
Abstract
Inflammatory bowel disease (IBD) is a chronic and recurrent inflammatory disease of the gastrointestinal tract, including two subtypes: Crohn’s disease (CD) and ulcerative colitis (UC). Metabolic disorders are important factors in the development of IBD. However, the evidence for the causal relationship between blood metabolites and IBD remains limited. A two-sample MR analysis was applied to evaluate relationships between 486 blood metabolites and IBD. The inverse variance weighted method was chosen as the primary MR analysis method. False discovery rate correction was used to control for false positives in multiple testing. Following complementary and sensitivity analyses were conducted using methods such as weight median, MR-egger, weighted mode, simple mode, Cochran Q test, and MR-PRESSO. Moreover, we performed replication, meta-analysis, Steiger test, and linkage disequilibrium score regression to enhance the robustness of the results. Additionally, we performed metabolic pathway analysis to identify potential metabolic pathways. As a result, we identified four significant causal associations between four blood metabolites and two IBD subtypes. Specifically, one metabolite was identified as being associated with the development of CD (mannose: odds ratio (OR) = 0.19, 95% confidence interval (CI) 0.08–0.43, P = 8.54 × 10–5). Three metabolites were identified as being associated with the development of UC (arachidonate (20:4n6): OR = 0.18, 95% CI 0.11–0.30, P = 2.09 × 10–11; 1, 5-anhydroglucitol: OR = 2.21, 95% CI 1.47–3.34, P = 1.50 × 10–4; 2-stearoylglycerophosphocholine: OR = 2.66, 95% CI 1.53–4.63, P = 5.30 × 10–4). The findings of our study suggested that the identified metabolites and metabolic pathways can be considered as useful circulating metabolic biomarkers for the screening and prevention of IBD in clinical practice, as well as candidate molecules for future mechanism exploration and drug target selection.
Similar content being viewed by others
Introduction
Inflammatory bowel disease (IBD) is a chronic and recurrent inflammatory disease of the gastrointestinal tract, including two subtypes: Crohn's disease (CD) and ulcerative colitis (UC). Worldwide, the prevalence of IBD is gradually increasing, especially in newly industrialized countries (China and India), with a prevalence of 0.5% in 2010, 0.75% in 2020, and projected to reach 1% in 2030. The increasing prevalence of the disease has led to an increase in hospitalization rates, which adds to the economic burden on health-care systems around the world1,2. Patients with IBD usually present clinically with chronic recurrent diarrhea, abdominal pain, and mucous bloody stools, and may develop serious complications such as intestinal perforation, intestinal obstruction, and anal fistulas3,4. However, clinically, the diagnosis of IBD lacks a gold standard and often requires a combination of endoscopic, histologic, and laboratory investigations, which makes it difficult to differentiate it from other nonspecific enterocolitis5. Furthermore, despite significant advances in the pharmacologic treatment of IBD with the introduction of novel biologic and small molecule therapies, patients pay a high price both financially and physically. Notably, these therapies do not cure IBD and may have life-threatening side effects. A considerable number of patients eventually require surgery after the treatment6,7,8. It highlights the need for new biodiagnostic markers and therapeutic strategies for IBD. However, the etiology and pathogenesis of IBD have not been fully elucidated. It is generally recognized that IBD is a persistent excessive immuno-inflammatory response resulting from the interaction of genetic, microbial and environmental factors9,10.
In recent years, the emergence of metabolomics as a component of systems biology has provided a new way to investigate the pathogenesis of diseases. There are increasing evidences that metabolic disorders are closely related to the development of IBD. For example, Scoville et al. found that 173 metabolites were significantly altered (27 increased and 146 decreased) by metabolomic analysis of serum collected from IBD patients and healthy controls. Most of the alterations occurred in lipids, amino acids, and energy-related metabolites11. Di'Narzo et al. found that serum from IBD patients was enriched in fibrinogen cleavage peptides as well as xanthine and tryptophan metabolites compared to healthy controls. Whereas fibrinogen cleavage polypeptide and diacylglycerols were significantly differentially expressed in CD versus UC patients12. However, the detailed pathophysiologic mechanisms of blood metabolites in IBD remain unclear. Moreover, due to limitations in sample size, confounding factors, and ethical issues, there are no prospective studies or randomized controlled trials (RCTs) to confirm their causal relationship. Therefore, there is an urgent need for a comprehensive and in-depth analysis of the causal role of blood metabolites in the pathogenesis of IBD.
With the development of high-throughput technologies, it is now possible to measure hundreds of circulating metabolites and genotype them in parallel in large populations13. Genome-wide association studies (GWAS) can provide molecular insight into the complex interplay of genetic factors and environmental factors in disease pathogenesis. To date, a considerable number of single nucleotide polymorphisms (SNPs) have been found to be strongly correlated with blood metabolites. However, there are still significant barriers to translating these genetic findings into the biological mechanisms of IBD development, requiring in-depth analyses to reveal the causal interactions of blood metabolites on IBD susceptibility.
Mendelian randomization (MR) analysis is a new and powerful epidemiological tool that has recently gained widespread use in disease etiology studies. MR assesses the causal impact of exposure on outcomes by selecting exposure-related SNPs as instrumental variables (IVs). In detail, this IVs alternative approach mimics the design of RCTs because SNPs are randomly assigned to offspring at conception, which largely avoids the effects of confounding factors14,15. Recently, GWAS was extended to metabolic phenotyping, producing a genetically determined metabolites (GDMs) atlas16. Inspired by the dataset of GDMs, we implemented a two-sample MR approach to (1) assess the causal effects of human blood metabolites on IBD; (2) explore whether there are common metabolites with causal effects on CD and UC; and (3) identify potential metabolic pathways that may contribute to the development of the IBD.
Materials and methods
Study design
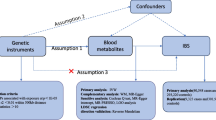
We systematically assessed the causal relationship between human blood metabolites and the risk of IBD by using a two-sample MR design. Compelling MR studies should meet 3 basic assumptions: (1) the genetic instruments should be directly associated with exposure (i.e., metabolites in this study); (2) the genetic instruments should be independent of outcome (i.e., IBD in this study) and independent of any known or unknown confounders; (3) the effect of IVs on outcome is mediated only by the exposure of interest. Genetic information for metabolites and IBD were obtained from separate GWAS datasets to avoid sample overlap. The study methods were compliant with the STROBE-MR checklist15. An overview of this MR study is shown in Fig. 1.

Overview of this Mendelian randomization (MR) analysis. IVW, inverse variance weighted; LD, linkage disequilibrium; LDSC, linkage disequilibrium score regression; SNPs, single nucleotide polymorphisms; WM, weighted median.
GWAS data for blood metabolites and IBD
The genome-wide association summary datasets of 486 metabolites associated with human genetic variation were obtained from the study by Shin et al.16. This is the most comprehensive analysis of human metabolites to date, with complete summary statistics available through the Metabolomics GWAS server (http://metabolomics.helmholtz-muenchen.de/gwas/). This GWAS analysis included 7824 adults and approximately 2.1 million SNPs from two European cohorts. In more detail, 1768 of these participants were from the KORA F4 study in Germany, and 6056 were from the UK Twin Study. Of the 486 metabolites, 309 were known metabolites, which can be grouped into eight categories: cofactors and vitamins, energy, amino acids, carbohydrates, lipids, nucleotides, peptides, and xenobiotic metabolisms, as documented in the Kyoto Encyclopedia of Genes and Genomes (KEGG) database. Another 177 were unknown metabolites whose chemical identity had not yet been determined.
To minimize bias due to racial differences and sample overlap, GWAS data for IBD from the FinnGen database (https://www.finngen.fi/en, R8 released in 2023) were used for the primary analysis17. In this data, UC and CD were strictly diagnosed by ICD codes and required KELA and at least 2 HDR. Specific information of participants, genotype platforms, data survey methods are available at the FinnGen website. The dataset used consisted of 1,665 patients with CD and 375,445 controls (phenocode: K11_CD_STRICT2), and 5,034 patients with UC and 371,530 controls (phenocode: K11_UC_STRICT2).
Selection of IVs
In order to fulfill the 3 basic assumptions of the MR study, we identified IVs associated with blood metabolites from multiple perspectives through rigorous screening conditions. First, considering the limited number of metabolite-related SNPs and referring to some previous studies, we set the significance threshold at p < 1 × 10–5 and the linkage disequilibrium (LD) r2< 0.1 within 500 kilobase (kb)18,19,20. Second, to eliminate the bias caused by poor quality IVs, we calculated the R2 and F-statistics for each SNP (the formulas are shown in Supplementary Table S1). In general, F-statistics < 10 were considered weak IVs and discarded21,22. Third, we extracted SNPs associated with metabolism from the results and discarded SNPs strongly associated with the results (p < 1 × 10–5). Fourth, we further harmonized SNPs for exposure and outcome and eliminated SNPs with palindromic effects and allelic incongruence. Finally, to avoid the effect of confounders, PhenoScanner V2 (http://www.phenoscanner.medschl.cam.ac.uk/) was used to test the assumption that IVs are independent of confounders by analyzing genome-wide significant relationships (P < 1 × 10–5). Through this process, we eliminated SNPs that were associated with known risk factors for IBD, such as depression, being a worrier, smoking, taking contraceptives, having sclerosing cholangitis23,24,25,26.
MR analysis and statistical analysis
The causal associations between blood metabolites and IBD for this MR analysis were primarily estimated using the inverse variance weighted (IVW) method. If there is no heterogeneity, the fixed-effect IVW method is used, and vice versa with random-effect. It is noteworthy that the IVW method can provide a consistent assessment of exposure causality when each variable satisfies all three assumptions of a valid instrumental variable27,28. To improve the stability and robustness of the results, we used four additional methods as complementary analyses to further evaluate the metabolite. The MR-Egger method can detect violations of the IVs assumptions and provide estimates of effects that are not affected by these violations29. The weighted median method is more tolerant of invalid IVs and produces plausible estimates when more than half of the weights correspond to valid IVs30. Moreover, simple mode and weighted mode methods were also applied as the complementary analyses31. In addition, we performed the following sensitivity analyses: (1) Cochran Q test was used to detect potential violations of the assumption by the heterogeneity of the association between individual IVs. Cochran Q test-derived p < 0.05 was considered as heterogeneity of the results32; (2) MR-Egger was utilized to estimate horizontal pleiotropy based on its intercept to ensure that genetic variants were independently associated with exposure and outcome33; (3) MR-PRESSO was applied to detect outliers and correct the horizontal pleiotropy34; (4) we performed leave-one-out analysis to assess whether the results were influenced significantly by a single SNP35. Statistical analyses were performed with R4.3.1 software, and MR analyses were performed with the MendelianRandomization package software package. MR-PRESSO was performed using the MRPRESSO package. False discovery rate (FDR) correction was used to control for false positives in multiple testing. According to previous studies, A statistically significant association was considered if the estimated causal effect of a given metabolite had a FDR < 0.136,37.
As a result, we rigorously screened blood metabolites potentially causally associated with IBD by multiple criteria; (1) significant p-value (FDRivw < 0.1) for primary analyses; (2) consistency in direction and magnitude of the five MR methods; (3) no heterogeneity or horizontal pleiotropy in the MR estimates; (4) MR estimates are not heavily confounded by individual SNPs.
Replication and meta-analysis
To fully assess the robustness of the candidate metabolites identified based on the above criteria, we replicated the IVW analysis in an additional IBD cohort from The International Inflammatory Bowel Disease Genetics Consortium. The cohort data are available at the IEU OpenGWAS database (https://gwas.mrcieu.ac.uk/). In this cohort, the CD group (GWAS ID: ieu-a-30) consisted of 5,956 European ancestry cases and 14,927 European ancestry controls, while the UC group (GWAS ID: ieu-a-32) consisted of 6,968 European ancestry cases and 20,464 European ancestry controls. The results of the meta-analysis of the two MR analyses allowed us to finally identify the blood metabolites causally associated with IBD.
Genetic correlation and direction validation
Previous studies have shown that MR results may have false positives due to genetic correlations between traits38. Although SNPs associated with IBD were excluded in the selection of IVs, combinations of SNPs not significantly associated with IBD may also contribute to the genetic risk of IBD. Linkage disequilibrium score regression (LDSC) can be used to estimate co-inheritance by performing chi-square statistics for two traits based on SNP. Therefore, genetic associations between identified metabolites and IBD were assessed by LDSC to determine whether causality is confounded by shared genetic architecture. In addition, we used the Steiger test to confirm whether the observed causalities were biased due to reversed causation. When a combination of SNPs was found to have no genetic risk for IBD compared to blood metabolites, the results indicated no bias in causal inference (Steiger p < 0.05)39.
Metabolic pathway analysis
Metabolic pathways were analyzed via the web-based MetaboAnalyst 5.0 (https://www.metaboanalyst.ca/)40. The functional pathway analyses module was used to identify potential metabolite pathways that may be relevant to the biological processes of IBD. The Kyoto Encyclopedia of Genes and Genomes (KEGG) database and the Small Molecule Pathway Database (SMPDB) were used in this study. Referring to previous studies, the significance level for pathway analysis was set at 0.1041,42.
Results
Selection of IVs
Of the 486 metabolites, 3–494 independent SNP component instrumental variables were collected in the CD group. Whereas in the UC group, 3–493 independent SNP component instrumental variables were collected. The F-statistics of the metabolite-related SNPs were all greater than the empirical threshold of 10, which indicated that all SNPs were sufficiently effective. Detailed data of IVs are shown in Supplementary Table S2 and S3.
Primary analysis
As shown in Fig. 2, 31 metabolites associated with CD were detected by the IVW method (p < 0.05), including 7 lipids, 5 amino acids, 3 carbohydrates, 2 peptides, 2 nucleotides, 2 xenobiotics and 10 unknown metabolites. Among the known metabolites, 10 metabolites were positively correlated with CD and remaining 11 metabolites were associated with a reduced risk of CD. However, after FDR correction, only glycochenodeoxycholate (FDRivw = 0.054), mannose (FDRivw = 0.041), and tryptophan betaine (FDRivw = 0.063) remained significant.

Mendelian randomization results for blood metabolites and the risks of CD based on the inverse variance weighted (IVW) method.
As shown in Fig. 3, 32 metabolites associated with UC were detected by the IVW method (p < 0.05), including 11 lipids, 8 amino acids, 4 xenobiotics, 1 carbohydrate, 1 peptide, 1 energy, 1 cofactor and vitamin, and 5 unknown metabolites. Among the known metabolites, 13 metabolites were positively correlated with UC and remaining 14 metabolites were associated with a reduced risk of UC. After FDR correction, only arachidonate (20:4n6) (FDRivw = 1.01 × 10–8), 2-stearoylglycerophosphocholine (FDRivw = 0.086), and 1,5-anhydroglucitol (FDRivw = 0.036) remained significant. Regrettably, we did not find metabolites with significant causal effect on both CD and UC.

Mendelian randomization results for blood metabolites and the risks of UC based on the inverse variance weighted (IVW) method.
Complementary and sensitivity analyses
Although the IVW method is highly effective in inferring causal relationships between exposures and disease outcomes, it is known to be susceptible to weak instrument bias. Therefore, we further performed complementary and sensitivity analyses to assess the robustness of causality. As shown in Fig. 4 and Table 1, we performed complementary analyses of meaningful associations obtained by the IVW method with the use of MR-Egger, weighted median, weighted mode, and simple mode methods. The results showed that the IVW method estimates were consistent with the direction and magnitude of MR-Egger, weighted median, weighted mode, and simple mode method estimates, suggesting that the causal relationship is reliable. As shown in Table 2, the results of Cochran Q test showed no heterogeneity. MR-Egger regression and global test of MR-PRESSO suggested that there was no horizontal pleiotropy in all associations. Moreover, the IVW method estimates are in the same direction as the MR-PRESSO method (Supplementary Table S4). In addition, leave-one-out analysis results supported that single SNP did not lead to MR estimation bias (Supplementary Fig. S1). In summary, after combining complementary and sensitivity analyses, we identified 6 metabolites (3 for CD and 3 for UC) that met stringent screening criteria.

Scatterplot of significantly associated (FDRivw < 0.1) and directionally consistent estimates between blood metabolites and IBD.
Replication and meta-analysis
To increase the persuasiveness of the estimates, we performed replication analysis using another IBD GWAS data to further validate our results. Combining the 2 GWAS datasets, a meta-analysis was performed for 6 metabolites that met stringent screening criteria. The results were as expected, and similar trends were observed for these 6 metabolites in another GWAS data (Fig. 5). In CD, in detail, genetic susceptibility for higher levels of mannose (OR = 0.27, 95% CI 0.15–0.49, P < 1 × 10–4) decreased risk of CD. The glycochenodeoxycholate (P = 0.236) and tryptophan betaine (P = 0.283) were not observed to be statistically significant, although consistent directions were shown in both MR analyses. Therefore, we discarded the glycochenodeoxycholate and tryptophan betaine. In UC, genetic susceptibility for higher levels of arachidonate (20:4n6) (OR = 0.28, 95% CI 0.12–0.63, P = 0.002) decreased risk of UC, while genetic liability for higher levels of 1,5-anhydroglucitol (OR = 1.95, 95% CI 1.44–2.63, P < 1 × 10–4), 2-stearoylglycerophosphocholine (OR = 1.95, 95% CI 1.04–3.66, P = 0.036) promoted susceptibility to UC.

Meta-analysis of the causal associations between metabolites and IBD.
Genetic correlation and direction validation
The results of the LDSC analysis showed weak evidence of a genetic correlation between 4 metabolites and IBD. Rg ranges from -0.8046 to 0.0682, the standard error from 0.0425 to 0.4230, and the p-value from 0.0527 to 0.9599 (p > 0.05). These results suggested that these shared genetic components did not confound the MR estimates (Supplementary Table S5). Furthermore, we further performed the Steiger test to examine whether there was reverse causality between metabolites and IBD. The results of Steiger do not support the existence of reverse causal effects between metabolites and IBD (Supplementary Table S6).
Metabolic pathway analysis
Based on the metabolites that passed the threshold of association by IVW (PIVW < 0.05), the metabolic pathway analysis identified 4 significant metabolic pathways in IBD (Supplementary Table S7). Specifically, our results show that the phenylalanine metabolism (P = 0.051) and ether lipid metabolism (P = 0.099) pathways were found to be associated with the pathogenetic process of CD, whereas arginine and proline metabolism (P = 0.008) and caffeine metabolism (P = 0.038) were considered to be associated with UC.
Discussion
In this study, we conducted a two-sample MR analysis to causally assess 486 blood metabolites and the risk of IBD through a rigorous MR design. We performed initial IVW analyses and identified a total of 63 causal relationships involving 59 metabolites and two IBD subtypes. Next, we performed complementary and sensitivity analyses on these metabolites. Then, to further ensure the reliability and stability of the results, we performed replication, meta-analysis, Steiger test, and LDSC. Ultimately, we identified four significant causal associations between four metabolites and two IBD subtypes. In addition, we detected four significant metabolic pathways involving two IBD subtypes. To our knowledge, this is the first MR study to systematically and comprehensively assess the causal role of human blood metabolites in IBD. Our study provides new insights into the role of gene-environment interactions in the pathogenesis of IBD and offers potential inspiration for further precise diagnosis and treatment.
In recent decades, IBD has been increasingly recognized as a metabolism-related disease, not only because it can coexist with a variety of metabolic disorders, but also because metabolites and metabolic pathways associated with IBD continue to be identified in metabolomic studies. Blood, feces, and synovial fluid are typical sample sources for metabolomics identification43. Among them, blood is considered a good source because it contains a large number of detectable metabolites and can be easily obtained in large samples, thus helping to screen for circulating markers of IBD risk. Moreover, blood metabolites provide a visual snapshot of biological mechanisms as they capture both endogenous and exogenous processes12,44. Metabolomics studies using serum/plasma have reportedly found altered metabolic profiles in IBD patients, and the most common metabolites are lipids, amino acids, and carbohydrates11,45,46. Despite the increasing number of studies on blood metabolism of IBD, there are still a lack of comprehensive and systematic studies to assess the causal relationship between IBD and blood metabolites, and thus the contribution to the precise diagnosis and treatment of IBD is limited. Therefore, we conducted a pivotal MR study with the aim of elucidating the causal relationship between blood metabolites and IBD and the metabolic pathways they may be involved in, thereby providing a reference direction for colorectal cancer screening and treatment.
Our findings suggested that genetic sensitivity to high levels of mannose can keep the body safe from CD. Mannose is a naturally occurring biologically active monosaccharide involved in glycoprotein metabolism and synthesis with anti-inflammatory and antioxidant activities. Metabolomics analyses showed that serum mannose concentrations were elevated in patients with CD, reflecting possible defects in the uptake, transport, or catabolism of endogenous mannose in cells47. A recent study showed that mannose could prevent endoplasmic reticulum stress induced by macrophage-secreted tumor necrosis factor-alpha (TNF-α) in intestinal epithelial cells by normalizing protein N-glycosylation, thus maintaining epithelial integrity. Moreover, mannose directly inhibited the production of TNF-α from macrophages by lowering the level of glyceraldehyde 3-phosphate and attenuates intestinal inflammation48. Another recent study found that mannose treatment attenuated intestinal barrier damage in IBD mouse models. The underlying mechanism may be that mannose limits cathepsin B release by enhancing lysosomal integrity, thereby preventing mitochondrial dysfunction and myosin light chainkinase (MLCK)-induced disruption of tight junctions in the context of intestinal epithelial injury. Notably, their experimental data demonstrated that mannose combined with mesalamine had a synergistic therapeutic effect on colonic inflammation in mice49. However, the direct mechanism of action of mannose in the treatment of IBD is unclear. Our results further support these findings and emphasize the importance of mannose as a protective factor in CD progression.
Arachidonate (20:4n6) is one of the polyunsaturated fatty acids found in cell membranes and is essential for maintaining cellular functions. Arachidonate can act as the substrate for the generation of a series of metabolic derivatives such as prostaglandins, thromboxanes, leukotrienes in response to cyclooxygenase, lipoxygenases, and cytochrome P450 epoxygenation. Metabolomics analysis showed that serum arachidonate levels were reduced in UC patients compared to healthy controls50. The role of arachidonate in UC remains incompletely elucidated. Several previous studies have shown that in mouse models of colitis, prostaglandin E2, a metabolic derivative of arachidonate, can exacerbate intestinal inflammation by increasing inflammatory cytokine secretion through promoting Th1 cell differentiation and Th17 cell proliferation51,52. Moreover, arachidonate can be metabolized to produce heparin A3 (HXA3), which promotes neutrophil migration into the lumen53. However, as studies progressed, some found that arachidonate could inhibit intestinal inflammation through the production of prostaglandins D2, poxyeicosatrienoic acids, 15-hydroxy eicosapentaenoic acid54,55,56. Notably, due to the complexity of the Arachidonate metabolic network, there are no convincing in vivo experimental data confirming a causal role between blood Arachidonate levels and UC. Our findings suggest that genetic susceptibility to high levels of Arachidonate plays a protective role in the development of UC, which needs to be further explored in in vivo experimental models.
We also confirmed genetic predisposition to higher levels of 1,5-anhydroglucitol and 2-stearoylglycerophosphocholine were detrimental to UC. To date, research on the relevance of 1,5-anhydroglucitol to UC is minimal. In patients with Glycogen Storage Disease type 1b (GSDIb), the accumulation of 1,5-anhydroglucitol-6-phosphate, which is produced by the metabolism of 1,5-anhydroglucitol in neutrophils, leads to neutropenia and neutrophil dysfunction57,58. Several recent studies have shown that the use of dapagliflozin or empagliflozin can induce IBD remission in patients with GSDIb by lowering plasma concentrations of 1,5-anhydroglucitol59,60. Therefore, it is reasonable to hypothesize that 1,5-anhydroglucitol can contribute to the development of UC by impairing neutrophil function. In contrast, reports on 2-stearoylglycerophosphocholine are extremely limited, with a few studies suggesting that 2-stearoylglycerophosphocholine may promote the development of severe COVID-1961. The causal role of 2-stearoylglycerophosphocholine on UC needs to be further explored.
Furthermore, in this study, we identified 4 metabolic pathways associated with the development of IBD, some of which are well documented in experimental studies for their role in the pathogenesis of IBD. Arginine is a semi-essential amino acid that participates in the urea cycle and arginine/proline metabolism. In humans, arginine is metabolized mainly by the arginase pathway to produce urea and ornithine; and by the nitric oxide synthase (NOS) pathway to produce citrulline and nitric oxide (NO)62. It is reported that NO has a protective role in colitis63. Moreover, ornithine is converted to proline by ornithine transaminase, which promotes the repair of the intestinal barrier during inflammation64. These data suggest that enhanced arginine and proline metabolism may be protective against UC. Caffeine is a natural chemical with stimulant effects. Previous studies have shown that caffeine can reduce the risk of UC and ameliorates colitis65,66. However, there are few experimental studies on the role of ether lipid metabolism and phenylalanine metabolism in the pathogenesis of IBD, which warrant further experimental exploration.
The present study has several strengths. First, the most significant strength of this study is that we covered a large number of genetic variables to analyze the relationship between blood metabolites and IBD. A total of 486 metabolites were covered in this study, which is of significant clinical research value. Second, rigorous MR analysis was used to discard unavoidable pitfalls of previous studies, such as confounding interference and reverse causality. In order to produce convincing estimates, a series of methods were implemented to ensure that violations of MR assumptions were eliminated to the greatest extent possible, according to the STROBE-MR Statement15. Specifically, the consistency of the five MR method estimates in direction and magnitude ensured the robustness of the results. At the same time, sensitivity analyses were conducted to exclude the effects of heterogeneity and pleiotropy on the estimates. Third, the reliability of the results was further validated by additional GWAS data used for repeated analyses and meta-analysis. Finally, we used LDSC to assess the genetic correlation between metabolites and IBD, which made the MR estimates more convincing.
There are some limitations to our study. First, all metabolite GWAS and IBD GWAS data were from European populations. Although this largely avoids population heterogeneity, MR results should be further validated in other populations to verify their generalizability with more GWAS data in future studies. Next, the number of metabolite-associated SNPs was limited. To address this issue, we set a slightly relaxed threshold for MR analysis, which is a common practice in other studies. Moreover, although we used PhenoScanner V2 to exclude IVs associated with confounders (such as depression, being a worrier, smoking, taking contraceptives, having sclerosing cholangitis), it was a limitation that numerous other IBD factors could not be fully accounted for. Finally, this study covers a relatively comprehensive metabolite profile, but the functions and mechanisms of some metabolites in the disease are not yet fully understood, which limits our interpretation of the results of this MR analysis. Our findings need to be validated by rigorous RCTs.
Conclusion
In conclusion, we identified four robust causal associations between four metabolites and two IBD subtypes. Through metabolic pathway analysis, four important metabolic pathways associated with IBD were identified. The finding of our study suggested that the identified metabolites and metabolic pathways can be considered as useful circulating metabolic biomarkers for the screening and prevention of IBD in clinical practice, as well as candidate molecules for future mechanism exploration and drug target selection.
Data availability
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
References
Buie, M. J. et al. Global Hospitalization trends for crohn’s disease and ulcerative colitis in the 21st Century: A systematic review with temporal analyses. Clin. Gastroenterol. Hepatol. Offic. Clin. Pract. J. Am. Gastroenterol. Assoc. https://doi.org/10.1016/j.cgh.2022.06.030 (2022).
Chen, X. et al. Evolving trends and burden of inflammatory bowel disease in Asia, 1990–2019: A comprehensive analysis based on the global burden of disease study. J. Epidemiol. Glob. Health https://doi.org/10.1007/s44197-023-00145-w (2023).
Flynn, S. & Eisenstein, S. Inflammatory bowel disease presentation and diagnosis. Surg. Clin. North Am. 99, 1051–1062. https://doi.org/10.1016/j.suc.2019.08.001 (2019).
Long, X. Q. et al. Bile acids and their receptors: Potential therapeutic targets in inflammatory bowel disease. World J. Gastroenterol. 29, 4252–4270. https://doi.org/10.3748/wjg.v29.i27.4252 (2023).
Banerjee, R., Pal, P., Mak, J. W. Y. & Ng, S. C. Challenges in the diagnosis and management of inflammatory bowel disease in resource-limited settings in Asia. Lancet Gastroenterol. Hepatol. 5, 1076–1088. https://doi.org/10.1016/s2468-1253(20)30299-5 (2020).
Seishima, R. et al. Effect of biologics on the risk of advanced-stage inflammatory bowel disease-associated intestinal cancer: A nationwide study. Am. J. Gastroenterol. 118, 1248–1255. https://doi.org/10.14309/ajg.0000000000002149 (2023).
Baumgart, D. C. & Le Berre, C. Newer biologic and small-molecule therapies for inflammatory bowel disease. N. Engl. J. Med. 385, 1302–1315. https://doi.org/10.1056/NEJMra1907607 (2021).
Ahmed, W. et al. Dual biologic or small molecule therapy for treatment of inflammatory bowel disease: A systematic review and meta-analysis. Clin. Gastroenterol. Hepatol. Offic. Clin. Pract. J. Am. Gastroenterol. Assoc. 20, e361–e379. https://doi.org/10.1016/j.cgh.2021.03.034 (2022).
Lee, M. & Chang, E. B. Inflammatory bowel diseases (IBD) and the microbiome-searching the crime scene for clues. Gastroenterology 160, 524–537. https://doi.org/10.1053/j.gastro.2020.09.056 (2021).
Guan, Q. A comprehensive review and update on the pathogenesis of inflammatory bowel disease. J. Immunol. Res. 2019, 7247238. https://doi.org/10.1155/2019/7247238 (2019).
Scoville, E. A. et al. Alterations in lipid, amino acid, and energy metabolism distinguish Crohn’s disease from ulcerative colitis and control subjects by serum metabolomic profiling. Metabol. Off. J. Metabol. Soc. 14, 17. https://doi.org/10.1007/s11306-017-1311-y (2018).
Di’Narzo, A. F. et al. Integrative analysis of the inflammatory bowel disease serum metabolome improves our understanding of genetic etiology and points to novel putative therapeutic targets. Gastroenterology 162, 828-843.e811. https://doi.org/10.1053/j.gastro.2021.11.015 (2022).
Kettunen, J. et al. Genome-wide study for circulating metabolites identifies 62 loci and reveals novel systemic effects of LPA. Nat. Commun. 7, 11122. https://doi.org/10.1038/ncomms11122 (2016).
Emdin, C. A., Khera, A. V. & Kathiresan, S. Mendelian randomization. Jama 318, 1925–1926. https://doi.org/10.1001/jama.2017.17219 (2017).
Skrivankova, V. W. et al. Strengthening the reporting of observational studies in epidemiology using Mendelian randomization: The STROBE-MR statement. Jama 326, 1614–1621. https://doi.org/10.1001/jama.2021.18236 (2021).
Shin, S. Y. et al. An atlas of genetic influences on human blood metabolites. Natu. Genet. 46, 543–550. https://doi.org/10.1038/ng.2982 (2014).
Kurki, M. I. et al. FinnGen provides genetic insights from a well-phenotyped isolated population. Nature 613, 508–518. https://doi.org/10.1038/s41586-022-05473-8 (2023).
Guo, M. N. et al. Human blood metabolites and lacunar stroke: A Mendelian randomization study. Int. J. Stroke Offic. J. Int. Stroke Soc. 18, 109–116. https://doi.org/10.1177/17474930221140792 (2023).
Yu, X. H., Cao, R. R., Yang, Y. Q. & Lei, S. F. Identification of causal metabolites related to multiple autoimmune diseases. Human Mol. Genet. 31, 604–613. https://doi.org/10.1093/hmg/ddab273 (2022).
Cai, J. et al. Genetic liability for prescription opioid use and risk of cardiovascular diseases: A multivariable Mendelian randomization study. Addiction 117, 1382–1391. https://doi.org/10.1111/add.15767 (2022).
Palmer, T. M. et al. Using multiple genetic variants as instrumental variables for modifiable risk factors. Stat. Methods Med. Res. 21, 223–242. https://doi.org/10.1177/0962280210394459 (2012).
Gill, D., Efstathiadou, A., Cawood, K., Tzoulaki, I. & Dehghan, A. Education protects against coronary heart disease and stroke independently of cognitive function: Evidence from Mendelian randomization. Int. J. Epidemiol. 48, 1468–1477. https://doi.org/10.1093/ije/dyz200 (2019).
Bisgaard, T. H., Allin, K. H., Keefer, L., Ananthakrishnan, A. N. & Jess, T. Depression and anxiety in inflammatory bowel disease: Epidemiology, mechanisms and treatment. Nat. Rev. Gastroenterol. Hepatol. 19, 717–726. https://doi.org/10.1038/s41575-022-00634-6 (2022).
Timmer, A., Sutherland, L. R. & Martin, F. Oral contraceptive use and smoking are risk factors for relapse in Crohn’s disease. The Canadian Mesalamine for Remission of Crohn’s Disease Study Group. Gastroenterology 114, 1143–1150. https://doi.org/10.1016/s0016-5085(98)70419-6 (1998).
Ananthakrishnan, A. N. et al. Lifestyle, behaviour, and environmental modification for the management of patients with inflammatory bowel diseases: An International Organization for Study of Inflammatory Bowel Diseases consensus. Lancet Gastroenterol. Hepatol. 7, 666–678. https://doi.org/10.1016/s2468-1253(22)00021-8 (2022).
Barberio, B. et al. Prevalence of primary sclerosing cholangitis in patients with inflammatory bowel disease: A systematic review and meta-analysis. Gastroenterology 161, 1865–1877. https://doi.org/10.1053/j.gastro.2021.08.032 (2021).
Burgess, S., Scott, R. A., Timpson, N. J., Davey Smith, G. & Thompson, S. G. Using published data in Mendelian randomization: A blueprint for efficient identification of causal risk factors. Eur. J. Epidemiol. 30, 543–552. https://doi.org/10.1007/s10654-015-0011-z (2015).
Burgess, S., Butterworth, A. & Thompson, S. G. Mendelian randomization analysis with multiple genetic variants using summarized data. Genet. Epidemiol. 37, 658–665. https://doi.org/10.1002/gepi.21758 (2013).
Bowden, J., Davey Smith, G. & Burgess, S. Mendelian randomization with invalid instruments: Effect estimation and bias detection through Egger regression. Int. J. Epidemiol. 44, 512–525. https://doi.org/10.1093/ije/dyv080 (2015).
Bowden, J., Davey Smith, G., Haycock, P. C. & Burgess, S. Consistent estimation in mendelian randomization with some invalid instruments using a weighted median estimator. Genet. Epidemiol. 40, 304–314. https://doi.org/10.1002/gepi.21965 (2016).
Hartwig, F. P., Davey Smith, G. & Bowden, J. Robust inference in summary data Mendelian randomization via the zero modal pleiotropy assumption. Int. J. Epidemiol. 46, 1985–1998. https://doi.org/10.1093/ije/dyx102 (2017).
Greco, M. F., Minelli, C., Sheehan, N. A. & Thompson, J. R. Detecting pleiotropy in Mendelian randomisation studies with summary data and a continuous outcome. Stat. Med. 34, 2926–2940. https://doi.org/10.1002/sim.6522 (2015).
Bowden, J. et al. Assessing the suitability of summary data for two-sample Mendelian randomization analyses using MR-Egger regression: The role of the I2 statistic. Int. J. Epidemiol. 45, 1961–1974. https://doi.org/10.1093/ije/dyw220 (2016).
Verbanck, M., Chen, C. Y., Neale, B. & Do, R. Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. Nat. Genet. 50, 693–698. https://doi.org/10.1038/s41588-018-0099-7 (2018).
Burgess, S. & Thompson, S. G. Interpreting findings from Mendelian randomization using the MR-Egger method. Eur. J. Epidemiol. 32, 377–389. https://doi.org/10.1007/s10654-017-0255-x (2017).
Glickman, M. E., Rao, S. R. & Schultz, M. R. False discovery rate control is a recommended alternative to Bonferroni-type adjustments in health studies. J. Clin. Epidemiol 67, 850–857. https://doi.org/10.1016/j.jclinepi.2014.03.012 (2014).
Li, P. et al. Association between gut microbiota and preeclampsia-eclampsia: A two-sample Mendelian randomization study. BMC Med. 20, 443. https://doi.org/10.1186/s12916-022-02657-x (2022).
O’Connor, L. J. & Price, A. L. Distinguishing genetic correlation from causation across 52 diseases and complex traits. Nat. Genet. 50, 1728–1734. https://doi.org/10.1038/s41588-018-0255-0 (2018).
Hemani, G., Tilling, K. & Davey Smith, G. Orienting the causal relationship between imprecisely measured traits using GWAS summary data. PLoS Genet. 13, e1007081. https://doi.org/10.1371/journal.pgen.1007081 (2017).
Pang, Z. et al. MetaboAnalyst 5.0: Narrowing the gap between raw spectra and functional insights. Nucleic Acids Res. 49, W388-w396. https://doi.org/10.1093/nar/gkab382 (2021).
Gu, Y. et al. Causality of genetically determined metabolites and metabolic pathways on osteoarthritis: A two-sample mendelian randomization study. J. Transl. Med. 21, 357. https://doi.org/10.1186/s12967-023-04165-9 (2023).
Yu, X. H. et al. Systematic evaluation for the causal effects of blood metabolites on osteoporosis: Genetic risk score and Mendelian randomization. Front. Public Health 10, 905178. https://doi.org/10.3389/fpubh.2022.905178 (2022).
Johnson, C. H., Ivanisevic, J. & Siuzdak, G. Metabolomics: Beyond biomarkers and towards mechanisms. Nat. Rev. Mol. Cell Biol. 17, 451–459. https://doi.org/10.1038/nrm.2016.25 (2016).
Wang, J. et al. Metabolomics window into the diagnosis and treatment of inflammatory bowel disease in recent 5 years. Int. Immunopharmacol. 113, 109472. https://doi.org/10.1016/j.intimp.2022.109472 (2022).
Kolho, K. L., Pessia, A., Jaakkola, T., de Vos, W. M. & Velagapudi, V. Faecal and serum metabolomics in paediatric inflammatory bowel disease. J. Crohn’s Colitis 11, 321–334. https://doi.org/10.1093/ecco-jcc/jjw158 (2017).
Murgia, A. et al. Italian cohort of patients affected by inflammatory bowel disease is characterised by variation in glycerophospholipid, free fatty acids and amino acid levels. Metab. Offic. J. Metabol. Soc. 14, 140. https://doi.org/10.1007/s11306-018-1439-4 (2018).
Schicho, R. et al. Quantitative metabolomic profiling of serum, plasma, and urine by (1)H NMR spectroscopy discriminates between patients with inflammatory bowel disease and healthy individuals. J. Proteome Res. 11, 3344–3357. https://doi.org/10.1021/pr300139q (2012).
Xiao, P. et al. Mannose metabolism normalizes gut homeostasis by blocking the TNF-α-mediated proinflammatory circuit. Cell. Mol. Immunol. 20, 119–130. https://doi.org/10.1038/s41423-022-00955-1 (2023).
Dong, L. et al. Mannose ameliorates experimental colitis by protecting intestinal barrier integrity. Nat. Commun. 13, 4804. https://doi.org/10.1038/s41467-022-32505-8 (2022).
Guan, S. et al. UPLC-QTOF-MS-based plasma lipidomic profiling reveals biomarkers for inflammatory bowel disease diagnosis. J. Proteom. Res. 19, 600–609. https://doi.org/10.1021/acs.jproteome.9b00440 (2020).
Sheibanie, A. F. et al. The proinflammatory effect of prostaglandin E2 in experimental inflammatory bowel disease is mediated through the IL-23–>IL-17 axis. J. Immunol. 178, 8138–8147. https://doi.org/10.4049/jimmunol.178.12.8138 (2007).
Yao, C. et al. Prostaglandin E2-EP4 signaling promotes immune inflammation through Th1 cell differentiation and Th17 cell expansion. Nature medicine 15, 633–640. https://doi.org/10.1038/nm.1968 (2009).
Mrsny, R. J. et al. Identification of hepoxilin A3 in inflammatory events: a required role in neutrophil migration across intestinal epithelia. Proc. Ntl. Acad. Sci. USA 101, 7421–7426. https://doi.org/10.1073/pnas.0400832101 (2004).
Rohwer, N. et al. Omega-3 fatty acids protect from colitis via an Alox15-derived eicosanoid. FASEB J. Offic. Publ. Feder. Am. Soc. Exp. Biol. 35, e21491. https://doi.org/10.1096/fj.202002340RR (2021).
Reisdorf, W. C. et al. Preclinical evaluation of EPHX2 inhibition as a novel treatment for inflammatory bowel disease. PloS One 14, e0215033. https://doi.org/10.1371/journal.pone.0215033 (2019).
Rajakariar, R. et al. Hematopoietic prostaglandin D2 synthase controls the onset and resolution of acute inflammation through PGD2 and 15-deoxyDelta12 14 PGJ2. Proc. Ntl. Acad. Sci. USA 104, 20979–20984. https://doi.org/10.1073/pnas.0707394104 (2007).
Veiga-da-Cunha, M. et al. Failure to eliminate a phosphorylated glucose analog leads to neutropenia in patients with G6PT and G6PC3 deficiency. Proc. Ntl. Acad. Sci. USA 116, 1241–1250. https://doi.org/10.1073/pnas.1816143116 (2019).
Visser, G. et al. Neutropenia, neutrophil dysfunction, and inflammatory bowel disease in glycogen storage disease type Ib: Results of the European Study on Glycogen Storage Disease type I. J. Pediatr. 137, 187–191. https://doi.org/10.1067/mpd.2000.105232 (2000).
Resaz, R. et al. The SGLT2-inhibitor dapagliflozin improves neutropenia and neutrophil dysfunction in a mouse model of the inherited metabolic disorder GSDIb. Mol. Genet. Metab. Rep. 29, 100813. https://doi.org/10.1016/j.ymgmr.2021.100813 (2021).
Rossi, A. et al. Crohn disease-like enterocolitis remission after empagliflozin treatment in a child with glycogen storage disease type Ib: A case report. Ital. J. Pediatr. 47, 149. https://doi.org/10.1186/s13052-021-01100-w (2021).
Huang, C. et al. Human serum metabolites as potential mediators from type 2 diabetes and obesity to COVID-19 severity and susceptibility: Evidence from Mendelian randomization study. Metabolites https://doi.org/10.3390/metabo12070598 (2022).
Morris, S. M. Jr. Arginine metabolism: Boundaries of our knowledge. The J. Nutr. 137, 1602s–1609s. https://doi.org/10.1093/jn/137.6.1602S (2007).
Yoshida, Y. et al. Role of inducible nitric oxide synthase in dextran sulphate sodium-induced colitis. Alimen. Pharmacol. Therap. 14(Suppl 1), 26–32. https://doi.org/10.1046/j.1365-2036.2000.014s1026.x (2000).
Singh, K. et al. L-arginine uptake by cationic amino acid transporter 2 is essential for colonic epithelial cell restitution. Am. J. Physiol. Gastrointestin. Liver Physiol. 302, 1061–1073. https://doi.org/10.1152/ajpgi.00544.2011 (2012).
Lee, I. A., Low, D., Kamba, A., Llado, V. & Mizoguchi, E. Oral caffeine administration ameliorates acute colitis by suppressing chitinase 3-like 1 expression in intestinal epithelial cells. J. Gastroenterol. 49, 1206–1216. https://doi.org/10.1007/s00535-013-0865-3 (2014).
Tanaka, K. et al. Coffee and caffeine intake reduces risk of ulcerative colitis: a case-control study in Japan. J. Gastroenterol. Hepatol. https://doi.org/10.1111/jgh.16439 (2023).
Acknowledgements
We thank all the consortium studies for making the summary association statistics data publicly available.
Funding
This work was supported by National Natural Science Foundation of China (Grant/Award Number: 81900466) and Hunan Provincial Natural Science Foundation of China (Grant/Award Number: 2020JJ5307).
Author information
Authors and Affiliations
Contributions
X.L., Y.Z. and M.L. conceived and designed the study and wrote the manuscript. X.L. and Z.L. acquired the data and analyzed the results. X.L. and L.X. proofread the results. X.X. and M.W. reviewed and edited the manuscript. All authors read and approved the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Long, X., Zhang, Y., Liu, M. et al. Causality of genetically determined blood metabolites on inflammatory bowel disease: a two-sample Mendelian randomization study. Sci Rep 14, 16361 (2024). https://doi.org/10.1038/s41598-024-67376-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-67376-0
- Springer Nature Limited