
Abstract
Marine microbial communities form the basis for the functioning of marine ecosystems and the conservation of biodiversity. With the application of metagenomics and metatranscriptomics in marine environmental studies, significant progress has been made in analysing the functioning of microbial communities as a whole. These molecular techniques are highly dependent on reliable, well-characterised, comprehensive and taxonomically diverse sequenced reference transcriptomes of microbial organisms. Here we present a set of 12 individual transcriptome assemblies derived from 6 representative diatom species from the northern Adriatic Sea grown under 2 environmentally relevant growth conditions (phosphate replete vs. phosphate deprived). After filtering the reads and assembly, an average number of 64,932 transcripts per assembly was obtained, of which an average of 8856 were assigned to functionally known proteins. Of all assigned transcripts, an average of 6483 proteins were taxonomically assigned to diatoms (Bacillariophyta). On average, a higher number of assigned proteins was detected in the transcriptome assemblies of diatoms grown under replete media condition. On average, 50% of the mapped proteins were shared between the two growth conditions. All recorded proteins in the dataset were classified into 24 COG categories, with approximately 25% belonging to the unknown function and the remaining 75% belonging to all other categories. The resulting diatom reference database for the northern Adriatic, focussing on the response to nutrient limitation as characteristic for the region and predicted for the future world oceans, provides a valuable resource for analysing environmental metatranscriptome and metagenome data. Each northern Adriatic transcriptome can also be used by itself as a reference database for the (meta)transcriptomes and gene expression studies of the associated species that will be generated in the future.
Similar content being viewed by others
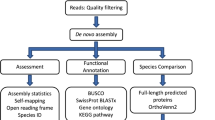


Introduction
Eukaryotic microbial communities are one of the key components for the functioning of marine ecosystems. Their combined metabolic activities drive a large range of oceanographic parameters, ecosystem functions and ecosystem services, they constitute the initial link in marine food webs, and their activity significantly contributes to biogeochemical nutrient cycling and oxygen production1,2,3,4. Members of these communities come from diverse eukaryotic lineages, present within each phylogenetic supergroup of eukaryotes2. Despite their ecological importance, much work remains to be done to understand the intricate ecological roles of these communities in marine environment. Diatoms (Bacillariophyta) represent an important group of marine eukaryotic microbial communities. In diverse marine environments diatoms are responsible for significant amounts of primary production and often, e.g. in the northern Adriatic, significantly contribute to the overall phytoplankton abundance as well5,6,7,8. Availability of nutrients and physiological response to nutrient limitation stand as the basic prerequisite for the survival, adaptation and success of diatoms in dynamic marine environments. Response of diatom species to nutrient stress have been extensively studied9,10,11,12,13,14,15,16,17,18,19,20 but still, diatom physiological responses to nutrient deficiency are poorly understood.
Application of high-throughput sequencing (omics) molecular techniques like metabarcoding, metagenomics and metatranscriptomics in marine environmental studies enabled significant progress in analysing the taxonomic, biological and functional diversity of microbial communities. The number of studies on the physiology and diversity of marine microbial communities using omics is increasing. Nevertheless, there is a lack of high-quality molecular resources needed for the complex analyses and interpretation of the generated (omics) data. These omics molecular techniques are highly dependent on the availability, coverage and curation of reference libraries or annotated sequence libraries as is the case for metatranscriptomics as well21,22. As far as the marine microbial community is concerned, eukaryotes in general and non–model organisms are particularly poorly represented among the available molecular omics resources21,23,24,25,26. Local, regional transcriptome datasets of these underrepresented microbial organisms form the core of successful omics data management, and are the base for any improvement in the resolution of environmental meta-omics analyses. Compared to other groups of marine microbial eukaryotes, many fully or partially sequenced and annotated genomes are available for diatoms27,28,29,30,31,32,33,34,35 or are being generated as part of ongoing sequencing projects (100 Diatom Genomes, https://jgi.doe.gov/csp-2021-100-diatom-genomes/). Nevertheless, available diatom genomes unsatisfactorily cover diatom diversity and genome sequencing still requires demanding efforts in its realization. Currently, reliable, well-characterised, comprehensive and taxonomically diverse sequenced reference transcriptomes stand as the most useful resource in metatranscriptomics analysis.
In 2014, Keeling et al. published a comprehensive database of the Marine Microbial Eukaryotic Transcriptome Sequencing Project (MMETSP) comprising 678 transcriptome assemblies from 405 species of marine microbial eukaryotes (including 71 diatom species) across various experimental conditions23. MMETSP database substantially expanded the available gene expression information of marine microbial eukaryotes and enhanced the understanding of the functional potential of diverse lineages of marine microbial eukaryotes, serving as the first molecular resource for some of these lineages23. In addition to the comprehensive MMETSP database, many individual studies of diatom transcriptomes have contributed significantly to the understanding of diatom physiology and related transcriptomes20,30,36,37,38,39,40,41,42,43,44. Specific regional transcriptome reference databases might allow to address regional peculiarities of transcripts and annotations and are expected to provide a higher functional and taxonomic resolution of the (regional) physiological patterns studied. At the same time, regional databases have global utility and significance for species-specific gene expression and biogeographic studies of those.
The northern Adriatic (NA) is a semi-enclosed, shallow coastal area of the Adriatic Sea and the northernmost part of the Mediterranean basin. The NA is exposed to a strong but fluctuating nutrient input, primarily from the Po River, one of the largest freshwater input in the entire Mediterranean45. The input from the Po River creates steep spatio-temporal ecological gradients in the northern Adriatic17,18,46,47 which together with the current circulation creates dynamic ecological conditions for the growth and succession of phytoplankton. In the NA, phytoplankton life strategies are mainly determined by light availability, temperature and the species' ability to cope with phosphorus limitation17,18,19,48,49,50,51. As the Po River is characterized by a much higher concentration of nitrogen, compared to phosphorus52 the species living and thriving in the northern Adriatic are used to unbalanced N/P ratio. Su et al. 2023., reported levels of alkaline phosphatase activity in the northern Adriatic to be the highest levels measured so far53. This is one of the central mechanisms to cope with phosphate limitation and one of the driving forces in the NA. The NA phytoplankton is largely dominated by diatoms, and species from the genera Skeletonema, Pseudo-nitzschia and Chaetoceros are known to be an important component of the NA phytoplankton community with some species occasionally forming very high abundances and toxic blooms8,48,54,55,56,57,58,59,60,61,62. The characterisation of the NA diatom community was mainly studied using classical light microscopy methods for species identification and physiological experiments8,19,48,61,63 while studies using high-throughput sequencing methods of molecular species identification and functional characterisation are still rare for the field64,65,66. Transcriptome and metatranscriptome studies of the diatom community are lacking for the NA.
Here, we present the first regional reference transcriptome database of diatom species isolated and cultivated from the northern Adriatic. A set of 12 individual transcriptome assemblies derived from 6 representative diatom species isolated from the northern Adriatic and grown under 2 experimental growth conditions (replete (F/2) vs. phosphorus deprived (P-limited)) was produced (Table 1). A detailed description of the creation, editing, curation and analysis of such a database is presented. To our knowledge, the reference transcriptome database created here is the first such valuable molecular ecology resource for the diatom community and, moreover, specific to the NA.
Materials and methods
Sampling monoclonal cultures establishment
Sampling for the isolation of monoclonal cultures was carried out at one coastal station (RV001) and two offshore stations (RV004, SJ101) in the northern Adriatic (Table 1, Fig. 1). At each station, vertical bottom—surface phytoplankton net (opening diameter 60 cm, length 2 m, mesh size 50 μm) hauls were performed. The diatom species were identified by light microscopy (Zeiss AxioObserver, Zeiss Oberkochen, Germany) and manually isolated from living net samples using Pasteur pipettes. Monoclonal cultures of the species were established by isolating single cells or single chain. Cells were grown in F/2 medium67, and incubated at 16 °C and 75 μmol photons m−2 s−1 in a 12:12 h light:dark photoperiod. The established monoclonal batch cultures were assigned to the phytoplankton cell culture collection of the Center for Marine Research (Table 1). Experimental in vitro incubations of the cultures were harvested for DNA extraction when the exponential growth phase was reached.

Sampling area. Stations RV001, RV004 and SJ101 where vertical net hauls for the isolation of the species were performed is marked.
Molecular species identification
Three barcodes were used for the molecular species identification: V4 region of the small subunit (18S) ribosomal RNA gene, D1–D3 region of the large subunit (28S) ribosomal RNA gene and 5’end region of the ribulose 1,5-bisphosphate carboxylase large subunit (rbcL) (Supplementary Table 1). 30 mL of each cell culture was filtered on 1.2 µm cellulose filters (Merck Milipore) and frozen at − 80 °C until further processing. Genomic DNA was isolated using the DNeasy Plant Mini Kit (Qiagen) according to the manufacturer's instructions. PCR amplification was performed using DreamTaq DNA polymerase (Thermofisher Scientific). The reaction mixture (25 μL) contained 10 µL H2O, 12.5 µL DreamTaq Master Mix (2X), 0.75 µL of each primer (10 uM) and 1 µL genomic DNA (c ≤ 5 ng/µL). PCR reactions were performed in the SimpliAmp Thermal Cycler (Applied Biosystems). PCR conditions were as follows: an initial denaturation step of 5 min at 95 °C, 33 cycles of 40 s at 95 °C, 40 s at 52 °C and 1 min at 72 °C, and a final extension step of 5 min at 72 °C. The PCR-amplified products were purified using the NucleoSpin Gel and PCR clean-up mini kit (Macherey–Nagel) according to the manufacturer's instructions. The purified PCR products were sequenced at Macrogen Europe (The Netherlands). The barcoding sequences of the cultivated diatom species were stored in GenBank under the Accession Numbers: PP838189-PP838194, PP839066-PP839071 and PP839974-PP839978. Geneious software68 was used to align the sequences (from both ends) and further phylogenetic analyses were performed. BLAST was used for searching and comparison with the NCBI GenBank database69,70 (Fig. 2).

Most common diatom Adriatic plankton species phylogenetic tree. Species used in this study are indicated with red letters.
In-vitro experimental incubations
Cultures for in-vitro experimental incubations were prepared as described in Smodlaka Tankovic et al.19. In brief, 2 mL of each established monoclonal batch culture (Table 1) were inoculated into 200 mL of the chosen growth medium (F/2 or P-limited). Nutrient-rich conditions (F/2) were simulated with medium F/267. Dissolved inorganic P limitation stress (P-limited) was simulated using a P-limited medium (F/2 medium without added sodium hydrogen phosphate). Both growth media were prepared using seawater from the northern Adriatic, which was stored in the dark for 2 months and sterilized by double filtration through 0.22 μm pore size cellulose filter (Merck Millipore) and boiling in a microwave oven71. Prepared F/2 and P-limited cultures were incubated in climate chamber (Memmert ICH110, Germany) with a light–dark cycle of 12:12 h in sterile 250 mL vented culture flasks (easy flasks, Nunclon, Denmark) at 16 °C and irradiance of 75 μmol photons m−2 s−1. Cultures were incubated until the abundance of at least 105 cells/L was reached for all cultures. At the end of incubation, 40–200 mL of each culture was filtered on 1.2 µm cellulose filters (Merck Millipore, Germany). Filtered culture volumes were chosen according to reached abundances, so that for the two growth conditions (F/2 and P-limited) of each culture the same number of cells was collected on the filter. Filters were stored at − 80 °C for the total RNA isolations.
Isolation of the total RNA and transcriptome sequencing
Total RNA was isolated using a PureLink Mini kit (Invitrogen) with on-column Pure link DNase (Invitrogen) treatment, according to manufacturer instructions and adapted to filter isolations. Filters were immersed in 2.5 mL of lysis buffer and cells were removed from filter by pipetting, vortexing, and 5 min RT incubation. Lysate was centrifuged on 12,000×g for 5 min. Supernatants were transferred in clean falcon tubes and further processed in steps of RNA column binding, washing and elution. Total RNA was stored at − 80 °C until sent for transcriptome sequencing. Total RNA quality check, library preparations and sequencing were performed by Macrogen Europe (The Netherlands). RNA quality check was performed using Agilent 2200 TapeStation System (Agilent Technologies, United States). Library preparation and sequencing was conducted using TruSeq Stranded mRNA LT Sample Prep Kit and NovaSeq 6000 PE Illumina technology.
Bioinformatics workflow
Paired-end reads (150 bp) were filtered for rRNA using SortMeRNA (v4.3.6) software72 with smr_v4.3_default_db.fasta as a reference and quality trimmed using Trimmomatic (v0.39)73 with settings for sliding window being 5:20 and minimum length of 50 bp. rRNA filtered and quality trimmed reads were assessed using FASTQC (v0.11.8) (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/) and MULTIQC (v1.13) software (https://github.com/ewels/MultiQC). Transcripts were de novo assembled from filtered reads using Trinity software (v2.15.0)74 and default settings. Protein sequences were predicted using TransDecoder (v5.7.0) software (https://github.com/TransDecoder/TransDecoder) for open reading frame (ORF) prediction with default settings. Transcript quantification was conducted using Salmon software (v1.9.0)75 with index constructed for predicted protein sequences of each sample separately. Protein sequences with zero counts were filtered out and excluded from further analysis. Functional annotation of predicted protein sequences was performed using eggNOG-mapper76 via BioBam Omix box software (OmicsBox—Bioinformatics Made Easy, BioBam Bioinformatics, March 3, 2019, https://www.biobam.com/omicsbox) for eggNOG77, KEGG78 and GO79 database annotations. Only sequences reaching both e-value and bitscore thresholds of 1 × 10–5 and 50, respectively, were considered successfully annotated.
eggNOG v 5.0 (Evolutionary genealogy of genes: Non-supervised orthologous groups), KEGG (Kyoto encyclopedia of genes and genomes) and GO (Gene ontology) are publicly available databases that classify genes into orthologues groups (OGs) and provide functional descriptions. GO classifies OGs into three broader functional categories; molecular function (MF), cellular component (CC) and biological process (BP)80. KEGG classifies OGs into broader functional categories in the context of KEGG molecular networks, namely, pathway maps, BRITE hierarchies and modules81 while eggNOG uses COG database broader functional categories system82.
Via eggNOG mapper algorithm for taxonomy prediction we assigned proteins their taxonomic origin. Only protein sequences corresponding to taxonomic level Bacillariophyta were used for the functional annotation analyses. Of all the Bacillariophyta OGs in the eggNOG database, 70% are functionally annotated, while the remaining 30% represent diatom OGs with unknown functions. Thus, functional annotation via eggNOG-mapper facilitates the distinction between diatom proteins with known functions and those with unknown functions, i.e., proteins identified in diatom genomes but whose functions are yet to be determined. To assign protein to a broader functional category a COG functional categories system was used82.
In order to compare our transcriptomes with the transcriptomes of the same species in the MMETSP database, we used files generated in Johnson et al.83. Seven Skeletonema marinoi transcriptomes (sample IDs: MMETSP0319, MMETSP0320, MMETSP0918, MMETSP0920, MMETSP1039, MMETSP1040, MMETSP1428) and four Chaetoceros curvisetus transcriptomes (sample IDs: MMETSP0716, MMETSP0717, MMETSP0718, MMETSP0719) from different growth conditions were found in MMETSP dataset. We downloaded predicted protein sequences (https://zenodo.org/records/257026), quantification files (https://zenodo.org/records/257145) and contig ID name maps (https://zenodo.org/records/3247846). Transcriptomes metadata was found in NCBI Biosample database for Bioproject accession number PRJNA231566 (Table 2). Similar to our dataset, MMETSP predicted protein sequences were generated using TransDecoder software and quantification files using Salmon software. Protein sequences were functionally annotated following the same bioinformatic workflow as for NA transcriptomes, using eggNOG-mapper. Proteins corresponded to transcripts with zero counts were filtered from the dataset. Transcriptomes under sample IDs MMETSP0716 and MMETSP0717 were excluded from analysis due to insufficient number of predicted proteins (6) and missing quantification file.
Results
Dataset structure
Transcriptome sequencing of six diatom species grown in two phosphate conditions (F/2 and P-limited) resulted in a dataset that consisted of on average 43,261,377 reads/sample (Table 3). After rRNA filtering, on average 31,334,595 mRNA reads/ sample remained (Supplementary Table 2). Additionally, approximately 2% of poor-quality mRNA reads were removed from each sample through Trimmomatic quality trimming (Supplementary Table 2). Overall, after pre-processing steps the dataset consisted of 365,178,904 reads (average 30,431,575 reads/sample) (Table 2). Trinity de novo assembly constructed 779,184 transcripts (on average 64,932 transcripts/sample) with an average length of 731 bp (Table 3). Obtained transcripts coded for 422,974 predicted protein sequences (on average 35,248 proteins/sample) (Supplementary Table 3). When proteins corresponding to transcripts with zero counts (erroneous or redundant transcripts) were filtered out from the dataset, the finalized dataset of the northern Adriatic diatom transcriptomes consisted of 106,274 protein sequences (average 8,856 protein sequences/sample) (Table 3).
Dataset analysis
To further analyse the final dataset, the protein sequences were functionally annotated using eggNOG-mapper software to obtain the eggNOG description, GO and/or KO terms for each protein sequence as well as taxonomic affiliation. Using eggNOG-mapper and after reaching the e-value and bitscore thresholds (see “Methods”), 97,271 protein or 91.5% of all protein sequences in the final dataset could be successfully annotated by using the searched databases (on average 8106 protein sequences/ sample). For 9003 or the remaining 0.5% of all protein sequences (750 protein sequences/sample) we haven’t found annotation passing the given e-value and bitscore tresholds (Fig. 3). After functional annotation with eggNOG- mapper, successfully annotated protein sequences could be divided in two categories, those that received at least one functional annotation and those corresponding to proteins with an unknown function. In total, 75,682 protein sequences (on average 6307 proteins/sample) received at least one functional annotation and 21,589 protein sequences (on average 1799 protein sequences/sample) are corresponding to proteins of unknown function (Fig. 3). Most protein sequences of the final dataset were assigned an eggNOG description (77.3%). Approximately half of proteins (51.5%), were not only assigned an eggNOG description but also associated with a KO or GO term or both. Notably, many protein sequences annotated with KO or GO received more than one term, while all eggNOG annotated protein sequences received only one eggNOG description. 34% of KO annotated protein sequences received more than one KO term, while 96.4% of all GO annotated protein sequences received more than one GO term. Overall, a total of 75,188 protein sequences were assigned an eggNOG description, 47,987 were assigned a KO term, and 30,081 were assigned a GO term (Supplementary Table 3).

Representation of functionally annotated (blue), function unknown (orange) and protein sequences with no annotations. Piechart represents percentages of protein sequences corresponding to each annotation level (group) in the final dataset. Barchart represents total number or protein sequences in each sample of the final dataset separately.
The highest number of annotated protein sequences (referred to as proteins in the following text) was found in the CIM1063 (Thalassiosira sp.) culture grown under P-limited conditions, while the CIM827 (C. protuberans) culture grown in F/2 had the lowest number of annotated proteins. For most cultures, a higher number of annotated proteins was associated with F/2 growth conditions. Only CIM827 (C. protuberans) and CIM1063 (Thalassiosira sp.) had more annotated proteins in P-limited condition (Fig. 3). The highest number of proteins received functional annotation based on eggNOG description (on average 6266 proteins/sample), followed by KO annotations (on average 3999 proteins/sample) and GO annotations (on average 2507 proteins/sample) (Supplementary Table 3).
Since NA diatom cultures used for experimental incubations and transcriptome sequencing were not axenic cultures, a part (on average 20% proteins/sample) of annotated protein sequences was assigned to taxonomic groups other than diatoms (Bacillariophyta). The number of non-diatom annotated proteins didn’t notably differ between the cultures or the two growth conditions. The majority of non-diatom annotated proteins in the final dataset were assigned to Eukaryota (15,764 proteins) and Bacteria (3666). The Eukaryota group included transcriptome sequences of eukaryotic taxa (e.g. heterotrophic protists) that do not belong to the Bacillariophyta, as well as sequences that were classified as Eukaryota at the lowest taxonomic level. In total, around 80% of all annotated protein sequences corresponded to diatoms (Bacillariophyta) and the remaining 20% to Eukaryota (16%), Bacteria (around 4%) and Archaea (less than 1%) (Fig. 4). The only exception was sample CIM950 P-limited (C. curvisetus) that had more proteins assigned to Bacteria than to Eukaryota, 14% and 9%, respectively. Protein sequences annotated as Bacillariophyta will be referred to as the diatom dataset in subsequent text.

Proportion of annotated proteins assigned to different taxonomic groups/levels (Bacillariophyta, Eukaryota, Bacteria, Archea and Viruses).
It total, the diatom dataset contains 77,796 sequences (on average 6483 proteins/sample). In this dataset 58,714 proteins (on average 4893 proteins/sample) received at least one functional annotation and 19,082 proteins (on average 1590 proteins/sample) are corresponding to diatom proteins of unknown function. Most protein sequences of the diatom dataset were assigned an eggNOG description (around 75%) (Supplementary Table 4). The majority of these proteins (approximately 68%), were not only assigned an eggNOG description but also associated with a KO or GO term or both. Also, most sequences were assigned more than one KO and GO term. 34.9% of KO annotated sequences received more than one KO term, while 96.7% of all GO annotated sequences received more than one GO term. Overall, a total of 58,295 protein sequences in the diatom dataset were assigned an eggNOG description, 38,453 were assigned a KO term, and 25,470 were assigned a GO term (Supplementary Table 4).
Diatom dataset analysis (Functional analysis)
To further analyse the final dataset according to functional annotation, only diatom assigned protein sequences were used (the diatom dataset) (Fig. 5). Unique annotations were obtained when protein sequences of the diatom dataset were merged according to identical eggNOG description/KO term/GO term. All that resulted in 21,458 proteins with unique annotationbased on eggNOG description, 21,926 based on KO term and 16,524 based on GO term (Supplementary Table 4). The highest number of unique annotations was found in CIM1063 (Thalassiosira sp.) culture grown under P-limited conditions, while the CIM827 (C. protuberans) culture grown in F/2 had the lowest number of unigenes (Supplementary Table 4). For most cultures, the higher number of unique annotations were associated with F/2 growth condition (Supplementary Table 4). Only CIM827 (C. protuberans) and CIM1063 (Thalassiosira sp.) had more unique annotations in the P-limited condition (Supplementary Table 4). For most cultures, merging the identical KO term gave the highest number of unique annotations (on average 1827 unique annotations/sample) (Supplementary Table 4). Following was the unique annotations obtained by merging the identical eggNOG description (1788 unique annotations/sample) (Supplementary Table 4). Only cultures CIM827 (C. protuberans) and CIM1063 (Thalassiosira sp.) grown under F/2 conditions and cultures CIM950 (C. curvisetus) and CIM1008 (P. mannii) grown under P-limited conditions had more unique annotations based on eggNOG description than on KO term (Supplementary Table 4).

Representation of annotated protein sequences in each culture and growth condition after functional annotation of the NA transcriptome dataset: total number of protein sequences (green), successfully annotated sequences (passing e-value and bitscore threshold)—final dataset (yellow) and successfully annotated protein sequences assigned to best taxonomic level Bacillariophyta—diatom dataset (blue).
To further analyse the diatom dataset, we used unique annotations obtained by merging the unique eggNOG annotations. On average, 50% of unique annotations were shared between cultures grown under different conditions (F/2 and P-limited) while 27.1% and 22.7% were specific to F/2 and P-limited, respectively (Fig. 6). Cultures CIM827 (C. protuberans) and CIM1063 (Thalassiosira sp.) were characterised by a notably higher number of unique annotations (1496 and 1207 respectively) found only in P-limited cultures. In contrast, the number of unique annotations specific for F/2 was notably higher (1780) in culture CIM950 (C. curvisetus) than in the other cultures (Fig. 6). For culture CIM843 (S. marinoi), the highest number of shared unique annotations (2270) was found between the two growth conditions (Fig. 6). Overall, 222 unique annotations were shared among all diatom species under F/2 conditions, and 255 unique annotations were shared under P-limited conditions, respectively.

Comparison of the eggNOG annotated protein structure in diatom dataset between different cultures and growth conditions.
COG categories82 were utilized to assign proteins to broader functional categories. Diatom proteins annotated with eggNOG description were classified into 24 out of 26 possible COG categories (Fig. 7). Across most cultures, the "function unknown" category consistently had the highest number of proteins, followed by the "posttranslational modification, protein turnover, chaperones" category and the "signal transduction mechanisms" category. Notably, only culture CIM950 P-limited (C. curvisetus) exhibited the highest protein count associated with the "Translation, ribosomal structure and biogenesis" category.

Representation of COG categories distrubution in diatom dataset across all cultures. x-axis—number of all detected proteins that passed e-value and bitscore treshold and were assigned to Best taxonomic level Bacillariophyta. y-axis—COG categories foun in diatom dataset. (Out of 26 COG categories 24 were found, missing functions: General function prediction only and Mobilome: prophages, transposons).
MMETSP database comparison
To compare the results with diatom transcriptome datasets available in the MMETSP database, analysis of the MMETSP diatom transcriptome datasets was conducted according to the pipeline for transcriptome analysis applied for the NA transcriptomes (described in Methods).
De novo assembly constructed on average 31,878 transcripts/sample for S. marinoi MMETSP dataset and on average 8816 transcripts/sample for C. curvisetus MMETSP dataset with average transcript length of 815 and 567 bp, respectively (Table 4). In comparison, S. marinoi NA transcriptomes had on average 36,630 transcripts/sample with an average length of 860 bp while C. curvisetus NA transcriptomes had on average 67,421 transcripts/sample with an average length of 624 bp (Table 4). Based on the number of assembled transcripts, S. marinoi MMETSP and NA transcriptomes were not very different. The average number of assembled transcripts for MMETSP S. marinoi transcriptomes was 31,878 transcripts/sample and NA S. marinoi transcriptomes had 50,374 (CIM843 F/2) and 22,886 (CIM843 P-limited) transcripts assembled (Table 4). The length of assembled transcripts varied from 606 (MMETSP0920) to 1011 (CIM843 F/2) with the average length being 825 bp (Table 4). The number of assembled transcripts in C. curvisetus transcriptomes however, did differ. The average number of assembled transcripts of C. curvisetus MMETSP transcriptomes was 8816 and for C. curvisetus NA transcriptomes, 67,421 (Table 4). The length of the transcripts varied from 350 (CIM950 P-limited) to 652 (MMETSP0718) (Table 4). The number of predicted protein sequences also varied in all cultures. Based on predicted protein sequence numbers, there is no notable difference between the NA and MMETSP transcriptomes. For obtained transcripts an average of 14,539 protein sequences/sample was predicted in S. marinoi MMETSP dataset and an average of 4160 protein sequences/sample in C. curvisetus MMETSP dataset. In comparison, the NA dataset had on average 13,444 predicted protein sequences/sample for S. marinoi transcriptomes and 5335 predicted protein sequences/sample for C. curvisetus transcriptomes (Table 4). These numbers applied to number of protein sequences after the elimination of those corresponding to transcripts with zero counts. S. marinoi transcriptome with the highest amount of predicted protein sequences (20,681) was MMETSP0918 generated in F/2 condition (Table 4). NA S. marinoi transcriptome generated in F/2 condition (CIM843 F/2) had 16,193 predicted protein sequences (Table 4). The C. curvisetus transcriptome with the highest number of predicted proteins was CIM950 F/2 with 9715 predicted protein sequences followed by MMETSP0718 (ASW -NO3) with 4934 predicted protein sequences (Table 4). Overall, the transcriptome from S. marinoi generated in F/2 -Si condition had the lowest number of predicted protein sequences, only 444. Despite this number being notably different from other S. marinoi transcriptomes, it appears that the average number of predicted protein sequences varies more between the two species than within different conditions for the same species. Specifically, the difference is notable in the number of predicted proteins being one order of magnitude greater for S. marinoi compared to C. curvisetus.
After functional annotations with eggNOG mapper, the percentage of protein sequences with annotations passing e-value and bitscore thresholds (see “Methods”) also didn’t differ greatly when comparing MMETSP and NA transcriptomes. For almost all samples it exceeded 90%. The percentage of protein sequences that received at least one functional annotation (eggNOG description, KO, GO) was 75% on average for MMESTP and NA transcriptomes of S. marinoi. The remaining 25% corresponded to proteins of unknown function (Table 5). For C. curvisetus MMETSP and NA transcriptomes the average percentage of protein sequences that received at least one functional annotation was 89% and 85%, respectively (Table 5). The remaining percentage of proteins with unknown function was smaller than in S. marinoi, 11% and 15% respectively (Table 5). Similar to NA transcriptomes, the highest number of annotations per sample was based on eggNOG description following by KO and GO term (Table 5). In both MMETSP and NA transcriptomes 99% of proteins with at least one functional annotation received an eggNOG description, 59–80% received a KO term and 21–55% received a GO term (Table 5).
We applied eggNOG best taxonomic level prediction algorithm to assign best taxonomic levels to predicted protein sequences. S. marinoi transcriptomes from both MMETSP and NA datasets had 83–85% of proteins assigned to Bacillariophyta (diatoms), 12–14% assigned to Eukaryota, 1–3% assigned to Bacteria and less than 1% assigned to Archaea and Viruses (Fig. 8). Best taxonomic levels of C. curvisetus transcriptomes for both datasets had slightly less amount of diatom proteins, 76–78% for samples MMETSP0718, CIM950 F/2 and CIM950 P-limited. These results are quite consistent with all NA transcriptomes with diatom proteins ranging from 77 to 84% per sample. Transcriptome MMETSP0719 had the lowest amount of diatom proteins, only 22%. The highest amount of proteins in that sample corresponded to best taxonomic level Archaea. This sample was treated with nocodazone, a known microtubule inhibitor that might have a negative effect on diatom growth and metabolic activity.

Comparison of proportions of annotated proteins assigned to different taxonomic groups/levels (Bacillariophyta, Eukaryota, Bacteria, Archea and Viruses) in S. marinoi and C. curvisetus MMETSP and NA transcriptomes.
To further analyse S. marinoi and C. curvisetus MMETSP and NA datasets according to functional annotation, only diatom assigned protein sequences were utilized. Unique annotations were obtained when protein sequences of the diatom dataset were merged according to identical eggNOG description/KO term/GO term. The highest amount of unique annotations for most cultures was reached by merging the unique KO term, followed by eggNOG and GO unique annotations (Supplementary Table 5). The highest number of unique annotations was found in S. marinoi transcriptome MMETSP1428, 2731 eggNOG, 2916 KO and 2021 GO unique annotations (Supplementary Table 5). The transcriptome with the lowest amount of unique annotations was C. curvisetus MMETSP0719 with the lowest number of diatoms assigned proteins (nocodazone treatment) (Supplementary Table 5). The average number of unique annotations per sample for S. marinoi MMETSP transcriptomes was 2131 eggNOG, 2220 KO and 1513 GO unique annotations (Supplementary Table 5). The average number of unique annotations per sample for S. marinoi NA transcriptomes was 2523 eggNOG, 2649 KO and 2003 GO unique annotations (Supplementary Table 5). C. curvisetus transcriptomes CIM950 P-limited and MMETSP0718 had numbers of unique annotations comparable with those of S. marinoi while CIM950 P-limited and MMETSP0719 had notably lower number of unique annotations (Supplementary Table 5).
Diatom proteins that received an eggNOG description were classified into 24 out of 26 possible COG categories. The majority of diatom proteins were assigned an "Unknown function" after COG categorization. Subsequently, in most cultures, the categories with the highest representation after "Unknown function" were "Posttranslational modification, protein turnover, chaperones" and "Signal transduction mechanisms” (Fig. 7). The COG functional categories did not reveal any patterns of expression considering condition or species.
Discussion
The raw data obtained in this study consisted of 519,136,526 strand-specific pair-end Illumina reads for 12 individual RNA samples generated from northern Adriatic diatom cultures grown in two conditions (Table 3). The condition of P-limited media was indicative of the typical environmental parameters of the northern Adriatic since the life strategies of phytoplankton in the northern Adriatic are significantly influenced by the limitation of phosphorus17,18,49. The applied bioinformatic pipeline resulted in a finalised dataset of the northern Adriatic diatom transcriptomes consisting of 106,274 protein sequences with an average of 8856 protein sequences per sample (Table 3). To inspect the dataset, eggNOG-mapper was applied to assign functional and taxonomic annotation to protein sequences. In total, 97,271 proteins or 91.5% of the proteins in final dataset were successfully annotated (Fig. 3). Distinguishing proteins with annotations revealed two categories: those receiving at least one functional annotation (eggNOG description, KO, or GO term) and those with unknown function but previously identified in sequenced genomes. In total, around 78% of all annotated proteins fall into the first category, while the remaining 22% fall into the second category (Fig. 3). Out of all annotated protein sequences, 77% were assigned an eggNOG description, while KO and GO term were assigned to 49% and 31% of annotated protein sequences, respectively (Supplementary Table 3). This distribution aligns with expectations, considering that the eggNOG database (v.5) is the most comprehensive, with 4.4 million orthologous groups (OGs) distributed across 379 taxonomic levels77. In characterising the NA reference database, the aim was to highlight all three annotation methods, since the use of new omics data is still quite rare and represents a new research approach. Therefore, detailed annotation at this stage would allow for more constructive and easier improvements and/or comparisons of omics research in the future. Working with a single unique annotation per protein may seem more straightforward when studying general physiological responses. However, incorporating multiple annotations may offer better resolution for functional annotation when investigating specific proteins. For instance, some proteins can perform multiple functions in different cell compartments and be part of more than one metabolic pathway. Nevertheless, the choice of a specific database should not solely depend on the number of annotations but also on the biological question, considering that all mentioned databases categorize proteins differently (refer to “Methods”; “Bioinformatic workflow”).
In addition to functional annotation, the pipeline which incorporated the eggNOG- mapper feature to assign the best taxonomic levels to searched protein queries, identified the taxonomic origin of transcripts and proteins. The results of the taxonomic assignment were consistent among all samples. 77–84% of functionally annotated proteins (final dataset) were assigned a best taxonomic level Bacillaryophyta (diatom dataset) (Fig. 8). On average 16% of functionally annotated proteins were assigned only to Eukaryota, 4% to Bacteria and less than 1% to Bacteria and viruses (Fig. 4). These results indicated the need for taxonomic filtering of transcriptomes, especially if transcriptomes are intended as a reference for taxonomic annotation of, for example, metatranscriptome samples. It is also important to highlight that proteins assigned to best taxonomic level Eukaryota could “hide” some diatom genes/proteins not yet annotated for diatoms, but being highly conserved among eukaryotes they were identified by eggNOG on a higher taxonomic level.
The efficacy of functional annotation of protein sequences assigned to best taxonomic level Bacillariophyta remained consistent with the final dataset. Within the diatom dataset, 75% of all proteins received at least one functional annotation, while 25% corresponded to diatom proteins of unknown function but was identified in sequenced diatom genomes. In the diatom dataset, as in the final dataset (all taxonomic levels), the majority of protein sequences received an eggNOG description (75%), followed by KO (49%) and GO terms (33%) (Supplementary Table 4). The functional annotation of diatom transcriptomes reflects the abundance of known and characterized diatom transcripts, proteins, and genes in the reference databases used for annotation. The segment of the dataset corresponding to proteins of unknown function but present in diatom genomes is yet to be experimentally characterized. Nevertheless, even without the provided annotation, these proteins have the potential to serve as a "transcriptome fingerprint" for the specific diatom culture and growth condition in future (meta)transcriptome studies. Progress in diatom transcript annotation will inevitably further improve the coverage of annotation in the NA dataset.
The diatom genera (Pseudo-nitzchia, Skeletonema and Chaetoceros) included in our regional transcriptome database (isolated from the northern Adriatic Sea) have been studied in previous environmental19,48,55,84,85 and in vitro experimental studies19 and their ecological importance and ability to cope with different environmental conditions has been demonstrated. Confronted with unfavourable environmental conditions phytoplankton reacts with physiological acclimation and even genetic adaptation63. Among other adaptation mechanisms they often remodel lipids. Some examples of this adaptation on physiological level are reported by Martin et al., where it was found that Thalassiosira pseudonana under P-limited condition replaces phospholipids phosphatidylcholine (PC) by the nitrogen-containing betaine lipid diacylglyceryl-carboxyhydroxymethyl-choline (DGCC) and phosphatidylglycerol (PG) by sulfolipid sulfoquinovosyldiacylglycerol (SQDG)86. While Abida et al., found Phaeodactilum tricornutum to adapt to adverse nutrient starvation conditions by replacing phospholipids with non-phospholipids87. These changes in environmental conditions also significantly affect protein biosynthesis and the level of phosphorylated metabolites that can affect a number of metabolic functions, including growth and the ability to photosynthesize. A high proportion of phospholipids and relatively constant composition of lipids in Leptocylindrus species cultured in nutrient replete and phosphate depleted conditions, suggest their evolutionary adaptation to phosphate scarcity63. They apparently developed mechanisms by which they take organic phosphate from the environment, as confirmed by alkaline phosphatases (AP) activity measurements. The molecular mechanisms underlying aforementioned adaptations and physiological responses still remain unclear. Understanding those mechanisms in vitro and in situ heavily relies on the availability of reference dataset like the ones described here and on the in-depth analysis of the condition-specific datasets. In addition, species-specific transcriptomes in the reference database improve the annotation of species targeted transcripts in metatranscriptomes, similar to how an additional growth condition for the species would enable the recognition of condition-specific transcripts. At the same time, the regional character of the reference database could allow to consider regional specificities of transcripts and enrich species-specific physiological patterns, gene expression and biogeographical studies.
The MMTSP database encompasses transcriptomes from 194 isolates representing 71 diatom species, originating from diverse locations across the world's oceans and cultivated under various conditions (Bioproject accession number PRJNA231566). Within the genus Chaetoceros, transcriptomes were available for eight distinct species, while for the genera Pseudo-nitzchia, Skeletonema, and Thalassiosira, transcriptomes for six species were available. Our NA dataset introduced three novel species: Chaetoceros protuberans, Chaetoceros danicus, and Pseudo-nitzchia mannii. Notably, for Chaetoceros curvisetus and Skeletonema marinoi, our dataset included transcriptomes specifically generated under phosphate deprived condition (P-limited), significantly expanding the available information (Tables 1 and 2). Furthermore, our dataset marked a significant milestone as the first transcriptome dataset of diatom species isolated from the Adriatic Sea and generated under environmentally relevant conditions for the area, as shown before. While the MMETSP dataset contained seven transcriptomes for Skeletonema marinoi isolates, two of them being from Adriatic Sea isolates (MMETSP1039 and MMETSP1040), the here presented NA dataset uniquely included transcriptomes under P-limited conditions for this species (CIM843) (Tables 1 and 2). With P-limited transcriptomes present, NA reference dataset significantly contributes to nutrient related diatom studies, since transcriptomes simulating phosphate deprived conditions in the MMETSP are scarce for species within the genera Chaetoceros, Skeletonema, Pseudo-nitzchia, and Thalassiosira. Only two species of the genus Thalassiosira (T. rotula MMETSP0912 and T. gravida MMETSP0494) grew under conditions (only 0.4 uM phosphate in F/2 medium) similar to the P-limited medium of NA cultures.
As highlighted in the MMETSP metadata available in the ENA archive (https://www.ebi.ac.uk/ena/browser/view/PRJNA231566), numerous researchers aimed to supply axenic and uni-algal total RNA extracts for sequencing, although this goal was not consistently achieved. This was also demonstrated by the number of non-diatom sequences retrieved in our reanalysis. Similarly, our extracts appeared to contain non-diatom DNA, prompting us to utilize the eggNOG-mapper software for functional annotation and taxonomic origin prediction. Addition of taxonomic origin prediction of genes (proteins) enabled us to differentiate the taxonomic origins of genes within our dataset. As the MMETSP transcriptomes, similar to the NA transcriptomes, did contain sequences not assigned to diatoms, we utilized the eggnog-mapper feature to determine the best taxonomic levels for proteins. The results of taxonomic assignment proved to be consistent in both MMETSP and NA datasets, with the percentage of proteins assigned to the best taxonomic level Bacillariophyta ranging from 76 to 85% in MMETSP and 77–85% in NA transcriptomes (Fig. 8). Taxonomic filtering of reference transcriptomes appears to be highly advisable, especially for samples that are not rendered axenic, particularly when treatments are expected to adversely impact diatom growth and/or transcriptional activity. When MMETSP and NA transcriptomes of each diatom species were compared, number of assembled transcripts, predicted proteins and functionally annotated proteins were similar. It appeared that the average number of predicted protein sequences varied more between the species than within different conditions for the same species. The number of predicted proteins was one order of magnitude greater for S. marinoi compared to C. curvisetus. Still, the transcriptomes S. marinoi MMETSP0920 and C. curvisetus CIM950 P-limited had notably less predicted protein sequences than all other transcriptomes. Although surprising, since these two samples had high number of assembled transcripts, the smallest average transcript lengths (606 and 350, respectively) of the two samples might explain the lower number of predicted protein sequences (Table 4). Evidence about the parameters such as the number of assembled transcripts and transcript length, and how these parameters affect the number of predicted proteins, all describe database quality. Described quality supports the usage of NA transcriptomes as a reference. We would expect that the higher the total number of transcripts and the longer the transcript, the more successful the protein prediction would be. However, the available reference studies that characterise the good quality of a transcriptome reference database are still missing and to perform a valid statistical analysis and test different parameters effect on database quality requires a more extensive transcriptome dataset for each species and growth condition than is available within NA and MMETSP. The differences between taxa in transcript lengths and total number of transcripts may be due to differences in sample quality that occur during the methodological steps of RNA isolation, library preparation and sequencing. To a certain extent, these differences are to be expected as different taxa have different morphological and physiological characteristics that could influence these methodological steps. Growth conditions could also affect expression, which is characterised by the quality and quantity of transcripts. A comprehensive benchmarking study would be needed to include and evaluate all these parameters and methodological steps for the reference database quality assessment.
Annotated proteins were for both reference datasets (MMETSP and NA) divided in two categories: those receiving at least one functional annotation (eggNOG description, KO, or GO term) and those with unknown function but previously identified in sequenced genomes. For all S. marinoi transcriptomes, the average percentage of proteins with at least one functional annotation was 75% (Table 5). In contrast, C. curvisetus transcriptomes exhibited a higher percentage, surpassing 80% in all samples (Table 5). This difference may indicate a limitation in the searched databases for functional annotation, particularly concerning the number of species with sequenced genomes. The predominant source of annotations across all samples was eggNOG description (99%), followed by KO (59–80%) and GO term (21–55%). This distribution aligns with expectations, considering that the eggNOG database (v.5) is the most comprehensive one77,80,81 (Table 5). The functional analysis of diatom datasets demonstrated consistency in both MMETSP and NA C. curvisetus and S. marinoi transcriptomes. Across all samples, the highest number of acquired unique annotations was observed for KO, followed by eggNOG and GO unique annotations. In addition, the distribution of proteins in COG categories in MMETSP C. curvisetus and S. marinoi transcriptomes does not exhibit notable differences from the NA dataset as well. Eventhough, MMETSP database is not the gold standard for the inclusion of new datasets in a reference database, MMETSP is nevertheless an impressively comprehensive database for microbial community research and is currently the only database of its kind available. Therefore, the comparison of the NA database with MMETSP to characterise the quality of the NA database and to point out common and/or unique features of both databases, serves as the landmark for the NA quality and supports the consistent usage of the two reference databases in the future.
Conclusion
The regional transcriptome database generated in this study consists of 12 individual transcriptome assemblies of diatom species isolated from the northern Adriatic and grown under two conditions representative of contrasting in situ conditions with high and low phosphate availability. Those conditions in situ, generate the highest alkaline phosphatase activities measurable in marine environments53. NA reference transcriptomes might be used to study current environmental conditions (P-limited), particularly in the northern Adriatic, but also in global ocean environments. As for the current/present conditions, reference database provides valuable data for interpretation and understanding of future/extreme scenarios88,89 as well88,89. The NA reference database raw data obtained is publicly available in the ENA archive under accession number PRJEB74140 together with the metadata required for further data analyses. The applied bioinformatic pipeline that encompasses eggNOG-mapper feature to assign functional annotations from several different databases resulted in a high rate of successfully annotated protein sequences, in total 97,271 or 91.5%. Distinguishing proteins with annotations revealed two categories: those receiving at least one functional annotation (eggNOG description, KO, or GO term) and those with unknown function but previously identified in sequenced genomes. Annotated protein sequences may serve to further enhance our understanding of diatom physiology while the segment of the dataset corresponding to proteins of unknown function remains to be experimentally characterized. These uncategorised proteins still have the potential to serve as a "transcriptome fingerprint" for the specific diatom culture, species and growth condition in future (meta)transcriptome studies. The eggNOG-mapper feature to assign best taxonomic levels to searched protein queries, shed light on the taxonomic origin of proteins. 77–84% of functionally annotated proteins were assigned a best taxonomic level Bacillariophyta. On average 16% of functionally annotated proteins were assigned to Eukaryota, 4% to Bacteria and less than 1% to Archaea and Viruses. These results proved to be consistent when compared with the MMETSP C. curvisetus and S. marinoi datasets, highlighting the need for taxonomic filtering of transcriptomes, especially if transcriptomes are intended as a reference for taxonomic annotation of, for example, metatranscriptome samples. Nevertheless, proteins assigned to best taxonomic level Eukaryota might still “hide” some highly conserved diatom genes/proteins and therefore should not be overlooked if transcriptomes are intended for diatom physiology studies. These sequences might be particularly novel diatom sequences that our study is adding to reference databases. Comprehensive diatom transcriptome assemblies included in the analysed NA dataset have the potential to significantly expand the knowledge accumulated for these genera/species and encourage/enable further research using omics methods. All the diatoms included in our study represent species with global distribution (www.gbif.org). We hence also expect the dataset to be instrumental in future research in the region but also in future research that aims at comparing metabolic capacities of those species with a spatial component as well as research around the world that involves the aforementioned species.
Data availability
The data for this study have been deposited in the European Nucleotide Archive (ENA) at EMBL-EBI under accession number PRJEB74140.
References
Falkowski, P. G., Fenchel, T. & Delong, E. F. The microbial engines that drive earth’s biogeochemical cycles. Science 320, 1034–1039. https://doi.org/10.1126/science.1153213 (2008).
Caron, D. A., Countway, P. D., Jones, A. C., Kim, D. Y. & Schnetzer, A. Marine protistan diversity. Ann. Rev. Mar. Sci. 4, 467–493 (2012).
De Vargas, C. et al. Eukaryotic plankton diversity in the sunlit ocean. Science 348, 1261605 (2015).
Carradec, Q. et al. A global ocean atlas of eukaryotic genes. Nat. Commun. 9, 373 (2018).
Falkowski, P. G. The role of phytoplankton photosynthesis in global biogeochemical cycles. Photosynth. Res. 39, 235–258 (1994).
Nelson, D. M., Tréguer, P., Brzezinski, M. A., Leynaert, A. & Quéguiner, B. Production and dissolution of biogenic silica in the ocean: Revised global estimates, comparison with regional data and relationship to biogenic sedimentation. Glob. Biogeochem. Cycles 9, 359–372 (1995).
Armbrust, E. V. The life of diatoms in the world’s oceans. Nature 459, 185–192 (2009).
Marić, D. et al. Phytoplankton response to climatic and anthropogenic influences in the north-eastern Adriatic during the last four decades. Estuar. Coast Shelf Sci. 115, 98–112 (2012).
Timmermans, K. R., Stolte, W. & de Baar, H. J. W. Iron-mediated effects on nitrate reductase in marine phytoplankton. Mar. Biol. 121, 389–396 (1994).
Greene, R. M., Geider, R. J. & Falkowski, P. G. Effect of iron limitation on photosynthesis in a marine diatom. Limnol. Oceanogr. 36, 1772–1782 (1991).
Milligan, A. J. & Harrison, P. J. Effects of non-steady-state iron limitation on nitrogen assimilatory enzymes in the marine diatom Thalassiosira weissflogii (Bacillariophyceae). J. Phycol. 36, 78–86 (2000).
Marchetti, A. & Harrison, P. J. Coupled changes in the cell morphology and the elemental (C, N, and Si) composition of the pennate diatom Pseudo-nitzschia due to iron deficiency. Limnol. Oceanogr. 52, 2270–2284 (2007).
Lin, Q. et al. Effects of fundamental nutrient stresses on the lipid accumulation profiles in two diatom species Thalassiosira weissflogii and Chaetoceros muelleri. Bioprocess Biosyst. Eng. 41, 1213–1224 (2018).
Wang, H. et al. Responses of marine diatom Skeletonema marinoi to nutrient deficiency: Programmed cell death. Appl. Environ. Microbiol. 86, e02460-e2519 (2020).
Lauritano, C., Orefice, I., Procaccini, G., Romano, G. & Ianora, A. Key genes as stress indicators in the ubiquitous diatom Skeletonema marinoi. BMC Genom. 16, 411 (2015).
Levitan, O. et al. Remodeling of intermediate metabolism in the diatom Phaeodactylum tricornutum under nitrogen stress. Proc. Natl. Acad. Sci. USA 112, 412–417 (2015).
Ivančić, I. et al. Survival mechanisms of phytoplankton in conditions of stratification-induced deprivation of orthophosphate: Northern adriatic case study. Limnol. Oceanogr. 57, 1721–1731 (2012).
Ivančić, I. et al. Alkaline phosphatase activity related to phosphorus stress of microphytoplankton in different trophic conditions. Prog. Oceanogr. 146, 175–186 (2016).
Tanković, M. S. et al. Insights into the life strategy of the common marine diatom Chaetoceros peruvianus Brightwell. PLoS ONE 13, e0203624 (2018).
Dyhrman, S. T. et al. The transcriptome and proteome of the diatom thalassiosira pseudonana reveal a diverse phosphorus stress response. PLoS ONE 7, e33768 (2012).
Weigand, H. et al. DNA barcode reference libraries for the monitoring of aquatic biota in Europe: Gap-analysis and recommendations for future work. Sci. Total Environ. 678, 499–524 (2019).
Rimet, F. et al. Diat barcode, an open-access curated barcode library for diatoms. Sci. Rep. 9, 15116 (2019).
Keeling, P. J. et al. The Marine Microbial Eukaryote Transcriptome Sequencing Project (MMETSP): Illuminating the functional diversity of eukaryotic life in the oceans through transcriptome sequencing. PLoS Biol. 12, e1001889 (2014).
Vasselon, V., Rimet, F. & Bouchez, A. Rsyst:: Diatom_rbcl_align_312bp Database: A Database Adapted to DNA Metabarcoding (version v7: 23-02-2018). (2018).
Niang, G. et al. METdb: A Genomic Reference Database for Marine Species (2020).
Groussman, R. D., Blaskowski, S., Coesel, S. N. & Armbrust, E. V. MarFERReT, an open-source, version-controlled reference library of marine microbial eukaryote functional genes. Sci. Data 10, 926 (2023).
Armbrust, E. V. et al. The genome of the diatom Thalassiosira pseudonana: Ecology, evolution, and metabolism. Science 1979(306), 79–86 (2004).
Bowler, C. et al. The Phaeodactylum genome reveals the evolutionary history of diatom genomes. Nature 456, 239–244 (2008).
Lommer, M. et al. Genome and low-iron response of an oceanic diatom adapted to chronic iron limitation. Genome Biol. 13, 1–21 (2012).
Galachyants, Y. P. et al. Sequencing of the complete genome of an araphid pennate diatom Synedra acus subsp. radians from Lake Baikal. Dokl Biochem. Biophys. 461, 84–88 (2015).
Tanaka, T. et al. Oil accumulation by the oleaginous diatom Fistulifera solaris as revealed by the genome and transcriptome. Plant Cell 27, 162–176 (2015).
Traller, J. C. et al. Genome and methylome of the oleaginous diatom Cyclotella cryptica reveal genetic flexibility toward a high lipid phenotype. Biotechnol. Biofuels 9, 1–20 (2016).
Mock, T. et al. Evolutionary genomics of the cold-Adapted diatom Fragilariopsis cylindrus. Nature 541, 536–540 (2017).
Ogura, A. et al. Comparative genome and transcriptome analysis of diatom, Skeletonema costatum, reveals evolution of genes for harmful algal bloom. BMC Genom. 19, 1–12 (2018).
Osuna-Cruz, C. M. et al. The Seminavis robusta genome provides insights into the evolutionary adaptations of benthic diatoms. Nat. Commun. 11, 3320 (2020).
Sapriel, G. et al. Genome-wide transcriptome analyses of silicon metabolism in Phaeodactylum tricornutum reveal the multilevel regulation of silicic acid transporters. PLoS ONE 4, e7458 (2009).
Shrestha, R. P. et al. Whole transcriptome analysis of the silicon response of the diatom Thalassiosira pseudonana. BMC Genom. 13, 499 (2012).
Yang, Z. K. et al. Systems-level analysis of the metabolic responses of the diatom Phaeodactylum tricornutum to phosphorus stress. Environ. Microbiol. 16, 1793–1807 (2014).
Cruz de Carvalho, M. H., Sun, H. X., Bowler, C. & Chua, N. H. Noncoding and coding transcriptome responses of a marine diatom to phosphate fluctuations. New Phytol. 210, 497–510 (2016).
Harke, M. J., Juhl, A. R., Haley, S. T., Alexander, H. & Dyhrman, S. T. Conserved transcriptional responses to nutrient stress in bloom-forming algae. Front. Microbiol. 8, 1279 (2017).
Amato, A. et al. Grazer-induced transcriptomic and metabolomic response of the chain-forming diatom Skeletonema marinoi. ISME J. 12, 1594–1604 (2018).
Harðardóttir, S. et al. Transcriptomic responses to grazing reveal the metabolic pathway leading to the biosynthesis of domoic acid and highlight different defense strategies in diatoms. BMC Mol. Biol. 20, 1–14 (2019).
Thangaraj, S. & Sun, J. Transcriptomic reprogramming of the oceanic diatom Skeletonema dohrnii under warming ocean and acidification. Environ. Microbiol. 23, 980–995 (2021).
Zhou, L. et al. Transcriptomic and metabolic signatures of diatom plasticity to light fluctuations. Plant Physiol 190, 2295–2314 (2022).
Degobbis, D. & Gilmartin, M. Nitrogen, phosphorus, and biogenic silicon budgets for the northern Adriatic Sea. Oceanol. Acta 13, 31–45 (1990).
Zavatarelli, M., Raicich, F., Bregant, D., Russo, A. & Artegiani, A. Climatological biogeochemical characteristics of the Adriatic Sea. J. Mar. Syst. 18, 227–263 (1998).
Zavatarelli, M., Baretta, J. W., Baretta-Bekker, J. G. & Pinardi, N. The dynamics of the Adriatic Sea ecosystem: An idealized model study. Deep Sea Res. Part I(47), 937–970 (2000).
Marić Pfannkuchen, D. et al. The ecology of one cosmopolitan, one newly introduced and one occasionally advected species from the Genus Skeletonema in a highly structured ecosystem, the Northern Adriatic. Microb. Ecol. 75, 674–687 (2018).
Ivančić, I. et al. Alkaline phosphatase activity in relation to nutrient status in the northern Adriatic Sea. Mar. Ecol. Prog. Ser. 378, 27–35 (2009).
Ivančić, I. et al. Phytoplankton and bacterial alkaline phosphatase activity in the northern Adriatic Sea. Mar. Environ. Res. 69, 85–94 (2010).
Ivančić, I., Kraus, R., Najdek, M. & Cozzi, S. Ecological importance of alkaline phosphatase activity in changing marine environmental conditions. Water 13, 2750 (2021).
Grilli, F. et al. Seasonal and interannual trends of oceanographic parameters over 40 years in the northern Adriatic Sea in relation to nutrient loadings using the EMODnet chemistry data portal. Water 12, 2280 (2020).
Su, B. et al. A dataset of global ocean alkaline phosphatase activity. Sci. Data 10, 205 (2023).
Giulietti, S., Romagnoli, T., Campanelli, A., Totti, C. & Accoroni, S. Ecology and seasonality of Pseudo-nitzschia species (Bacillariophyceae) in the northwestern Adriatic Sea over a 30-years period (1988–2020). Mediterr. Mar. Sci. 22, 505–520 (2021).
Godrijan, J., Marić, D., Tomažić, I., Precali, R. & Pfannkuchen, M. Seasonal phytoplankton dynamics in the coastal waters of the north-eastern Adriatic Sea. J. Sea Res. 77, 32–44 (2013).
Dermastia, T. T. et al. Ecological time series and integrative taxonomy unveil seasonality and diversity of the toxic diatom Pseudo-nitzschia H. Peragallo in the northern Adriatic Sea. Harmful Algae 93, 101773 (2020).
Bernardi Aubry, F., Acri, F., Bastianini, M., Finotto, S. & Pugnetti, A. Differences and similarities in the phytoplankton communities of two coupled transitional and marine ecosystems (the Lagoon of Venice and the Gulf of Venice-Northern Adriatic Sea). Front. Mar. Sci. 9, 974967 (2022).
Aubry, F. B., Berton, A., Bastianini, M., Socal, G. & Acri, F. Phytoplankton succession in a coastal area of the NW Adriatic, over a 10-year sampling period (1990–1999). Cont. Shelf Res. 24, 97–115 (2004).
Cerino, F., Fornasaro, D., Kralj, M., Giani, M. & Cabrini, M. Phytoplankton temporal dynamics in the coastal waters of the north-eastern Adriatic Sea (Mediterranean Sea) from 2010 to 2017. Nat. Conserv. 34, 343–372 (2019).
Cabrini, M., Fornasaro, D., Cossarini, G., Lipizer, M. & Virgilio, D. Phytoplankton temporal changes in a coastal northern Adriatic site during the last 25 years. Estuar. Coast Shelf Sci. 115, 113–124 (2012).
Marić, D. et al. Blooms of the potentially toxic diatom Pseudo-nitzschia calliantha Lundholm, Moestrup & Hasle in coastal waters of the northern Adriatic Sea (Croatia). Estuar Coast Shelf Sci 92, 323–331 (2011).
Viličić, D., Marasović, I. & Mioković, D. Checklist of phytoplankton in the eastern Adriatic Sea. Acta Bot. Croat. 61, 57–91 (2002).
Kužat, N. et al. Morpho-physiological adaptations of Leptocylindrus aporus and L. hargravesii to phosphate limitation in the northern Adriatic. Sci. Rep. 12, 2687 (2022).
Schroeder, A. et al. DNA metabarcoding and morphological analysis: Assessment of zooplankton biodiversity in transitional waters. Mar. Environ. Res. 160, 104946 (2020).
Lin, Y. et al. Plankton diversity in Anthropocene: Shipping vs. aquaculture along the eastern Adriatic coast assessed through DNA metabarcoding. Sci. Total Environ. 807, 151043 (2022).
Grižančić, L. et al. A metabarcode based (species) inventory of the northern Adriatic phytoplankton. Biodivers. Data J. 11, 6947 (2023).
Guillard, R. R. L. Culture of phytoplankton for feeding marine invertebrates. In Culture of Marine Invertebrate Animals: Proceedings—1st Conference on Culture of Marine Invertebrate Animals Greenport (eds Smith, W. L. & Chanley, M. H.) 29–60 (Springer US, 1975).
Kearse, M. et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649 (2012).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990).
Benson, D. A. et al. GenBank. Nucleic Acids Res. 46, D41 (2018).
Keller, M. D., Bellows, W. K. & Guillard, R. R. L. Microwave treatment for sterilization of phytoplankton culture media. J. Exp. Mar. Biol. Ecol. 117, 279–283 (1988).
Kopylova, E., Noé, L. & Touzet, H. SortMeRNA: Fast and accurate filtering of ribosomal RNAs in metatranscriptomic data. Bioinformatics 28, 3211–3217 (2012).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Grabherr, M. G. et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29, 644–652 (2011).
Patro, R., Duggal, G., Love, M. I., Irizarry, R. A. & Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 14, 417–419 (2017).
Huerta-Cepas, J. et al. Fast genome-wide functional annotation through orthology assignment by eggNOG-Mapper. Mol. Biol. Evol. 34, 2115–2122 (2017).
Huerta-Cepas, J. et al. EggNOG 5.0: A hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 47, D309–D314 (2019).
Kanehisa, M. & Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 28, 27–30 (2000).
Consortium, G. O. The Gene Ontology (GO) database and informatics resource. Nucleic Acids Res. 32, D258–D261 (2004).
Consortium, T. G. O et al. The Gene Ontology knowledgebase in 2023. Genetics 224, iyad031 (2023).
Kanehisa, M., Furumichi, M., Sato, Y., Kawashima, M. & Ishiguro-Watanabe, M. KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res. 51, D587–D592 (2023).
Galperin, M. Y., Makarova, K. S., Wolf, Y. I. & Koonin, E. V. Expanded Microbial genome coverage and improved protein family annotation in the COG database. Nucleic Acids Res. 43, D261–D269 (2015).
Johnson, L. K., Alexander, H. & Brown, C. T. Re-assembly, quality evaluation, and annotation of 678 microbial eukaryotic reference transcriptomes. Gigascience 8, giy158 (2019).
Grbin, D. et al. Multigene phylogeny and morphology of newly isolated strain of Pseudo-nitzschia mannii Amato & Montresor (Adriatic Sea). Diatom Res. 32, 127–131 (2017).
Tanković, M. S. et al. Experimental evidence for shaping and bloom inducing effects of decapod larvae of Xantho poressa (Olivi, 1792) on marine phytoplankton. J. Mar. Biol. Assoc. U. K 98, 1881–1887 (2018).
Martin, P., Van Mooy, B. A. S., Heithoff, A. & Dyhrman, S. T. Phosphorus supply drives rapid turnover of membrane phospholipids in the diatom Thalassiosira pseudonana. ISME J. 5, 1057–1060 (2011).
Abida, H. et al. Membrane glycerolipid remodeling triggered by nitrogen and phosphorus starvation in Phaeodactylum tricornutum. Plant Physiol. 167, 118–136 (2015).
Kemena, T. P., Landolfi, A., Oschlies, A., Wallmann, K. & Dale, A. W. Ocean phosphorus inventory: Large uncertainties in future projections on millennial timescales and their consequences for ocean deoxygenation. Earth Syst. Dyn. 10, 539–553 (2019).
Randolph-Flagg, N. G. et al. Phosphate availability and implications for life on ocean worlds. Nat. Commun. 14, 2388 (2023).
Acknowledgements
We thank the crew of R/V Vila Velebita and R/V Burin for help during sampling and the Ruđer Bošković Institute, Center for Marine Research in Rovinj for long term data on phytoplankton and oceanography of the northern Adriatic. We thank Laboratory for Evolutionary Ecology members: Marija Fornazar, Denis Skalic and Victor Stinga Perusco for help during the field sampling, for maintaining instruments and laboratory equipment and also for help during the lab work. The results were supported by the Croatian Science Foundation (UIP-2020-02-7868 ADRI Life and UIP-2014-09-6563 P-limited) as well as by the H2020 program project JERICO-S3.
Author information
Authors and Affiliations
Contributions
Mia Knjaz wrote the manuscript, preformed molecular work, analyzed data and presented the results. Ana Baricevic helped with the development of molecular methodology, followed the student lab work, and worked with data analyses and manuscript preparation. Natasa Kuzat performed isolation and curation of the cultures. Mirta Smodlaka Tankovic planed the experiment, helped with measurements and discussed the results. Lana Grizancic helped with sampling, field work, molecular work and helped with discussing the results. Ivan Vlasicek helped with sampling, analyzed in situ phytoplankton samples, worked with isolation and curation of the cultures. Ivan Podolosak helped with sampling, data handling. Tjasa Kogovsek helped with sampling and discussion of results Martin Pfannkuchen planed the experiment, helped with the analysis of the results, pipeline development, discussed the results helped with manuscript planning and writing. Daniela Maric Pfannkuchen planed the work and supervised the work. All Authors contributed to data analysis, the writing and reviewing of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Knjaz, M., Baricevic, A., Tankovic, M.S. et al. First regional reference database of northern Adriatic diatom transcriptomes. Sci Rep 14, 16209 (2024). https://doi.org/10.1038/s41598-024-67043-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-67043-4
- Springer Nature Limited