despite the insights gained, the genomic underpinnings of homogeneity and adaptability, particularly through the lens of runs of homozygosity (ROH), remain under-explored. ROH, long homozygous regions traceable to a common ancestor, serve as crucial markers for assessing genome-wide inbreeding (Froh) and elucidating the genetic basis of homogeneity within the Moroccan sheep population19. This study, therefore, aims to dissect the ROH patterns across five Moroccan sheep breeds (Beni Guil, Ouled Djellal, D’man, Sardi, Timahdite and Admixed population) to unravel the historical events leading to their current homogenous genetic makeup. The “Admixed” sheep, refers to the local sheep population, which does not conform to the established phenotypes of known native breeds, suggesting these animals are likely hybrids derived from multiple genetic lineages.
Results
Population structure
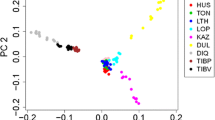
As expected, the analysis of the first two principal components (PC1 and PC2) revealed no clear separation among the breeds. In total, PC1 and PC2 (Fig. 1a) accounted for 11.18% of the total variance observed among the populations, resulting in the breeds merging into a single notable cluster along PC1. The configuration of PC1 and PC3 (Fig. 1b) formed a “V” shape, mirroring the global sheep population structure previously outlined by Kijas et al.20. This observed pattern, however, did not serve to distinguish between the sheep breeds being studied. The wide spread of Admixed population indicates high level of within-breed genetic heterogeneity than other breeds.

Population structure of six sheep breeds from Morocco as defined by principal component analysis. (a) PC1 versus PC2; (b) PC1 versus PC3.
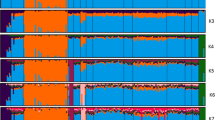
Figure 2a depicts the admixture software in a circular style. The cross-validation error achieved its minimal value at K = 2 (Supplementary Fig. S1). Like Fig. 1, there was no noticeable grouping or distinction across breeds. At a value of K = 2, there are two distinct clusters that are easily noticeable, but there is no one breed that looks to be uniform or homogenous. Remarkably, the admixture pattern remained consistent among all breeds, suggesting a significant degree of genetic similarity among the breeds being examined. As the value of K increased from 2 to 10, there was a proportional rise in the levels of mixing. Nevertheless, it was noted that there was a lack of homogenous breeds among the studied breeds across all K clusters. Figure 2b shows two distinct clusters of closely related breeds, agreeing with cross-validation results of K = 2. The first cluster, consisting of D’man, Ouled Djellal, and Beni Guil, exhibits a lower level of differentiation among its members, but is considerably dissimilar from the second cluster, which contains Timahdite and Sardi. The Admixed breed, as anticipated, is positioned at the center of the neighbor joining tree, indicating that it is highly admixed compared to all other breeds. These results can be interpreted alongside the FST (Supplementary Table 1). The highest pairwise FST was observed between Sardi and Dman (0.003, p < 0.001), while the lowest pairwise FST was observed between Ouled Djellal and Beni Guil (P -0.000, P > 0,05).

Population structure and relationship between breeds. (a): A circulat barchat showing ancestral member coefficients of each individual; (b): Neighbour joining tree showing relationship between breeds.
Runs of homozygosity and inbreeding coefficients
Across the 159 samples analyzed, the average ROH length was determined to be 1.83 Mb. Among the breeds studied, D’man displayed the highest frequency of ROH, whereas Beni Guil showed the lowest (Table 1). In terms of ROH segment frequency, the 1–6 Mb segments were most prevalent across all breeds, with the 6–12 Mb segments being significantly less common. Although both the Admixed and D’man breeds had a high frequency of short ROH segments, they were the only breeds that displayed long segments ranging from 24–48 mb, with each breed having just one such segment. The anticipated longest segments of over 48 Mb were absent in all breeds (Table 1). Chromosomal analysis revealed that chromosomes 1 and 2 harbored the highest frequency of SNPs within ROHs, while chromosomes 25 and 26 had the least (Fig. 3a). The Admixed and D’man breeds exhibited the highest frequencies of SNPs within ROHs, in contrast to Beni Guil, which had the lowest (Fig. 3b). The mean inbreeding coefficients obtained in all breeds were generally moderate (mean FROH = 0.051 ± 0.077, mean FHOM = 0.043 ± 0.079 and FGRM = 0.040 ± 0.080). The FROH values ranged between 0.106 ± 0.110 (D’man) and 0.009 ± 002 (Beni Guil), FHOM ranged between 0.098 ± 0.113 and 0.001 ± 0.001 while FGRM ranged between 0.002 ± 0.004 and 0.097 ± 0.114 -same breeds. (Table 1 and Fig. 4a). All the three inbreedings coefficients were significantly (p < 0.05) highly correlated with each other (FROH-FHOM; 0.997, FGRM-FROH;0.999, FGRM-FHOM;0.999). When examining inbreeding levels per chromosome across breeds, the Admixed and D’man breeds demonstrated higher inbreeding levels per chromosome compared to others. Generally, the inbreeding levels per breed mirrored the ROH patterns previously described (Fig. 4b). To determine the potential timeframe in which genetic homogeneity may have originated, two distinct programs for determining effective population size were used and their outcomes were subsequently compared. Supplementary Fig. 2 displays the Ne for every generation, ranging from the most distant to the most recent generation. The SNEP results revealed a decrease in effective population size across all breeds, with a sudden fall occurring between the 350th and 250th generations up to the most recent generation. Nevertheless, the GONE program’s findings revealed varying Ne throughout generations, with all breeds experiencing various bottlenecks. Importantly, the presence of several crossings might serve as a possible signal of genetic admixing.

Number of SNPs in runs per chromosome. (a): Cumulative frequency for all breeds; (b): Frequency per breed.

Genomic inbreeding coefficients based on ROH. (a): distribution of FROH; (b): breed-specific mean FROH per chromosome.
ROH islands and gene annotation
ROH islands were defined as genomic regions showing homozygosity in at least 80% of the animals within a specific breed under study. This threshold was initially applied to all 159 animals, leading to the identification of ROH islands only in the Beni Guil breed. To mitigate any potential bias, we applied this threshold to each breed rather than all at once. This adjustment revealed multiple ROH islands across different breeds, with a varying count of SNPs (Table 2). With the exception of Ouled Djellal, which had ROH islands on chromosomes 1 and 5, with the greatest SNP count inside these islands, all other breeds demonstrated ROH islands solely on chromosome 5. Notably, the Timahdite breed did not present any SNPs under this criteria.
Thus, to reduce bias in identifying runs of homozygosity (ROH) islands, we adopted the quantile method for their determination of ROH Islands in Timahtide. SNPs within the highest 99.99% frequency were categorized as key SNPs of interest.
Further, to enhance the reliability of the results (genomic regions identified) by the ROH approach, the integrated haplotype score (iHS) method, implemented in the Rehh package in R21 within R software, was used to scan the chromosomes/genomic regions that had the largest number of ROH islands. And in this case, only Chr1 and 5 were scanned using this method. “Gene-hunting” and annotation was conducted by manual searches in two complementary databases- NCBI (https://www.ncbi.nlm.nih.gov) and ensemble (https://www.ensembl.org). Gene annotation was done using ARS-UI_Ramb_v3.0 as reference genome. Annotation and gene hunting was done in phases, first phase used ROH islands, and it centered on all identified Islands on Chr5, spanning the genomic regions from the lowest SNP site at 107.3 Mb to the highest SNP site at 107.9 Mb. The second phase focused on the Ouled Djellal breed, specifically annotating Islands found on Chr1, covering a genomic range from 131.1 Mb to 131.8 Mb. Third phase involved scanning the integrated haplotype homozygosity scores (iHS) and delineating regions with outlier SNPs, christened regions of interest (RoI). RoIs were identified by scanning through the genome in sliding windows (window size = 1 mb—similar to the one used in idendifying ROH), and regions that fullifilled the following criteria were selected: “minimum number of markers = 4, minimum number of extremal markers = 4, minimum percentage of extremal markers among the markers = 0 and a log.p value threshold = 3”. The genes identified under ROH islands are detailed in Table 2, while those identified by iHS method are shown in Suppplementary Tables S2 and S3. All the ROH Islands on Chr5 were found to be similar across all breeds, resulting in the identification of two genes: TRNAW-CCA and HS3ST1. However, the breed Ouled Djellal had its ROH Islands on both Chr1 and Chr5. These ROH Isands on Chr1 revealed four genes: ATP5PF, GABPA, JAM2, and MRPL39. The results of ROH Islands were consistent with the iHS results, that is, RoI on Chr5 were similar across all breeds. However, the RoI on Chr1 varied among breeds, yielding different genes with Admixed having 52, Beni Guil having 16, D’man having 30, Ouled Djellal having 30, Sardi having 50, and Timahdite having 43 genes. All genes identified in Chr1 were unique to each breed, with only 16 genes being common to all breeds. Among them, all the four genes detected in ROH Islands were also detected by iHS method. Only common genes identified in all breeds per chromosome were further analyszed dowmstream to understand their biological functions. As a result, Metascape (https://metascape.org/)22 was used for gene ontology term enrichment analysis, whereas GenMania (https://genemania.org/)23 was used to construct a gene–gene interaction network. Furthermore, description of gene functions was enhanced by a comprehensive analysis of the available research articles.
The genes found on Chr1 are mostly associated with “response to stimulus”, “localization”, and “metabolic processes”, (Supplimentary Fig. S4a). Additionally, the gene interaction analysis revealed that SOD1, SLAMF9, RTP4, CLDN1 and PRKAA2 exhibit strong connectivity suggesting their vital role in response to stimuli (Supplimentary Fig. S4b). The study revealed that most genes located on Chr5 in sheep are significantly associated with important biological processes, such as the “immune system process”, “regulation of biological process”, and “positive regulation of biological process” (Supplementary Fig. S5a). The gene–gene interaction network (Supplementary Fig. S5b) indicates a substantial connection between SLC39A1, IL23A, CAST, IL5, IL13, and IL4, suggesting their vital function in the survival of these breeds.
Discussion
This study involved the analysis of whole genome sequencing from 159 sheep breeds that were sampled from various geographical regions of Morocco. As per the guidelines provided by FAO24, the number of samples recommended for genetic characterization studies depends on the density of the markers. It is recommended to have a minimum of 15 samples for higher density markers and 20 samples for medium density markers in order to obtain reliable results. However, Ouchene-Khelifi et al.14 used at most 22 samples and atleast 8 samples per breed in to study the “genetic homogeneity of North African goats” using 50 K SNP data. Since our study used whole genome sequencing, we are confident that the results obtained here are reliable and provides a basis of understanding the insights into the genomic homogeneity of Moroccan sheep breeds.
Previous research has consistently highlighted the genetic homogeneity among native small ruminants in North African countries, including Morocco11,16,17,25. This study aimed to delve deeper into the characteristics of this homogeneity in sheep through the lens of runs of homozygosity (ROH). Our findings affirm the genetic homogeneity of Moroccan indigenous sheep breeds, as consistently shown in Figs. 1 and 2 alongside FST values (Supplimentary Table S1), supporting previous documentation and observations. Figure 1a,b demonstrate that there were no evident clusters seen whether based on breed or geographical origin. PC1, PC2, and PC3 account for just 16% of the genetic diversity seen between populations, suggesting a weak population structure. Although the D’man and Sardi breeds showed some divergence along PC1, the Ouled Djellal breed, interestingly positioned at the center, suggested its pivotal role in fostering genetic similarity across these breeds. Historical accounts, as discussed by Belabdi et al.25, support the influence of the Ouled Djellal breed on the observed genetic structure. The authors highlighted that the unsupervised crossbreeding of other native breeds with Ouled Djellal could have caused the genetic admixture. Similar genetic uniformity has been reported in North African goats14,16, reflecting the shared domestication and migration histories of sheep and goats, hence the observed homogeneity in both species is unsurprising.
Analyzing the ROH, we observed varying segment lengths across breeds, with a predominance of shorter segments (1–6 mb) and a notable scarcity of longer segments (24–28 mb) in only two breeds, while segments longer than 48mb were absent in all. Notably, the D’Man breed exhibited the highest genomic inbreeding based on ROH coefficient (FROH = 10%) among the breeds studied, with a significant presence of ROH segments within the 6–12 mb and 12–24 mb ranges, indicating a degree of inbreeding higher than initially anticipated. Noteworthy to mention that the Admixed breed, despite its hypothetical genetic mixture, exhibited high ROH compared to standardized breeds challenging conventional expectations. This could theoretically result from bottleneck effects, selection pressures, or sampling biases. Sampling biase may be the possible cause of large number of ROHs in Admixed populations since the sample size of the Admixed population was relatively higher than the other breeds which might have introduced another layer of complexity in analyzing and comparing genetic structures. No significant variations were observed in genomic inbreeding coefficients (FROH, FHOM and FGRM) in all breeds and FROH across chromosomes within individual breeds. All the calculated inbreeding coefficnents were significantly and highly correlated (r > 0.9). The significant high correlation between molecular inbreeding values might be attributed to the fact that the greater amount of inbreeding captured was associated to ROH26, and it has also been shown that as Ne decrease, the similarity between molecular inbreeding coefficients increase27. The ROH segments profile (deficiency of longer ROH segments) suggests that significant genetic homogenization events among these breeds occurred approximately 250–350 generations ago, translating to roughly about 750 to 1050 years ago, assuming a sheep generation interval of three years. Indeed, the analysis of ROH segment profiles, together with the examination of Ne trends across generations using GONE and SNEP programs provides evidence that genetic homogeneity happened between 250 and 350 generations back. For instance, Supplementary Fig. S2 reveals that Ouled Djellal, Sardi, and Timahdite saw an increase in their Ne from around 300–250 generations ago. These increases were marked by peaks and flactutations, with many trend lines intersecting each other, possibly indicating genetic intermixing. Admixed population had a continuously stable pattern, with a notable increase in genetic exchange occurring around from 300 generations ago. This further substantiates the existence of gene exchange at that particular time period. Similar conclusions may be drawn from Supplementary Fig. S3, which demonstrates that all populations underwent a progressive decrease in Ne until around 300 to 250 generations ago which was followed by a rapid decline. The findings align with the chronological sequence of historical events and policies under the French rule, as emphasized by Belabdi et al.25, which might be the potential reason for the genetic homogeneity of small ruminants in North Africa.
To discern potential selection signatures within homozygous regions, we delineated ROH Islands. Notably, the Ouled Djellal breed, originally from Algeria, was distinguished by having unique ROH islands on Chr1, contrasting with all other breeds which presented their Islands on Chr5. The inclusion of the Ouled Djellal breed among predominantly Moroccan breeds, emphasizes the transboundary nature of genetic influences and the importance of considering geographical and historical contexts in understanding genetic diversity and uniformity in regional livestock populations. This observation led us to hypothesize that Chr5 might harbor critical genes relevant to domestication, evolutionary processes, and/or adaptability while Chr1 might be the chromosome contributing to the genetic diversity between breeds. A complementary haplotype-based method (iHS) was used to scan for signatures of selection on both Chr1 and Chr5 in all breeds.
Targeted gene searches were performed manually in both ensemble (https://www.ensembl.org/) and NCBI databases (https://www.ncbi.nlm.nih.gov/).
Within the ROH islands spanning 107.3 M to 107.9 M on Chr5, we identified two genes: the transfer RNA tryptophan (anticodon CCA; TRNAW-CCA) and Heparan sulfate glucosamine 3-O-sulfotransferase 1 (HS3ST1) for all breeds while iHS method yielded 16 genes (same genes for all breeds) among them were genes identified under ROH Island. Gene Ontology term enrichment performed in Metascape (https://metascape.org/) revealed that immunity system and regulation of biological processes are the major pathways that were over-represented by all genes found by both ROH islands and iHS methods (Supplementary Fig. S5). Efficient response to immunity is one of the most important traits of adaptation to environmental circumstances28,29,30, whereas efficient control of biological processes guarantees that physiological activities are kept within ideal parameters, which is crucial for the survival of animals in challenging environments as their misregulation can lead to genome instability and disease31,32. Gene interaction netword as developed using GeneMANIA (https://genemania.org/) showed that genes SLC39A1, IL23A, CAST, IL5, IL13, and IL4 are highly connected suggesting their crucial role in immune response and adaptation. Interleukins family genes have highly been linked to immune response28,33 while SLC39A1 gene apart from being responsible for immunity34, it is also linked with maintaining of homeostasis35 which is also a crucial biological function for adaptation. We hypothesize that genes identified under ROH Island on Chr5 are conserved in all populations and that Chr5 is more homogeneous than other chromosomes. Only genes identified under ROH Island are discussed below.
TRNAW-CCA has been associated with body weight in Merino sheep36,37 and with tail fatness in sheep adapted to semi-arid environments38. Additionally, it has been identified as a gene coding for proteins influential in plasma urea concentrations39. HS3ST1 gene, on the other hand, has been linked to feed efficiency through transcriptome analysis40,41 and is suggested to play a role in immunity across species as well42.
The Ouled Djellal breed’s ROH islands contained genes such as ATP5PF, GABPA, JAM2, and MRPL39 on Chr1. However, the iHS method produced different numbers of genes in different populations. Notably, the genes found on ROH Island in Oulled Djellal were also among the genes identified by the iHS method, with just 16 genes in common. The gene ontology term enrichment analysis of the common genes showed that the most over-represented biological function was the “response to stimuli”. Response to stimuli is a complex process that encompasses numerous chemical and signaling channels pathways. Genetic diversity has a vital role in altering the expression of genes in response to stimuli. This variation can influence the initiation of certain genes and the formation of intricate molecular networks in response to external stimuli43. The studies conducted by Liu et al.44 and Lee et al.45 highlight the significance of certain stimuli, such as hormones, stressors, darkness, and mechanostimulation, in initiating the binding of regulatory proteins to genes that respond to stimuli. These findings indicate that various stimuli might cause specific genetic reactions, resulting in changes in the control of gene expression and subsequent transcriptional responses. Gene network analysis revealed that genes SOD1, SLAMF9, RTP4, CLDN1,and PRKAA2, exhibit strong connectivity, suggesting their crucial functions within the response to stimuli. SOD1 gene for instance is linked to response to exidative stress, adaptation to plateau and also response to hypoxia46,47,48. The SLAMF9 gene belongs to the Signaling Lymphocyte Activation Molecule Family, and it has been extensively researched in relation to several aspects of sheep adaptability. The study conducted by Zeng et al.49 emphasized the function of SLAMF9 in inhibiting liver inflammation by reducing the expression of TLR4 on macrophages. This suggests that SLAMF9 is involved in immune responses and inflammatory processes. It is evident that genetic variability among these breeds is significantly contributed by Chr1. The genes mapped under ROH Islands on Chr1 are discussed below.
ATP5PF is known for its involvement in energy metabolism50, GABPA has been related to milk production traits51, JAM2 has been implicated in immunity to gastrointestinal parasites52, and MRPL39 is associated with mitochondrial function and aerobic metabolism53. This compilation of genes within ROH islands underscores their potential importance in the adaptation and physiological characteristics of these sheep breeds.
This research delved into the distribution of runs of homozygosity (ROH) across the genomes of five indigenous sheep breeds in Morocco, uncovering a significant genomic uniformity among them. Our analysis indicates that this homogeneity stems from inbreeding events occurring between 250 to 350 generations ago, rather than recent breeding practices. ROH islands, or genomic hotspots, emerge through natural selection and can illuminate regions under selective pressure. Except for the Ouled Djellal breed, which displayed unique hotspots on chromosome 1, the detected ROH hotspots across the other breeds were predominantly located on chromosome 5. This consistent localization suggests that chromosome 5 may harbor essential genes crucial for adaptation to arid environments. Notably, study showed that Chr5 is highly homogenous and both ROH Island and iHS methods identified same genes in all breeds under study. The most important biological function of the identified genes on Chr5 was responsible for immunity, a vital adaptation trait in the face of climate change. On the contrary, genomic regions on Chr1 varied between breeds suggesting that Chr1 contributes to the genetic variability seen between breeds. The most over-represented biological function by genes identified in Chr1 was response to stimuli which is also a crucial adaptation trait in the face of climate change. This research not only contributes to the understanding of ROH distribution across breeds but helps design and implement native sheep breeding and conservation strategies in Morocco. Future research, incorporating a broader sample size and utilizing the pangenome for reference, is recommended to further elucidate these breeds’ genomic landscapes and adaptive mechanisms.
Methods
Sample description and data management
This study analyzes whole genome sequence data from 159 sheep samples collected from Morocco, with their distribution illustrated in Fig. 5.It was developed using QBIS.

Distribution of the studied breeds and runs of homozygosity segments across Morocco.
The breeds examined include Admixed (n = 72), Beni Guil (n = 6), D’man (n = 30), Ouled Djellal (n = 8), Sardi (n = 27), and Timahdite (n = 16). A significant portion of the samples for this research were previously analyzed in Ouhrouch et al.11. The methods for sample collection and processing are detailed in Ouhrouch et al.11. For identification purposes, samples named after specific breeds were sourced from individuals exhibiting characteristic physical features of those breeds. The Admixed breeds in Morocco are identified as breeds that have adapted locally without displaying the phenotypic features of any known standard breeds, believed to be a product of interbreeding among various indigenous breeds.
The processing of whole genome sequence data was carried out using PLINK 1.9, focusing exclusively on Single Nucleotide Polymorphisms (SNPs) and excluding insertions and deletions (indels) from the analysis. To ensure a high-quality dataset for subsequent investigation, we applied several filtering criteria. We began by discarding variants located on unassigned and sex chromosomes. We also excluded SNPs with a call rate of less than 90% within any given breed, as well as those SNPs with a minor allele frequency (MAF) less than 0.05. This filtering process led to the removal of 36,012 variants failing to meet the MAF threshold and an additional 6,382 variants due to missing genotype information. We also exclude any individual samples based on close familial relationships, such as parent–offspring and full-sibling pairs. After applying these criteria, All samples and 19.7 million SNPs passed the filtres for downstream analysis.
Population structure
To explore the genetic relationships and spatial arrangements among different sheep breeds, we executed an unsupervised principal component analysis (PCA) on all the breeds, including only the variants that successfully underwent the quality control process54. Despite the potential effects of data imbalance on PCA outcomes, we believe that leveraging whole genome sequence data effectively captures significant genetic variations across these breeds. The PCA analysis was carried out using PLINK 1.9, applying the --pca flag. The subsequent PCA plots were generated utilizing the tidyverse package55, in R software56 which integrates tools for both data visualization and manipulation. The level of differiantiation between breeds was determined by computing FST using StAMPP package57 implemented in R56. Neighnour network was developed by split tree (https://mybiosoftware.com/splitstree-compute-phylogenetic-networks.html)58 while determination of co-ancestry membership and admixture level was performed by ADMIXTURE 1.3 software (https://dalexander.github.io/admixture/)59 .
Detection of runs of homozygosity and inbreeding
Following the methodology outlined by Meyermans et al.19 for identifying runs of homozygosity (ROH), we employed PLINK 1.9 ( https://www.cog-genomics.org/plink/)60 with specific configurations, opting out of linkage disequilibrium (LD) pruning. Our approach utilized a sliding window method for ROH detection, employing the -homozyg flag. We set the SNP calling threshold at 0.05, required a minimum of 50 homozygous SNPs within each window, allowed up to 3 heterozygous SNPs, and established a minimum SNP density of 10 SNPs per kb. The maximum permissible gap for homozygosity was 1 mb, with a maximum missingness threshold of 1. To thoroughly assess the lengths of ROHs, we analyzed them at three scales: 100, 500, and 1000 kb, to encompass both short and long ROHs effectively. ROHs were categorized into five length segments: 0–6, 6–12, 12–24, 24–28, and > 48 bp. For additional analyses, such as pinpointing SNPs within ROHs, identifying high-frequency SNPs in each breed, and calculating genome-based inbreeding coefficients (Froh), we utilized the deterctRUNs package61. Three inbreeding coefficients, FROH and FHOM, and FGRM were computed and compared. FROH quantifies the level of autozygosity, which denotes the presence of DNA segments that are homozygous because they are inherited identically from both parents. FHOM, on the other hand, measures the overall homozygosity of a population or individual by evaluating the frequency of individuals with two identical alleles at a specific gene locus throughout the genome62 while FGRM measures inbreeding using the genomic relationship matrix, considering overall genetic relatedness26,63. FROH was calculated using detectRun package61 implemented in R software56 whereas the rest were calculated using GCTA (Genome-wide Complex Trait Analysis) program64,65. The correlation between inbreeding coefficients was calculated using R software56.The identification of ROH islands was carried out using the detectRUNS package implemented in R software (https://www.r-project.org/)56, adhering to the criteria that these islands are defined as genomic regions where a certain percentage of individuals within a breed possess SNPs in ROH. Our approach involved a two-phase process. Initially, we examined all samples for the presence of ROH islands, applying an 80% threshold to identify common regions across the sample set. The subsequent phase focused on pinpointing breed-specific ROH islands.
Data availability
The data supporting this article can be freely and openly accessed https://ftp.ebi.ac.uk/pub/databases/nextgen/ovis/variants/genus_snps/.
References
Field, C. B. et al. Intergovernmental panel on climate change. Climate change 2014: Impacts, adaptation, and vulnerability. In contribution of working group II to the fifth assessment report of the intergovernmental panel on climate change. Cambridge University Press Cambridge/New York. (2014)
Collier, R. J., Renquist, B. J. & Xiao, Y. A 100-year review: Stress physiology including heat stress. J. Dairy Sci. 100, 10367–10380 (2017).
McManus, C. M. et al. Heat stress effects on sheep: Are hair sheep more heat resistant?. Theriogenology 155, 157–167 (2020).
Ciani, E. et al. On the origin of European sheep as revealed by the diversity of the Balkan breeds and by optimizing population-genetic analysis tools. Genet. Sel. Evol. 52, 25 (2020).
Her, C. et al. Broad maternal geographic origin of domestic sheep in Anatolia and the Zagros. Anim. Genet. 53, 452–459 (2022).
Taberlet, P. et al. Are cattle, sheep, and goats endangered species?. Mol. Ecol. 17, 275–284 (2008).
Muigai, A. W. T. & Hanotte, O. The Origin of african sheep: Archaeological and genetic perspectives. Afr. Archaeol. Rev. 30, 39–50 (2013).
MacDonald, K. C. (2006) The origins of African livestock: indigenous or imported? in The Origins and Development of African Livestock, Routledge, 2–17
FAOSTAT. FAOSTAT. Preprint at http://www.fao.org/faostat/en/#data/QA/visualize (2023).
World Population Review. Africa Population. https://worldpopulationreview.com/continents/africa-population (2023).
Ouhrouch, A. et al. Genomic uniqueness of local sheep breeds from morocco. Front. Genet. 12, 723599 (2021).
Ibnelbachyr, M., Benjelloun, B. & El Amiri, B. Local knowledge on Moroccan Siroua sheep management assists its resilience to climate change. Efficiency and resilience of forage resources and small ruminant production to cope with global challenges in Mediterranean areas”. A. López-Francos, M. Jouven, C. Porqueddu, H. Ben Salem, A. Keli, A. Araba and M. Chentouf 215–220 (2021).
Gaouar, S. B. S., Kdidi, S. & Ouragh, L. Estimating population structure and genetic diversity of five Moroccan sheep breeds by microsatellite markers. Small Rumin. Res. 144, 23–27 (2016).
Ouchene-Khelifi, N.-A. et al. Genetic homogeneity of North-African goats. PLoS One 13, e0202196 (2018).
Abdoli, R., Zamani, P., Mirhoseini, S. Z., Ghavi Hossein-Zadeh, N. & Nadri, S. A review on prolificacy genes in sheep. Reprod. Domest. Anim. 51, 631–637 (2016).
Benjelloun, B. et al. Mitochondrial DNA polymorphism in Moroccan goats. Small Rum. Res. 98, 201–205 (2011).
Kandoussi, A., Badaoui, B., Boujenane, I., Piro, M. & Petit, D. How have sheep breeds differentiated from each other in Morocco? Genetic structure and geographical distribution patterns. Genet. Sel. Evol. 53, 1–13 (2021).
El Fadili, M., Michaux, C., Detilleux, J. & Leroy, P. L. Genetic parameters for growth traits of the Moroccan Timahdit breed of sheep. Small Rumin. Res. 37, 203–208 (2000).
Meyermans, R., Gorssen, W., Buys, N. & Janssens, S. How to study runs of homozygosity using PLINK? A guide for analyzing medium density SNP data in livestock and pet species. BMC Genom. 21, 1–14 (2020).
Kijas, J. W. et al. Genome-wide analysis of the world’s sheep breeds reveals high levels of historic mixture and strong recent selection. PLoS Biol. 10, e1001258 (2012).
Gautier, M. & Vitalis, R. rehh: An R package to detect footprints of selection in genome-wide SNP data from haplotype structure. Bioinformatics 28, 1176–1177 (2012).
Zhou, Y. et al. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 10, 1523 (2019).
Franz, M. et al. GeneMANIA update 2018. Nucl. Acids Res. 46, W60–W64 (2018).
Ajmone-Marsan, P., Boettcher, P. J., Ginja, C., Kantanen, J. & Lenstra, J. A. Genomic characterization of animal genetic resources: practical guide. (Food & Agriculture Org., 2023).
Belabdi, I. et al. Genetic homogenization of indigenous sheep breeds in Northwest Africa. Sci. Rep. 9, 7920 (2019).
Ghoreishifar, S. M. et al. Genomic measures of inbreeding coefficients and genome-wide scan for runs of homozygosity islands in Iranian river buffalo Bubalus bubalis. BMC Genet. 21, 16 (2020).
Keller, M. C., Visscher, P. M. & Goddard, M. E. Quantification of inbreeding due to distant ancestors and its detection using dense single nucleotide polymorphism data. Genetics 189, 237–249 (2011).
Pernthaner, A., Cole, S., Morrison, L. & Hein, W. R. Increased expression of interleukin-5 (IL-5), IL-13, and tumor necrosis factor alpha genes in intestinal lymph cells of sheep selected for enhanced resistance to nematodes during infection With<i>trichostrongylus colubriformis</I>. Infect. Immun. 73, 2175–2183 (2005).
Xiang, R., Oddy, V. H., Archibald, A. L., Vercoe, P. E. & Dalrymple, B. P. Epithelial, metabolic and innate immunity transcriptomic signatures differentiating the rumen from other sheep and mammalian gastrointestinal tract tissues. PeerJ. 4, e1762 (2016).
Wanjala, G., Bagi, Z. & Kusza, S. Meta-analysis of mitochondrial DNA control region diversity to shed light on phylogenetic relationship and demographic history of African sheep (Ovis aries) breeds. Biology (Basel) 10, 762 (2021).
Roychoudhury, S. et al. Endogenous oxidized DNA bases and APE1 regulate the formation of G-quadruplex structures in the genome. Proc. Natl. Acad. Sci. 117, 11409–11420 (2020).
Sauer, M. & Paeschke, K. G-Quadruplex unwinding helicases and their function <i>in Vivo</I>. Biochem. Soc. Trans. 45, 1173–1182 (2017).
Akdis, M. et al. Interleukins, from 1 to 37, and interferon-γ: Receptors, functions, and roles in diseases. J. Allergy Clin. Immunol. 127, 701-721.e70 (2011).
Ma, X. et al. SLC39A1 overexpression is associated with immune infiltration in hepatocellular carcinoma and promotes its malignant progression. J. Hepatocell Carcinoma 9, 83–98 (2022).
Jeong, J. & Eide, D. J. The slc39 family of zinc transporters. Mol. Asp. Med. 34, 612–619 (2013).
Al-Mamun, H., Clark, S., Kwan, P. & Gondro, C. Genome-wide association study on body weight reveals major loci on OAR6 in Australian merino sheep. In 10th world congress on genetics applied to livestock production (Asas, 2014).
Dakhlan, A. Gene by birth type interaction in merino lamb. Proc. Assoc. Advmt. Anim. Breed. Genet. 22, 45–48 (2018).
Baazaoui, I., Bedhiaf-Romdhani, S., Mastrangelo, S. & Ciani, E. Genome-wide analyses reveal population structure and identify candidate genes associated with tail fatness in local sheep from a semi-arid area. Animal 15, 100193 (2021).
de Souza, T. C. et al. Genome-wide association for plasma urea concentration in sheep. Livest. Sci. 248, 104483 (2021).
Halli, K., Bohlouli, M., Schulz, L., Sundrum, A. & König, S. Estimation of direct and maternal genetic effects and annotation of potential candidate genes for weight and meat quality traits in a genotyped outdoor dual-purpose cattle breed1. Transl. Anim. Sci. 6, txax022 (2022).
Suárez-Vega, A. et al. Feed efficiency in dairy sheep: An insight from the milk transcriptome. Front. Vet. Sci. 10, 1122953 (2023).
Kalaiyarasi, K. & Elayadeth-Meethal, M. Selection for tolerance to haemo protozoan diseases in ruminants. Sustain. Livest. Prod.: Prospect. Innov. 19 (2023).
Gat-Viks, I. et al. Deciphering molecular circuits from genetic variation underlying transcriptional responsiveness to stimuli. Nat. Biotechnol. 31, 342–349 (2013).
Liu, C. et al. Arabidopsis ARGONAUTE 1 binds chromatin to promote gene transcription in response to hormones and stresses. Dev. Cell 44, 348-361.e7 (2018).
Lee, D., Polisensky, D. H. & Braam, J. Genome-wide identification of touch- and darkness-regulated arabidopsis genes: A focus on calmodulin-like and <i>XTH</I> Genes. New Phytol. 165, 429–444 (2004).
Andersen, P. M. Amyotrophic lateral sclerosis associated with mutations in the CuZn superoxide dismutase gene. Curr. Neurol. Neurosci. Rep. 6, 37–46 (2006).
Yurchenko, A. A. et al. High-density genotyping reveals signatures of selection related to acclimation and economically important traits in 15 local sheep breeds from Russia. BMC Genom. 20, 1–19 (2019).
Wang, W. et al. Deep genome resequencing reveals artificial and natural selection for visual deterioration, plateau adaptability and high prolificacy in chinese domestic sheep. Front. Genet. 10, 300 (2019).
Zeng, X. et al. Combined deficiency of SLAMF8 and SLAMF9 prevents endotoxin-induced liver inflammation by downregulating TLR4 expression on macrophages. Cell Mol. Immunol. 17, 153–162 (2018).
Zhuang, Y., Chai, J., Abdelsattar, M. M., Fu, Y. & Zhang, N. Transcriptomic and metabolomic insights into the roles of exogenous β-hydroxybutyrate acid for the development of rumen epithelium in young goats. Anim. Nutr. 15, 10–21 (2023).
Saridaki, A. et al. Combined haplotype blocks regression and multi-locus mixed model analysis reveals novel candidate genes associated with milk traits in dairy sheep. Livest Sci. 220, 8–16 (2019).
Berton, M. P. et al. Genomic regions and pathways associated with gastrointestinal parasites resistance in Santa Inês breed adapted to tropical climate. J. Anim. Sci. Biotechnol. 8, 73 (2017).
Hui, W. et al. Gene expression profile in the liver of sheep infected with cystic echinococcosis. PLoS One 11, e0160000 (2016).
Maćkiewicz, A. & Ratajczak, W. Principal components analysis (PCA). Comput. Geosci. 19, 303–342 (1993).
Wickham, H. & Wickham, M. H. Package tidyverse. Easily Install and Load the ‘Tidyverse (2017).
Team, R. C. 2020. R: A Language and environment for statistical computing. R foundation for statistical computing, Vienna, Austria: Available at: https://www.R-project.org/.[Google Scholar] (2019).
Pembleton, L. W., Cogan, N. O. I. & Forster, J. W. St AMPP: An R package for calculation of genetic differentiation and structure of mixed-ploidy level populations. Mol. Ecol. Resour. 13, 946–952 (2013).
Huson, D. H. & Bryant, D. Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 23, 254–267 (2006).
Alexander, D. H., Shringarpure, S. S., Novembre, J. & Lange, K. Admixture 1.3 software manual. Los Angeles: UCLA Human Genetics Software Distribution (2015).
Purcell, S. et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Human Genet. 81, 559–575 (2007).
Biscarini, F., Cozzi, P., Gaspa, G. & Marras, G. detectRUNS: Detect runs of homozygosity and runs of heterozygosity in diploid genomes. (2018).
Villanueva, B. et al. The value of genomic relationship matrices to estimate levels of inbreeding. Genet. Sel. Evol. 53, 42 (2021).
Mastrangelo, S. et al. Genomic inbreeding estimation in small populations: Evaluation of runs of homozygosity in three local dairy cattle breeds. Animal 10, 746–754 (2016).
McQuillan, R. et al. Runs of homozygosity in European populations. Am. J. Human Genet. 83, 359–372 (2008).
Yang, J., Lee, S. H., Goddard, M. E. & Visscher, P. M. GCTA: A tool for genome-wide complex trait analysis. Am. J. Human Genet. 88, 76–82 (2011).
Acknowledgements
This study was supported by the bilateral S&T cooperation programme within the project “Genetic characterization of native sheep in Carpathian basin and Morocco as a potential factor for climate change adaptation” from the National Development, Research and Innovation Fund (2021-1.2.4-TÉT-2021-00014) and also supported by the University of Debrecen Program for Scientific Publication. Thanks to Hungarian Eötvös State Scholarship and Stipendium Hungaricum Scholarship from Tempus Public Foundation for supporting GW studies.
Funding
Open access funding provided by University of Debrecen.
Author information
Authors and Affiliations
Contributions
S.K., B.B. concieved the study, participated in design and coordination, peformed statistical analysis and wrote the original draft, G.W. participated in data analysis, visualization and wrote the original draft. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kusza, S., Badaoui, B. & Wanjala, G. Insights into the genomic homogeneity of Moroccan indigenous sheep breeds though the lens of runs of homozygosity. Sci Rep 14, 16515 (2024). https://doi.org/10.1038/s41598-024-67558-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-67558-w
- Springer Nature Limited