
Abstract
Leaf scald, caused by Xanthomonas albilineans, is a severe disease affecting sugarcane worldwide. One of the most practical ways to control it is by developing resistant sugarcane cultivars. It is essential to identify genes associated with the response to leaf scald. A panel of 170 sugarcane genotypes was evaluated for resistance to leaf scald in field conditions for 2 years, followed by a 1-year greenhouse experiment. The phenotypic evaluation data showed a wide continuous distribution, with heritability values ranging from 0.58 to 0.84. Thirteen single nucleotide polymorphisms (SNPs) were identified, significantly associated with leaf scald resistance. Among these, eight were stable across multiple environments and association models. The candidate genes identified and validated based on RNA-seq and qRT-PCR included two genes that encode NB-ARC leucine-rich repeat (LRR)-containing domain disease-resistance protein. These findings provide a basis for developing marker-assisted selection strategies in sugarcane breeding programs.
Similar content being viewed by others


Introduction
Sugarcane is an important commercial crop belonging to the Poaceae family. It is cultivated in tropical and subtropical regions worldwide1. Being a dual-purpose crop, it accounts for 85% of global sugar production and 40% of bioenergy feedstock for ethanol production2. As the third-largest global sugarcane producer, China contributes 5% of the total sugar production worldwide (FAO, http://www.fao.org/faostat). Despite its economic significance, sugarcane is susceptible to various pathogens, resulting in yield losses, sucrose reduction, and economic setbacks3. Xanthomonas albilineans is a xylem-intruding bacterium that causes sugarcane leaf scald, a severe disease in 66 countries4. When the pathogen colonizes the plant’s vascular system, it forms a white, narrow, and sharply defined stripe. This infection ultimately leads to complete wilting and necrosis in the diseased leaves, posing a severe threat to susceptible sugarcane genotypes5. A recent report of a leaf scald outbreak in southern Mexico indicated a 3 to 15% incidence of leaf scald in the infected fields6. Leaf scald was first reported in China in 2016 and has subsequently spread and escalated into an epidemic, resulting in a severe threat to sugar production5,7,8. The most cost-effective and environmentally friendly strategy to manage the permanent risk of leaf scalds is to develop resistant cultivars9,10. However, the troublesome aspect of resistance evaluation is that symptom expression is strongly affected by environmental conditions11. The unstable symptom expression results in an inability to detect susceptibility accurately; thus, multiple field trials are needed. Furthermore, utilizing molecular markers associated with resistance can significantly contribute to marker-assisted sugarcane breeding programs. Genetic markers, especially DNA markers, have become essential and efficient tools in developing disease resistance12 and are more conducive to developing new varieties with stable resistance than traditional breeding methods13.
Sugarcane has a complex and large genome (approximately 10 Gb) because sugarcane is derived from interspecific hybridization between the high-sugar species Saccharum officinarum and the wild species S. spontaneum, followed by several backcrosses with S. officinarum, making genetic and genomic studies of sugarcane cultivars challenging14,15. However, target enrichment sequencing is an ideal and cost-effective approach for sequencing hundreds of putatively single-copy nuclear regions in multiple samples16. SNP genotyping platforms are based on target enrichment sequencing, which is very flexible regarding the number of SNP markers. The Axiom sugarcane 100K SNP is an efficient genetic tool for high-throughput genotyping in highly polyploid sugarcane17. The Axiom sugarcane 100K SNP arrays were designed using a large number of single nucleotide polymorphisms (SNPs) markers discovered from sugarcane genomes using the sorghum (Sorghum bicolor) genome as reference18,19, which is a closely related crop containing 95.2% identical coding regions with sugarcane20. Most of the markers were developed using mapping populations derived from bi-parental crosses. In constructing a genetic linkage map, a 3:1 (resistant: susceptible) segregation was observed in the sugarcane cultivar “R570” for rust resistance, indicating the possible presence of dominant resistance genes21. Eight QTLs associated with leaf scald response were identified, explaining 5.23% to 16.93% of the phenotypic variance22. Eighteen QTLs regulated resistance to sugarcane yellow leaf virus, segregating the two mapping populations containing 27 disease-resistant genes17. These successful examples provide feasibility for marker-assisted selection in sugarcane. In addition, genome-wide association studies (GWAS) have also become a popular tool for identifying the genetic factors underlying marker-trait associations and specific genomic regions responsible for the essential genes associated with various crop diseases and complex traits 23,24,25.
To investigate candidate loci and genes related to resistance against leaf scald, we genotyped 170 sugarcane germplasms using an Axiom Sugarcane 100K SNP array. The genotypes were evaluated for leaf scald resistance in field conditions over 2 years (2021–2022), followed by one greenhouse experiment. We used five association models of GAPIT26 to identify SNP loci associated with leaf scald resistance. Building on this, we explored candidate resistance genes by integrating gene annotation information with the publicly available transcriptome. Our study describes promising results that will accelerate marker-assisted selection for improving leaf scald resistance in sugarcane breeding programs and delineate prospective targets for cloning novel resistant genes.
Results
Phenotypic evaluation for leaf scald
The disease incidence (IC) and index (DI) of leaf scald in each experiment have been listed in Supplementary Table 1. Analysis of variance for IC and DI indicated that the genotypes (G), environment (E), and G × E effects were highly significant (p < 0.001; Supplementary Table 2). The IC distribution of sugarcane genotypes ranged from 0.00 to 21.43%, and the DI distribution ranged from 0.00 to 12.00 (Table 1). IC and DI values were continuous in all environments, indicating that the traits were quantitative. The broad-sense heritability (HB2) for IC and DI over two consecutive years in the field were 0.58 and 0.70, respectively. Moreover, HB2 for IC and DI of the greenhouse experiment were 0.72 and 0.84, respectively, indicating that genetic factors played an important role (Table 1).
Although the correlation between IC and DI of the cultivars was significant between field and greenhouse studies, the actual differences in the average IC and DI among genotypes were much more minor in the field trial than in the greenhouse trial (Supplementary Table 1). The phenotyping results of most genotypes were similar. Thirty-two genotypes were symptomless or immune in all three environments. The genotype CP97-1602 was the most severe in all three environments. Specific genotypes showed inconsistent phenotypes across the three environments. For instance, seven genotypes (4–91, 4–195, 5–77, 4–117, GT07-713, GT02-1156, 2–65) were asymptomatic in a field environment (2022), but showed typical symptoms in greenhouse environments (Supplementary Table 1).
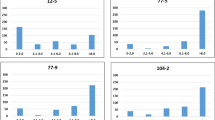
The distribution of the raw phenotypic data for leaf scald was visualized using the histogram, which showed continuous distribution variation and left skewed in each experiment of 170 genotypes (Supplementary Fig. 1). To achieve normality of residuals, BLUP values were calculated by applying a mixed linear model, and the data from the greenhouse experiment were transformed using an inverse sine square root transformation. Ultimately, eight data sets were used for subsequent GWAS analysis, including the value of genotype by year interaction, BLUP, and transformed phenotypic data from the greenhouse (Fig. 1, Supplementary Table 3). Correlations between IC and DI within transformed data ranged from 0.73 to 0.99. Pearson’s correlation coefficient among different datasets indicated a highly significant (p < 0.001) correlation (Fig. 1).

Genotypic correlation between disease severity evaluated in different experiments. The diagonal panels show histograms for each environment trait. The lower and upper triangular panels show scatter plots and Pearson correlation coefficients between pairs of environment traits, respectively. ***Indicates significance at 0.001 levels. The X and Y-axis represent the numerical values.
Population structure
A total of 100,097 SNPs were filtered to generate 26,787 high-quality SNPs for subsequent analysis. These SNPs were distributed across ten sorghum chromosomes, with the highest number of SNPs on chromosome 1 (Fig. 2). The pairwise kinship matrix heatmap of the 170 genotypes revealed three significant subgroups, with their relationships displayed along the diagonal line with a few large blocks of closely related individuals (Supplementary Fig. 2). Moreover, cross-validation (CV) error was used to predict the most appropriate value for K (the optimal number of populations in the dataset). According to admixture clustering, a break in the slope was observed at K = 3, showing the lowest CV error (Supplementary Fig. 3). For the phylogenetic tree, lines with different colors indicated different subgroups, where genotypes 4–26 and 8–91 of Group II were clustered into Group I (Fig. 3A). Principal component analysis (PCA) plot for all genotypes clearly showed that samples from the same subgroups clustered together (Fig. 3B). The admixture plots showed results for K = 3, the lowest cross-validation (CV) error (Fig. 3C), revealing a high degree of admixture in many genotypes, with 23 (35%) genotypes in group I, 57 (93.44%) genotypes in group II, and 27 (62.8%) genotypes in group III, respectively, mixed with another group. In particular, most genotypes in groups II and III were mixed. This result may be due to these genotypes inherited from a common ancestry. According to the clustering, kinship analysis, phylogenetic tree construction, and PCA analyses, parental origin was the primary factor determining this panel’s classification diversity.

Distribution of high-quality SNPs over ten sorghum chromosomes.

Phylogenetic and population structure analysis of 170 sugarcane genotypes based on 26,787 SNP markers. (A) Phylogenetic tree of the 170 genotypes; (B) scatterplot based on principal component analysis; (C) stacked bar plots of ADMIXTURE results for K = 3.
Genome-wide association analysis
Five GWAS models were tested, and QQ plots for each trait were compared to assess the optimal model for controlling false-positive associations due to the structure and relatedness (Supplementary Fig. 4). A total of 107 SNPs were identified, of which 41 were by GLM, 35 by MLM, 12 by MLMM, 7 by SUPER, and 12 by BLINK (Table 2, Supplementary Table 4). After analysis, most SNPs were overlapping; thirteen significant SNPs were associated with the response to leaf scald (Table 3, Fig. 4). Eight SNPs were detected in multi-environments and -association models, including S1_25897077, S1_61002332, S2_72384593, S5_14567373, S6_50836650, S6_60340985, S6_60594020, S7_65315409. The other five SNPs were detected in a single environment or association model. The thirteen candidate SNPs were located on seven sorghum chromosomes (Chr01, Chr02, Chr04, Chr05, Chr06, Chr07, Chr10). Most were located on Chr02 and Chr06, each containing three SNPs (Table 3). The phenotypic variation explained (PVE) by a single SNP ranged from 4.28 to 17.98%; eight SNPs had a PVE greater than 10% (Table 3). These significant SNPs and their − log10 p-values were visualized in the Manhattan plots (Fig. 4).

Manhattan plots of GWAS results showing significant SNPs associated with leaf scald resistance in sugarcane genotypes diversity panel. The X-axis shows the distribution of SNPs across 10 chromosomes, while the Y-axis shows the Bonferroni corrections threshold. The black dashed horizontal line depicts the uniform significance threshold (− Log10 p-value = 5.73).
Candidate gene for leaf scald resistance
Of the thirteen significant SNPs, only one SNP (S10_8458899) was identified in the intergenic spacer region. The remaining 12 significant SNP markers were located within genes and were considered candidate genes (Table 4). SNP analysis revealed diverse variant classifications, including four missense variants, two three-prime UTR variants, three intron variants, and three synonymous variants. The SNP with the largest log10 (p) value found in this study was marker S1_25897077, situated within the gene BTB-POZ (gene symbol) sequence, predicted to code a BTB-POZ and MATH domain. In addition, SNP markers S1_61002332 and S2_71293304 were found within genes coding for an Oxoprolinase (OXO) and Tetratricopeptide repeat (TPR)-like superfamily protein (TPR), respectively. Two SNPs (S5_14567373 and S6_50836650) were identified inside the gene encoding for putative proteins with LRR and NB-ARC domains, known for their essential role in disease resistance. Finally, SNP marker S7_65315409 was located within gene SLDMS (gene symbol), predicted to code for an S-adenosyl-l-methionine-dependent methyltransferase superfamily protein. Moreover, the CDS sequences of the sorghum genome were BLASTed to the S. spontaneum and S. officinarum genome separately. Twelve sorghum candidate genes exhibited 30 and 102 homologous genes in S. spontaneum and S. officinarum, respectively (Supplementary Table 5). We conducted a detailed analysis of candidate genes, integrating GWAS-identified genes with differentially expressed genes (DEGs) from RNA-seq. Among the twelve candidate genes, the six genes (NHH, TPR, PCN, BTB-POZ, OXO, SLDMS) were found to show relatively higher expression levels in resistant cultivar (LCP 85–384), gene PBP showed a shallow level or no expression in both susceptible cultivar (ROC20) and resistant cultivar (LCP 85–384) (Fig. 5A). In addition, we analyzed the differential expression of twelve genes in both cultivars at different inoculation time points, and the results show that five genes displayed significant differential expressions at least at a one-time point in both susceptible cultivar (ROC20) and resistant cultivar (LCP 85–384) (Supplementary Table 6).

The expression pattern of the candidate genes observed in (A) transcriptome and (B) quantitative reverse transcription-PCR (qRT-PCR). Each column of (A) represents a time point after bacterial inoculation in one cultivar; S-susceptible cultivar (ROC20); R-resistant cultivar (LCP 85–384); 0, 24, 48, and 72 indicates hours post-inoculation. For (B), the abscissa represents cultivar (ZZ1, GT46) and treatment (CK, un-inoculated; LSD, inoculated), and the ordinate represents the relative expression of genes calculated by the 2 − ΔΔCT method. Different letters (a, b, and c) represent statistically significant differences (ANOVA with Turkey post-hoc analysis, 5% level). The mean ± S.D. of the biological replicates is presented.
Validation of candidate gene expression
A complete list of Ct values, including reference genes and candidate genes, is given in Supplementary Table 8. The quality of primers was tested by examining the formation of primer dimers based on the dissolution curves. All primer pairs with a single high peak demonstrated no nonspecific amplification (Supplementary Fig. 5). Six candidate genes were selected for qRT-PCR validation. Under normal conditions without Xanthomonas albilineans stress, there are four genes (ARK, LRR, UK1, and NB-ARC) showed significant differences in the relative expression levels of the resistant (ZZ1) and susceptible (GT46) genotypes. In contrast, the genes BTB-POZ and PCN showed no significant difference in the relative expression levels. The genotypes ZZ1 did not show a significant difference in the relative gene expression level of genes ARK and NB-ARC compared with control when subjected to inoculation treatment. However, there was a difference in the relative expression levels in GT46 compared with control when subjected to inoculation treatment of these genes. One of the genes coded LRR domains and gene BTB-POZ were upregulated in resistant genotype ZZ1. Another gene, NB-ARC, was expressed significantly higher in the susceptible genotype GT46 than in the resistant genotype ZZ1. The gene PCN encoded a P-loop containing superfamily protein of triphosphate hydrolases, which was significantly downregulated in resistant genotype ZZ1. In addition, the genes involved in coded LRR and PCN were not differentially expressed after inoculation in GT46 (Fig. 5B). This further proves that the six putative genes were closely associated with the response to leaf scald.
Discussion
Severe yield losses in China have been reported due to leaf scald disease5,8. Consequently, future outbreaks of leaf scalds represent a persistent danger to yield and quality stability. It is urgent to breed resistant varieties due to the rapid evolution of virulent races of the pathogen. Genome-wide association mapping (GWAS) is a powerful approach that facilitates identifying genes regulating several traits. To develop the genetic markers linked to the leaf scald resistance for marker-assisted selection in the breeding programs, we used the genetic data available for 170 genotypes tested for leaf scald resistance in three different environments. Our results could help understand the resistance mechanism of leaf scald and accelerate the sugarcane breeding process toward combating this disease.
A high phenotypic variation was observed within the 170 genotypes. We calculated BLUP values through two consecutive years in the field to reduce the environmental impact. The frequency distribution for BLUP values followed a biased normal distribution, similar to the previous studies27,28. Despite significant genotype-by-environment interaction, the correlation of leaf scald severity across different experiments was high, indicating the significance of the evaluation method for leaf scald. The high heritability highlighted the importance of genetic factors and the reliability of the phenotypes. These findings collectively suggested that the observed phenotypic variation could be attributed to the differences at the genotypic level.
SNP microarray-based genotyping is widely used in crop genetic studies, including genome-wide association analysis (GWAS), linkage disequilibrium map, genome selection, and population structure18,29,30. Due to the lack of a high-quality sugarcane genome as a reference in earlier studies, the researchers developed an Axiom Sugarcane 100K SNP microarray using sorghum as a reference genome, which was applied to QTL localization and GWAS analysis of sugarcane traits17. The LD caused by population structure could promote false positive detection in GWAS analysis31,32. To avoid these false associations, the models consider covariates (population structure matrix and/or kinship matrix) to adjust the association tests on markers. When analyzing the population structure of genotypes, we found that subgroups were directly related to their parental origin, which is consistent with other findings33,34.
Thirteen SNP markers associated with the response to leaf scald were observed, with eight SNPs stably detected across multi-environments and -association models. These SNPs linked to resistance against leaf scald hold potential applications in sugarcane breeding programs for marker-assisted selection. In addition, these SNPs could be used to develop a Kompetitive allele-specific PCR (KASP) assay, a technique well evidenced in wheat35,36,37. GWAS analysis identified SNPs in deciphering disease mechanisms and the SNPs within the gene, especially the genes associated with the LD regions that might be involved in plant disease resistance31,32. However, current studies on LD decay distances in sugarcane have shown distances of 5 kb, 10 kb, 50 kb, and even 3.5 Mb 38,39,40,41. In this investigation, only the gene containing significant SNP was regarded as candidate genes due to the inaccuracy of determining the LD decay distance owing to the small number of SNPs. Moreover, we aligned the coding sequence (CDS) of each candidate gene with the S. spontaneum genome (AP85–441) and the S. officinarum genome, and the result showed that sorghum candidate genes had homologous genes in both S. spontaneum and S. officinarum. Therefore, these consistent corresponding homologous genes indicated that the SNP array hybridization results were accurate and reliable to a certain extent.
Genes identified by GWAS did not explain the gene’s contribution to the phenotype, necessitating a combination of gene expression patterns42. In this study, the expression levels of the candidate genes were analyzed using publicly available leaf scald disease-related transcriptomes. Six overlapping genes were generated after combining the GWAS-identified and differentially expressed genes (DEGs) of RNA-seq. Furthermore, differentially expressed candidate genes in resistant and susceptible varieties provide ideal molecular markers for identifying loci associated with leaf scald resistance in breeding. Therefore, all six genes were verified for resistance response to leaf scald in resistant and susceptible genotypes using qRT-PCR experiments. After functional annotation and categorization of candidate genes, we found that many genes are directly or indirectly associated with plant disease resistance. In a previous study, the broad-spectrum resistance genes contained a nucleotide-binding site and leucine-rich repeat domains- similar to the ones identified in this study43. Activated NB-LRRs represent a tip of the signaling cascade that triggers defense responses and not the causal genes defining the resistance alone44,45. Pathogens often have different infection strategies, while plants regulate their defense responses based on the nature of the pathogen and its pathogenic mechanisms. The genes PCN identified in the present study might play a negative regulatory role in leaf scald resistance. Moreover, the GWAS results will be used to analyze further resistant genes’ functions and regulatory mechanisms against leaf scald.
In conclusion, this study addressed the critical challenge of identifying resistant genes against leaf scald in sugarcane. Through evaluating a diverse panel of 170 sugarcane genotypes, thirteen significant SNPs associated with the response to leaf scald were discovered, with eight demonstrating stability across various environments and models. Combined with the data from the GWAS, transcriptome, and qRT-PCR, six candidate genes involved in the response to Xanthomonas albilineans, including two genes encoding NB-ARC leucine-rich repeat (LRR)-containing domain disease-resistance protein. All candidate SNPs and genes will provide a solid basis for implementing marker-assisted selection strategies in sugarcane breeding programs, offering a promising strategy for effective disease control and variety improvement.
Materials and methods
Plant materials
A total of 170 genotypes (local and abroad) were collected and kept in the sugarcane breeding base of Guangxi University. These core germplasm, including breeding lines, landraces, and commercial hybrids, have been selected after years of continuous evaluation for various traits. These genotypes were originated from 10 origins, namely United States of America (9), Australia (1), Sichuan (1), Yunnan (3), Guangdong (6), Fujian (9), Guangxi (135), Hainan (1) and Taiwan (5). Given different parental origins, 170 genotypes were subdivided into three distinct subgroups. Group I consists of 66 genotypes of the diversity panel, Group II consists of 61 progeny genotypes from the cross of ROC22 × CT103, and Group III consists of 43 progeny genotypes from the cross of ROC25 × YZ89-7. The list of genotypes is provided in Supplementary Table 1.
Field evaluation
Field trials were performed in Fusui, China (107° 31′ E, 22° 17′ N) in a randomized complete block design with three replicates. Each plot consisted of a single-row zone with a length of 5 m and a row spacing of 2 m. Every plot was planted in 80 buds evenly over 5 m and separated by 1 m for every genotype. The natural occurrences of leaf scald were recorded for two consecutive years (2021–2022). Disease assessments were performed in April of each year, using the previously published method9, and recorded until it stabilized.
Greenhouse evaluation
Evaluation of the leaf scald response of sugarcane to Xanthomonas albilineans was performed under a controlled greenhouse environment of the College of Agriculture, Guangxi University. The greenhouse experiments were set up using completely randomized blocks with three replicates. Each plot was 2 m long, 0.8 m wide, and spaced 1 m apart, and 40 buds were planted in each plot. The artificial inoculation was performed (in 2021) to verify the resistance of each genotype using the potent virulent strain of JG4346. The bacterial suspension was calibrated to 0.3 at OD600 using sterile distilled water. The sugarcane stalks were chopped into single buds, soaked in the bacterial solution for 3 h, and planted in a greenhouse at 28 °C with a relative humidity of 80%. The disease level was investigated using the previously published method9, and diseased plants were counted continuously from when the sugarcane grew to the 5-leaf stage (approximately one month after planting) until the disease level stabilized.
Phenotypic statistical analysis
The severity of the leaf scald was evaluated as disease incidence and index. The following formulas calculated two disease indices: (1) Disease incidence (IC) = (ND/T) × 100; (2) Disease index (DI) = (1 × FL + 2 × ML + 3 × CB + 4 × N + 5 × D)/(5 × T) × 100), where ND = Number of diseased stalks, FL = the number of stalks with one or two streak lines, ML = the number of stalks with more than two streak lines, CB = the number of stalks with leaf chlorosis or bleaching, N = the number of stalks with leaf necrosis, D = the number of dead stalks or stalks with abnormal side shoots, and T = the total number of stalks. Variance analysis (ANOVA) was performed using the R software “multicomp” package (v4.1.2; R Core Team 2021). Field data were analyzed using a mixed model ANOVA, while greenhouse data were analyzed using one-way ANOVA. A mixed linear model was used to calculate the best linear unbiased predictors (BLUPs).
The model used for data analysis was as follows:
where Yijk is the observation of the kth genotype in the ith year in the jth replicate, μ is the overall mean, Gk is the effect of the kth genotype, Ei is the effect of the ith year, Rj(i) is the effect of the jth replication nested on the ith year, EGik is the effect of the interaction between the ith year and kth genotype, and εijk is the effect of experimental error.
The broad-sense heritability (HB2) was estimated according to the formula:
where σg genetic variance, σge genetic and year interaction variance, σe error variance, ne the number of years, and nr the number of replicates.
All statistical models were fitted using the software package “asreml” within the R computing environment47. The heritability of the greenhouse experiment was calculated using the formula mentioned above. The phenotype data of the greenhouse experiment were directly transformed by arcsine square root to satisfy the model assumption of a normal distribution and input for the subsequent GWAS analyses. Correlation analysis of the different datasets was analyzed using the corrplot package in R software48.
Genotyping and population structure analysis
The panel was genotyped with an Axiom Sugarcane 100K SNP array developed by Affymetrix using the Affymetrix GeneTitan® system. The physical positions of all the SNPs were based on sorghum genome v3.1.1. The Axiom Sugarcane100K SNP array obtained 100,097 SNPs. SNPs with a missing rate higher than 0.2 and a minor allele frequency (MAF) less than 0.05 were filtered out, resulting in 26,787 high-quality SNP markers for downstream analysis17. Based on the SNP markers obtained after filtering, neighbor-joining (NJ) tree construction was performed using MEGA-X software (model: Kimura 2-parameter; bootstrap: 1000) and edited with iTOL49. Principal Component Analysis (PCA) was performed using the R software and visualized and edited using the “ggplot2” R package. All genotypes were classified into three groups according to parental origin and labeled with different colors in the evolutionary tree and PCA graphs. Population structure analysis was performed by admixture software50, and the obtained results were visualized using the “ggplot” and “pophelper” R package51,52. The kinship matrix (K) was plotted using GAPIT's default parameters26.
Genome-wide association analyses
Association analysis was performed using five association models of GAPIT, including the General Linear Model (GLM), Mixed Linear Model (MLM), Multiple-Locus Mixed Linear Model (MLMM), Settlement of MLM Under Progressively Exclusive Relationship (SUPER), and the Bayesian-information and Linkage-disequilibrium Iteratively Nested Keyway (BLINK) models53,54,55,56. The final trait values for the GWAS were derived from eight sets of data (Fig. 1, Supplementary Table 3). The first five PCA and Kinship results were the Q-Matrix and K-Matrix, respectively, as the variables were co-analyzed with genotype and phenotype combination. The results of GWAS were visualized using the “CMplot” R package (https://github.com/YinLiLin/R-CMplot). For GWAS results, a threshold of − log10 p-value ≥ 5.73 with Bonferroni correction (0.05/number of SNP markers) for each trait was considered significant for the association markers. The phenotypic variation explained by each SNP marker was calculated from the GAPIT26.
Identification of candidate genes
Candidate genes were identified and described based on the general feature format (GFF) file from the sorghum genome v3.1.1, using TBtools software57. To assess the available candidate gene sequences from multiple species, we aligned the coding sequence (CDS) of each gene in the sorghum genome with that of its orthologous gene in the S. spontaneum reference genome (AP85–441) and S. officinarum (http://sugarcane.zhangjisenlab.cn/sgd/html/download.html). S. spontaneum and S. officinarum genes were screened with an e-value < 1e−06 and similarity ≥ 90% to the sorghum genes. The transcriptome data (PRJNA549590) was downloaded from the National Center for Biotechnology Information (NCBI) (https://www.ncbi.nlm.nih.gov/). The transcriptome consisted of leaf samples of resistant (LCP 85–384) and susceptible (ROC20) sugarcane cultivars infected by Xanthomonas albilineans collected at four-time points (0, 24, 48, and 72 h post-inoculation) with three biological replicates for transcriptome sequencing 58. All the qualified reads were aligned to the sorghum genome v3.1.1 as a reference genome using HISAT2 (https://ccb.jhu.edu/software/hisat2). Each leaf sample’s estimated transcript abundance (FPKM) was calculated using feature counts software with default parameters. Following the transcript abundance (FPKM), the calculated mean fold-change in expression values of differently treated samples of candidate genes were displayed in a heatmap using TBtools software 57. The candidate genes with p-value ≤ 0.05 and an absolute log2 fold-change ≥ 1.0 were considered to have statistically significant differential expressions.
Validation of candidate genes by qRT-PCR
Based on the results of both GWAS and RNA-seq studies, six potential genes show significant differential expressions at least at one point time post-inoculation in both susceptible cultivar (ROC20) and resistant cultivar (LCP 85–384) and were chosen for qRT-PCR investigation. The CDS sequences of the candidate genes were extracted using TBtools software. Species-specific primer sets were designed using Vector NTI software; six pairs of specific primers for selected candidate genes and one primer pair for the reference gene, i.e., glyceraldehyde 3-phosphate dehydrogenase (GAPDH)59, are listed in Supplementary Table 7. The reaction efficiency of each pair of primers was determined by gradient dilution of the template. A five-gradient dilution of cDNA was used as a template to create the standard curves. Reaction efficiency was calculated from the slope of the standard curve using the formula: reaction efficiency = [10 (− 1/slope)] − 1 × 100. The reaction efficiency for each pair of primers was between 95 and 113% (Supplementary Table 7). For qRT-PCR, the leaf samples were collected from two genotypes (ZZ1 and GT46) at 1-month post-inoculation. ZZ1 is a resistant genotype identified in several studies, while the GT46 has been reported to be susceptible to leaf scald7. Samples were quickly packed into foil and frozen in liquid nitrogen within 10 s of collection. Total RNA was extracted using the TRIzol reagent (ThermoFisher), following the manufacturer’s instructions. The qRT-PCR assay was performed on a Lightcycler480 (Roche) Real-Time PCR Detection System using the ChamQ SYBR qPCR Master Mix (Vazyme, Q311-02). Each reaction contained 10 µL of the Master Mix, 0.4 µL each of the forward and reverse primers, 0.4 µL of ROX Reference Dye II, 1.0 µL of the cDNA sample, and distilled water to the final volume of 20 µL. The qRT-PCR conditions consisted of pre-denaturation at 95 °C for 5 min, followed by 40 cycles of 95 °C for 15 s, 60 °C for 25 s, and 72 °C for 25 s. At the end of the PCR cycles, melting curve analysis was performed to validate the specificity of the PCR product. Three independent biological replicates, each with three technical replicates, were analyzed respectively. The relative gene expression levels were calculated using the 2−ΔΔCt method60. The Ct values of internal reference genes were used to standardize the Ct values of target genes for all samples. Multiple comparisons were performed using one-way analysis of variance (ANOVA) followed by Turkey multiple comparison test.
We confirm that the use of plants in the present study complies with international, national, and institutional guidelines.
Data availability
Phenotypic data from the current study are included in the Supplementary Files, and genotypic data are available from the corresponding author upon reasonable request.
References
Wang, J. et al. Mapping sugarcane plantation dynamics in Guangxi, China, by time series Sentinel-1, Sentinel-2, and Landsat images. Remote Sens. Environ. 247, 111951 (2020).
Sindhu, R., Gnansounou, E., Binod, P. & Pandey, A. Bioconversion of sugarcane crop residue for value-added products—An overview. Renew. Energy 98, 203–215 (2016).
Comstock, J. C. Sugarcane diseases: Futuristic management strategies. Sugar Technol. 15, 1–2 (2013).
Rott, P. & Davis, M. J. Leaf scald. In A Guide to Sugarcane Diseases (ed. Rott, P.) 38–44 (La Librairie du Cirad, 2000).
Lin, L.-H. et al. Molecular detection and prevalence of Xanthomonas albilineans, the causal agent of sugarcane leaf scald in China. Crop Prot. 109, 17–23 (2018).
Cervantes-Romero, B. et al. Distribution, phylogeny, and pathogenicity of Xanthomonas albilineans causing sugarcane leaf scald in Mexico. Crop Prot. 150, 105799 (2021).
Zhang, R. Y. et al. First report of sugarcane leaf scald caused by Xanthomonas albilineans in the Province of Guangxi, China. Plant Dis. 101, 1541 (2017).
Ntambo, M. S. et al. Identification and characterization of Xanthomonas albilineans causing sugarcane leaf scald in China using multilocus sequence analysis. Plant Pathol. 68, 269–277 (2019).
Rott, P. et al. Resistance to leaf scald disease is associated with limited colonization of sugarcane and wild relatives by Xanthomonas albilineans. Phytopathology 87, 1202–1213 (1997).
Carine, N. A. et al. Sugarcane leaf scald disease in Côte d’Ivoire: Pathogenicity and biocontrol of Xanthomonas albilineans isolates. J. Exp. Agric. Int. 41, 1–11 (2019).
Champoiseau, P., Rott, P. & Daugrois, J.-H. Epiphytic populations of Xanthomonas albilineans and subsequent sugarcane stalk infection are linked to rainfall in Guadeloupe. Plant Dis. 93, 339–346 (2009).
Bollinedi, H. et al. Genome-wide association study reveals novel marker-trait associations (MTAs) governing the localization of Fe and Zn in the rice grain. Front. Genet. 11, 213 (2020).
Sahu, P. K. et al. Advancement in the breeding, biotechnological and genomic tools towards development of durable genetic resistance against the rice blast disease. Plants 11, 2386 (2022).
Garsmeur, O. et al. A mosaic monoploid reference sequence for the highly complex genome of sugarcane. Nat. Commun. 9, 2638 (2018).
Zhang, J. et al. Allele-defined genome of the autopolyploid sugarcane Saccharum spontaneum L.. Nat. Genet. 50, 1565–1573 (2018).
Mamanova, L. et al. Target-enrichment strategies for next-generation sequencing. Nat. Methods 7, 111–118 (2010).
You, Q. et al. Development of an axiom sugarcane100k SNP array for genetic map construction and QTL identification. Theor. Appl. Genet. 132, 2829–2845 (2019).
Song, J. et al. Natural allelic variations in highly polyploidy Saccharum complex. Front. Plant Sci. 7, 804 (2016).
Yang, X. et al. Mining sequence variations in representative polyploid sugarcane germplasm accessions. BMC Genom. 18, 594 (2017).
Wang, J. et al. RMeseiacrcrhoarctioclellinearity between autopolyploid sugarcane and diploid sorghum genomes. BMC Genom. 11, 261 (2010).
Daugrois, J. H. et al. A putative major gene for rust resistance linked with an RFLP marker in sugarcane cultivar ‘R570’. Theor. Appl. Genet. 92, 1059–1064 (1996).
Gutierrez, A. F., Hoy, J. W., Kimbeng, C. A. & Baisakh, N. Identification of genomic regions controlling leaf scald resistance in sugarcane using a bi-parental mapping population and selective genotyping by sequencing. Front. Plant Sci. 9, 877 (2018).
Wang, Y.-X. et al. Genome-wide identification and expression analysis of GRAS family transcription factors in tea plant (Camellia sinensis). Sci. Rep. 8, 3949 (2018).
Ahn, E. et al. Genome-wide association analysis of sorghum mini core lines regarding anthracnose, downy mildew, and head smut. PLoS ONE 14, e0216671 (2019).
Bainsla, N. K. et al. Genome-wide association study for spot blotch resistance in Afghan wheat germplasm. Plant Pathol. 69, 1161–1171 (2020).
Wang, J. & Zhang, Z. GAPIT Version 3: Boosting power and accuracy for genomic association and prediction. Genom. Proteom. Bioinform. 19, 629–640 (2021).
Gao, L., Turner, M. K., Chao, S., Kolmer, J. & Anderson, J. A. Genome-wide association study of seedling and adult plant leaf rust resistance in elite spring wheat breeding lines. PLoS ONE 11, e0148671 (2016).
Edae, E. A., Pumphrey, M. O. & Rouse, M. N. A genome-wide association study of field and seedling response to individual stem rust pathogen races reveals combinations of race-specific genes in North American Spring wheat. Front. Plant Sci. 9, 52 (2018).
Liu, Z., An, C., Zhao, Y. & Xiao, Y. Genome-wide identification and characterization of the CsFHY3/FAR1 gene family and expression analysis under biotic and abiotic stresses in tea plants (Camellia sinensis). Plants 10, 570 (2021).
Dalton-Morgan, J. et al. A high-throughput SNP array in the amphidiploid species Brassica napus shows diversity in resistance genes. Funct. Integr. Genom. 14, 643–655 (2014).
Newman, D. L., Abney, M., McPeek, M. S., Ober, C. & Cox, N. J. The importance of genealogy in determining genetic associations with complex traits. Am. J. Hum. Genet. 69, 1146–1148 (2001).
Müller, B. U., Stich, B. & Piepho, H.-P. A general method for controlling the genome-wide type I error rate in linkage and association mapping experiments in plants. Heredity 106, 825–831 (2011).
Gouy, M. et al. Genome-wide association mapping of agro-morphological and disease resistance traits in sugarcane. Euphytica 202, 269–284 (2015).
Barreto, F. Z. et al. A genome-wide association study identified loci for yield component traits in sugarcane (Saccharum spp.). PLoS ONE 14, e0219843 (2019).
Rasheed, A. et al. Development and validation of KASP assays for genes underpinning key economic traits in bread wheat. Theor. Appl. Genet. 129, 1843–1860 (2016).
Yi, X. et al. Development of a kompetitive allele-specific PCR marker for selection of the mutated Wx-D1d allele in wheat breeding. Plant Breed. 136, 460–466 (2017).
Zhao, Y. et al. Detection of candidate genes and development of KASP markers for Verticillium wilt resistance by combining genome-wide association study, QTL-seq and transcriptome sequencing in cotton. Theor. Appl. Genet. 134, 1063–1081 (2021).
Zhang, Q. et al. Genomic insights into the recent chromosome reduction of autopolyploid sugarcane. Nat. Genet. 54, 885 (2022).
O’Connell, A. et al. Combining genomic selection with genome-wide association analysis identified a large-effect QTL and improved selection for red rot resistance in sugarcane. Front. Plant Sci. 13, 1021182 (2022).
Zhang, B. et al. Genome-wide association study unravels quantitative trait loci and genes associated with yield-related traits in sugarcane. J. Agric. Food Chem. 71, 16815–16826 (2023).
Yang, X. et al. Target enrichment sequencing of 307 germplasm accessions identified ancestry of ancient and modern hybrids and signatures of adaptation and selection in sugarcane (Saccharum spp.), a ‘sweet’ crop with ‘bitter’ genomes. Plant Biotechnol. J. 17, 488–498 (2019).
Emilsson, V. et al. Genetics of gene expression and its effect on disease. Nature 452, 423–428 (2008).
Xie, Z. et al. A nucleotide-binding site-leucine-rich repeat receptor pair confers broad-spectrum disease resistance through physical association in rice. Philos. Trans. R. Soc. B Biol. Sci. 374, 20180308 (2019).
Zhang, H. et al. A rice LRR receptor-like protein associates with its adaptor kinase OsSOBIR1 to mediate plant immunity against viral infection. Plant Biotechnol. J. 19, 2319–2332 (2021).
Rose, L. E. et al. The maintenance of extreme amino acid diversity at the disease resistance gene, RPP13, in Arabidopsis thaliana. Genetics 166, 1517–1527 (2004).
Li, M. et al. Comparative genome analysis unravels pathogenicity of Xanthomonas albilineans causing sugarcane leaf scald disease. BMC Genom. 23, 671 (2022).
Butler, D. G., Cullis, B. R., Gilmour, A. R. & Gogel, B. J. {ASReml}-R Reference Manual (2009).
Wei, T. et al. Package ‘corrplot’’. Statistician 56(316), e24 (2017).
Letunic, I. & Bork, P. Interactive tree of life (iTOL) v4: Recent updates and new developments. Nucleic Acids Res. 47, W256–W259 (2019).
Alexander, D. H., Novembre, J. & Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 19, 1655–1664 (2009).
Francis, R. M. Pophelper: An R package and web app to analyze and visualize population structure. Mol. Ecol. Resour. 17(1), 27–32 (2017).
Warnes G. R. et al. gplots: Various R Programming Tools for Plotting Data. https://CRAN.R-project.org/package=gplots (2020).
Zhang, Z. et al. Mixed linear model approach adapted for genome-wide association studies. Nat. Genet. 42, 355–360 (2010).
Liu, X., Huang, M., Fan, B., Buckler, E. S. & Zhang, Z. Iterative usage of fixed and random effect models for powerful and efficient genome-wide association studies. PLoS Genet. 12, e1005767 (2016).
Wang, Q., Tian, F., Pan, Y., Buckler, E. S. & Zhang, Z. A SUPER powerful method for genome-wide association study. PLoS ONE 9, e107684 (2014).
Huang, M., Liu, X., Zhou, Y., Summers, R. M. & Zhang, Z. BLINK: A package for the next level of genome-wide association studies with both individuals and markers in the millions. GigaScience 8, 154 (2019).
Chen, C. et al. TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 13, 1194–1202 (2020).
Ntambo, M. S. et al. Comparative transcriptome profiling of resistant and susceptible sugarcane cultivars in response to infection by Xanthomonas albilineans. Int. J. Mol. Sci. 20, 6138 (2019).
Ling, H., Wu, Q., Guo, J., Xu, L. & Que, Y. Comprehensive selection of reference genes for gene expression normalization in sugarcane by real time quantitative RT-PCR. PLoS ONE 9, e97469 (2014).
Livak, K. J. et al. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) method. Methods 25, 402–408 (2001).
Acknowledgements
The authors thank all laboratory members for their valuable discussion.
Funding
This research was supported by the China Agricultural Research System of MFA and MARA (CARS 170109) and the Guangxi Science and Technology Major Project (Guike AA22117001 and AD20207020).
Author information
Authors and Affiliations
Contributions
MZ and JW conceived and designed experiments and coordinated research activities; YL and QY performed the experiments; PL and QY analyzed the data; YL and MZ wrote the draft manuscript; MZ, JH, WY, and JW revised the manuscript. All author(s) read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, Y., Lin, P., You, Q. et al. Identification of candidate single-nucleotide polymorphisms (SNPs) and genes associated with sugarcane leaf scald disease. Sci Rep 14, 16214 (2024). https://doi.org/10.1038/s41598-024-67059-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-67059-w
- Springer Nature Limited