
Abstract
Ardisia crispa(Myrsinaceae) is an ethnomedicine with horticultural and important medicinal values. Its morphology is complex, and its identification is difficult. We analyse the chloroplast genome characteristics and phylogenetic position of A. crispa to provide basic research data for the identification of A. crispa species and resource conservation. This study assemble and annotate the chloroplast genome of A. crispa and to compare it with the chloroplast genome within Ardisia. The A. crispa chloroplast genome is 156,785 bp in length, with a typical quadripartite structure containing 131 genes, including 86 protein-coding genes, 37 tRNA genes, and 8 rRNA genes; a total of 59 SSRs sites were identified, and the codon preference of this chloroplast genome is greater in A/U than in G/C, and leucine is the amino acid with the highest frequency of use. The chloroplast genomes of the nine Ardisia species are conserved in gene content and number, with more stable boundaries and less variation. In the phylogenetic tree, A. crispa is clustered on a branch with A. crispa var dielsii, and is closely related to A. mamillata and A. pedalis. In this study, we constructed and analyzed the chloroplast genome structure of A. crispa, and conducted phylogenetic analysis using the whole chloroplast genome sequence data of Ardisia plants, which is of great significance in understanding the genetic basis of A. crispa and adaptive evolution in Ardisia plants, and this will lay the foundation for the future research on A. crispa resource conservation and species identification.
Similar content being viewed by others


Introduction
Ardisia crispa (Thunb.) A. DC. is derived from Myrsinaceae, which is distributed in the south provinces of the Yangtze River Basin in China, and its root is used as a medicine, which is one of the basal species of the famous specialty Hmong medicine, Bazhuajinlong, in Guizhou1,2,3. Modern research shows that A. crispa is rich in triterpene glycosides, flavonoids, isocoumarins and other chemical constituents, with heat and pharyngeal, tendon activation and other efficacy, used for the treatment of sore throat, tonsillitis, nephritis, edema and other diseases2, known as the “laryngeal medicine”.
Traditional taxonomy considers Ardisia Swartz as belonging to the Myrsinaceae4,5, whereas in APG III6 the genus is associated with the Primulaceae family, and this opinion is also retained in APG IV7. Currently, the taxonomic identification of Ardisia is still based on traditional taxonomy, but the identification of species based only on morphological differences in plants and leaves is somewhat subjective and still lacks a scientific basis8. A. crispa medicinal herbs are mainly derived from the wild, and the morphology among its Ardisia species is complex and variable, while the phenomenon of homonymy and heteronymy is prevalent9, leading to confusion in the use of medication and making its efficacy reduced. However, the structural features of the plastid genome, functional classification, codon preference analysis, as well as the structural differences of the chloroplast genome, and the kinship relationship have not been reported, which limits the understanding of the genetic background, conservation of germplasm resources, and phylogenetic evolution of A. crispa, and also tends to lead to the difficulty of accurate identification of the mixed pseudo-products of the A. crispa medicinal herbs. Therefore, it is extremely important to accurately classify and identify medicinal herbs.
Chloroplasts are semi-autonomous organelles of plants and their genome is the second largest in plant cells10. The chloroplast genome of most plants consists of a large single copy (LSC) region, a small single copy (SSC) region, and two inverted repeats (IR)11. The uniparental inheritance, moderate mutation rate, and relative ease of sequencing make the chloroplast genome often considered a more efficient resource than the nuclear and mitochondrial genomes, and is commonly used for exploring the origin and evolution of plants, understanding the phylogenetic relationships of different taxonomic classes, and for species identification12. In recent years, with the increasing sophistication of high-throughput sequencing technologies, a large number of subspecies chloroplast genomes have been successfully assembled and annotated and analyzed. Currently, chloroplast genome analysis has been applied to achieve good results in the study of identification, genetic and phylogenetic relationships of Sabia, Phoebe, Vaccinium, Hibiscus rosa-sinensis, Dalbergia hainanensis, Litsea and Zingiber13,14,15,16,17,18,19, among other species.
In view of this, this study was conducted to sequence, assemble and annotate the whole chloroplast genome of A. crispa based on high-throughput sequencing technology to obtain the full-length sequence information of its chloroplast genome. Comparative analysis of the structural characteristics and phylogenetic relationships between A. crispa and other species of Ardisia using bioinformatics was carried out to provide data support for the studies on species and medicinal herbs identification, phylogeny and species conservation of A. crispa.
Results
Chloroplast genome assembly and annotation
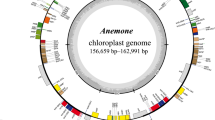
The structure of the A. crispa chloroplast genome is the same as that of most angiosperms, which is a cyclic double-stranded molecule with a typical quadratic structure (Fig. 1). Its total length is 156,785 bp, with a GC content of 37.0%. The length of the LSC region is 86,342 bp, the length of the SSC region is 18,417 bp, and the length of the IR region is 26,014 bp, and the GC contents of the LSC, IR and SSC regions are 35.0%, 43.0% and 30.1%, respectively. A total of 131 genes were annotated, including 86 protein-coding genes, 37 tRNA genes, 8 rRNA genes, 15 genes with 2 copies (rrn4.5, rrn5, rrn16, rrn23, trnA-UGC, trnI-CAU, trnI-GAU, trnL-CAA, trnN-GUU, trnR-ACG, trnV-GAC, rps12, rps7, rpl2, ndhB), and 21 genes had 1 intron (trnA-UGC(× 2), trnG-UCC, trnI-GAU(× 2), trnK-UUU, trnL-UAA, trnV-UAC, rps12(× 2), rps16, rpl16, rpl2(× 2), rpoC1, petB, petD, atpF, ndhA, ndhB(× 2), and 2 genes with 2 introns (ycf3, clpP) (Table 1.).

Chloroplast genome map of A. crispa. Genes shown inside the circle are transcribed clockwise, whereas genes outside are transcribed counterclockwise. The light gray inner circle shows the AT content, the dark gray corresponds to the GC content.
Repeat sequences analysis
The A. crispa chloroplast genome had 59 SSRs, including 44 single-nucleotide repeats, 5 dinucleotide repeats, 8 tetranucleotide repeats, 2 pentanucleotide repeats, and no trinucleotide or hexanucleotide repeats, and the types of SSRs were mainly A/T (41) (Fig. 2a). A total of 49 long repetitive sequences were detected, including 22 forward repeats, 26 palindromic repeats and 1 inverted repeat, and no complementary repetitive sequences were detected, in which the lengths of forward and palindromic repetitive sequences were concentrated in 30 ~ 49 bp (Fig. 2b). In total, 38 tandem repeats were detected, with a minimum length of 10 bp and a maximum length of 30 bp.

Repeat type and number of A. crispa chloroplast genome (a: SSR type b: long repeat type and frequency of use).
Codon analysis
The chloroplast codon statistics of A. crispa showed that the 86 protein-coding genes encode a total of 52,261 codons (Fig. 3). Of all the amino acid codons, three amino acids, leucine (Leu), serine (Ser), and arginine (Arg), were encoded by six codons with a frequency of 5218 (9.98%), 4779 (9.14%), and 3216 (6.15%), respectively, with Leu being the most frequently used, (Ser the next most frequently used, and tryptophan (Trp ) was the least used 688 (1.32%). Among all the codons, the most frequently used codon was AAA, with a frequency of 2182 times and an relative synonymous codon usage (RSCU) of 1.35, and the least frequently used codon was GCG, with a frequency of 221 times and an RSCU of 1.22. There were a total of 35 codons with an RSCU ≥ 1 in the chloroplast genome of A. crispa, among them, 8 codons ended with G/C, and 27 codons ended with A/U. In addition, the A. crispa chloroplast genome had an ENc value of 55.71, a CAI value of 0.652, and GC, GC1, GC2, and GC3 contents of 37.04%, 36.89%, 36.61%, and 37.62%.

Relative synonymous codon usage (RSCU) for protein-coding genes in A. crispa.
IR contraction and expansion in the chloroplast genome
Comparison of IR-LSC and IR-SSC boundaries in the chloroplast genomes of nine Ardisia species, the results showed that there were four boundaries in the chloroplast genome of Ardisia plants, and the genes at the boundaries and their lengths varied (Fig. 4). The A. crispa. and the remaining eight Ardisia plants chloroplasts had rpl22 genes on the left side of the complete genome LSC/IRb boundary (JLB), rpl2 genes on the right side, and rps19 genes across the JLB. Of these, 240 bp expansion of the rps19 gene into the LSC region was observed in A. crispa and A. crispa var. dielsii; 232 bp expansion in A. crispa var. amplifolia, A. crenata, A. crenata var. Bicolor, A. japonica, A. polysticta; and 69 bp expansion in A. gigantifolia and A. bullata. The extent of expansion of the IRb/SSC (JSB) boundary showed that, except for A. japonica, whose JSB boundary was located to the left of the ndhF gene, the ndhF genes of the other eight species of Ardisia plants spanned the JSB boundary, but the extent of the expansion varied slightly, with the expansion of 5 bp into the IRb region for A. crispa, A. crispa var. dielsii, A. gigantifolia, and A. bullata, and the remaining four species were expanded by 3 bp. The range of expansion of the SSC/IRa (JSA) boundary showed that the ycf1 genes of the nine Ardisia species straddled the JSA boundary, but the extent of expansion varied, ranging from 4600 bp to 4614 bp toward the SSC region. The extent of the LSC/IRa (JLA) boundary expansion showed that the trnH genes spanned the JLA boundary, in addition to the rps1 genes of A. crenata and A. polysticta, which were located at the JLA boundary.

Changes of IR/SC boundary of chloroplast genomes of nine Ardisia species.
Comparative chloroplast genomic and nucleotide diversity analyses
To assess the extent of differences in the chloroplast genome sequences of Ardisia, the full-length sequences of the chloroplast genomes of the nine Ardisia species were compared and analyzed in this study using A. crispa (OP762693) as the reference genome and the mVISTA online tool (Fig. 5). Nucleotide polymorphism analysis showed that the mean value of nucleotide diversity (Pi) in the nine Ardisia species was 0.00459. Five highly variable regions (trnT-psbD, ndhB-trnL, rpl32-trnL, trnL-ccsA, trnL-ndhB) were detected when Pi > 0.02, with one each on the IRa, IRb and LSC regions and two in the SSC region (Fig. 6).

Global alignment analysis of the nine Ardisia chloroplast genomes.

Comparative analysis of nucleotide diversity (Pi) values among the nine Ardisia species chloroplast genome sequences.
Phylogenetic analyses
To determine the phylogenetic position of A. crispa BI(bayesian inference) and ML(maximum likelihood) phylogenetic trees were constructed for a total of 29 chloroplast genome sequences of 22 Ardisia species in this study (Fig. 7). The results show that the phylogenetic trees constructed by the two methods have similar topologies and both have high support, differing only at certain nodes. There are two main branches of Ardisia in the ML tree, in branch I, A. japonica, A. solanacea and A. quinquegona have 89%, 72% and 67% node support respectively, and in branch II, A. replicata and A. fordii have 83% and 54% node support respectively. In the BI tree, Ardisia had several branches, two of them had 98% and 82% node support and the rest had 100%. The results showed that each genus is clustered into a single branch. A. crispa and A. crispa var. dielsii clustered, forming a sister relationship, and were more closely related to A. mamillata and A. pedalis. However, A. crispa var. amplifolia clustered on one branch with A. crenata var. bicolor, which is closely related to A. crenata.

Phylogenetic analysis based on chloroplast genome sequences.
Discussion
In this study, the sequencing, assembly and annotation of the chloroplast genome of A. crispa were completed. Similar to most angiosperms, the chloroplast genome of A. crispa has a typical quadripartite structure, with the GC content in the sequences of each region being IRs > LSC > SSC, which may be attributed to the presence of high GC content rRNA genes in the IR region, which is in agreement with the results of the previously reported studies of Paris mairei, A. crenata, A. crenata var. bicolor, A. crispa var. dielsii, and A. crispa var. amplifolia20,21,22. Repetitive sequences are widely present in chloroplast genomes, and their type, number, and distribution differ depending on the species or population13. They are widely used in studies of genetic variation, population structure and species identification, and play an important role in the structural rearrangement of the chloroplast genome23,24,25,26. Three types of A. crispa chloroplast genome interspersed repeats types were detected (forward, reverse, and palindromic), with forward and palindromic repeats being the most common types, and the repeat sequence lengths were all concentrated at 30–49 bp, which is consistent with the previous reports of A. crispa var. dielsii, A. crispa var. amplifolia, and Dendrobium devonianum, among others, which is consistent with most of the plants reported by previous authors22,27. A total of 59 SSRs were detected in this study, among which the highest number of single-nucleotide repeats were mainly composed of A or T. This also indicates that A or T are frequently used in base formation in the A. crispa chloroplast genome, and the SSRs of A. crispa chloroplast genome can potentially provide a basis for the development of molecular markers and identification for Ardisia species.
Codon preference is the unequal use of synonymous codons encoding the same amino acids by species. Codon usage bias is an important feature of genome evolution and is important for the study of molecular evolution and gene ectopic expression28. RSCU indicates the ratio of the actual usage frequency of a codon to its theoretical expected usage frequency. When RSCU = 1, it means that the frequency of codon usage is equal to the frequency of its synonymous codons and there is no usage preference; when RSCU > 1, it means that the codon usage preference is strong; when RSCU < 1, it means that the codon usage preference is weak. Compared to the first 2 bases, mutations in the 3rd position of codon bases are subject to weaker selective pressure and correlate with amino acid species. The codon’s 3rd base composition and content is one of the most important indicators of genomic preference, and higher plants tend to use codons ending in A/U29. In the codon preference analysis of the chloroplast genome of A. crispa, there were 35 codons with RSCU ≥ 1, among which 27 codons ended in A/U, which indicated that the synonymous codons of the chloroplast genome of A. crispa also preferred to end in A/U, similar to those of Phyllanthaceae30, Notopterygium31, Cinnamomum camphora32 and so on, and further verified the conclusion of the preference for codons that ended in A/U. The ENc value of A. crispa chloroplast genome was 55.71, indicating a weak preference. The GC and GC3 contents of A. crispa chloroplast genome were both less than 50%, indicating a codon preference for the use of A and T bases, which was similar to the results of Dendrobium devonianum33 and Glycyrrhiza eurycarpa34.
The chloroplast genome IR region is a common region in most higher plants, and its contraction and expansion is a common evolutionary phenomenon that is considered to be one of the main causes of chloroplast genome size variation35. The expansion of the IR region leads to changes in the copy number of the related genes. Due to the reverse repeatability of the region, complete genes or incomplete gene fragments are formed in the IR region on the other side. In this paper, we analyzed the chloroplast genome boundaries of nine species of Ardisia and found that the JLB and JLA boundaries differed greatly, while the JLB and JSA boundaries were relatively conserved. The ycf1 gene is missing in A. crispa, A. crispa var. dielsii, and A. crispa var. amplifolia, and the ycf1 gene located at the border of the JSB is a pseudogene36. It has been reported in the literature that differences in selective pressure on the ycf1 gene lead to differences in evolutionary rates37,38. The ndhF gene was present only in the SSC region in A. japonia, and the rps12 gene was present in the IRa region. The expansion of rps19 gene to the IRb region was obvious in A. bullata and A. gigantifolia, and the boundary expansion and contraction phenomenon existed as in the chloroplast gene boundary analysis of other plants of the same genus, and the chloroplast genome boundary analysis of the species of Rubia cordifolia39, Polygala sibirica40, and Triticum41 which had been already investigated also had the similar result of the change in the boundary of the chloroplast genome did not show regularity. Nucleotide diversity can be calculated to quantify differences in chloroplast genomes at the sequence level42. These regions may undergo accelerated nucleotide substitutions at the species level, suggesting their potential for use as molecular markers in plant identification and phylogenetic analysis43. The results of nucleotide polymorphism analysis in this study showed that the non-coding regions of the chloroplast genome sequences of the nine species of Ardisia species were highly variable, in which there were obvious differences in the spacer regions of the trnT-psbD, rpl32-trnL, trnL-ccsA, trnL-ndhB, and ndhB-trnL genes, and these regions of variability can provide the basis for the development of molecular markers, species identification, and DNA barcode screening of Ardisia.
The chloroplast genome has been shown to be successful in resolving phylogenetic relationships of different plant taxa44,45. In this study, we constructed BI and ML phylogenetic trees based on 29 chloroplast genomes with Lysimachia christinae (Primulaceae) as outgroup. It was found that A. crispa is closely related to A. crispa var. dielsii, and the two clustered on a single branch, however, they did not cluster with A. crispa var. amplifolia, suggesting that there is a high degree of intraspecific variation in Ardisia, which may be due to geographic factors. The A. crispa var. amplifolia clustered in one branch with A. crenata and A. crenata var. bicolorat 100% support, which is in agreement with the ITS and ITS2 sequence identifications reported in the literature46,47,48. It is similar to the results of previous studies that the A. crenata is more closely related to the A. crenata var. bicolor and A.polysticta22,44. The ML and BI phylogenetic trees in this construction have similar topologies, and species between genera are clustered on the same branch with high support. The phylogenetic tree shows that Primula of Primulaceae is closely related to Aegiceras and Myrsine of Myrsinoideae, which verifies that evolution is complex and variable and is in line with modern taxonomy merging the two family7. Even when sequence analyses based on chloroplast genomes show different evolutionary histories, taxonomists still take into account a variety of factors when determining taxonomic units in order to provide as accurate and comprehensive a classification system as possible.
Conclusion
In this study, we sequenced the chloroplast genome of A. crispa, comprehensively analyzed the chloroplast genome sequence, structure and characteristics of A. crispa, and explored the phylogenetic relationships of Ardisia. The results not only enrich the information of A. crispa chloroplast genome, but also provide a reference for the taxonomic identification and phylogenetic relationship of Ardisia, which is of great significance for the conservation of Ardisia germplasm resources and identification of germplasm resources.
Material and methods
Sample collection
In the study, the fresh young leaves of sample (Fig. 8) were collected from the Ceheng county, Guizhou Province, China, (coordinates: N105°47′29.73″, E24°59′59.51″; altitude: 933 m). It was identified as Ardisia crispa (Thunb.) A. DC.) by Associate Professor Yan Fulin of Guizhou University of Traditional Chinese Medicine. The voucher specimen (with collection numbers of YFL_2021040307) has been deposited in the Herbarium of Guizhou University of Traditional Chinese Medicine (GZYGH), Guizhou, China. The collection of plant materials complies with the wild plant protection regulations of the People’s Republic of China, and we obtained the permission of local authorities on forestry and the grassland bureau in Guizhou province in China.

The plant of Ardisia crispa and its growing environment (A): Growing environment (B): Living plants (C): Flowers).
DNA extraction and chloroplast genome sequencing
Total DNA was extracted from the fresh leaves of A. crispa by the modified CTAB method49. Sequencing was carried out on the Illumina HiSeq XTen to generate approximately 3 GB of sequence in total, at Beijing Genomics Institute (BGI, Wuhan, China).
Genome assembly and annotation
The filtered reads were assembled into a complete cp genome by the program GetOrganelle v 1.550. In this pipeline, the complete cp genome reads were extracted from total genomic reads and were subsequently assembled using SPAdes version 3.1051. The genes were annotated using PGA52 and Geneious 11.0.353 with the published complete cp genome of A. crenata (GenBank accession number: NC_059021) as the reference. Transfer RNAs (tRNAs) were confirmed by their specific structure predicted by tRNAscan-SE 2.054. The OGDRAW (https://chlorobox.mpimp-golm.mpg.de/OGDraw.html)55 was used to draw a detailed physical map of the A. crispa cp genome.
Chloroplast genome structural analysis
The online software REPuter (https://bibiserv.cebitec.unibielefeld.de/reputer) was employed to analyze forward (F), palindromic (P), reverse (R), and complement (C) repeats, with the following settings: Minimum repeat size of 3 bp and hamming Distance of 30 bp56. We used the default parameters in the online Tandem Repeats Finder (http://tandem.bu.edu/trf/trf.html)57 to search for tandem repeats in DNA sequences. The online software MISA58 was applied to predict SSRs with parameter thresholds set at 1, 2, 3, 4, 5 and 6, and nucleotide parameters of 10, 5, 4, 3, 3 and 3, and the distance between two SSRs was not less than 100 bp. Relative synonymous codon usage (RSCU) analysis of the chloroplast genome of A. crispa was performed using condon W (https://galaxy.pasteur.fr/?form=codonw) online software. By using CUSP online software (http://imed.med.ucm.es/EMBOSS/)59, we calculated the effective codon count (ENc), codon adaptation index (CAI), and counted the total codon GC content (GCall), the GC contents of positions 1, 2, and 3 (GC1, GC2, and GC3), and the GC content of position 3 of the synonymous codons (GC3s).
Genome comparison
The genes of the boundaries in the chloroplast genomes of the Ardisia species were compared and visually represented using IRscope60(https://irscope.shinyapps.io/ irapp/) to reveal contraction and expansion of the IR regions. The comparative analysis of the whole sequence identity of the chloroplast genomes was performed using mVISTA61 with the chloroplast genome of A.crispa (OP762693) as the reference sequence. The nucleotide variability (Pi) values of the nine species of the genus Ardisia were calculated using the Dnasp V562 software, the sliding window length of 600 bp and a step size set to 200 bp.
Phylogenetic analysis
A phylogenetic analysis was conducted based on chloroplast genomes from 29 species, including those of the one A. crispa sequenced and assembled in this study and another 28 downloaded from GenBank. Twenty-nine complete plastid sequences were aligned using MAFFT v7.01763. Using MEGA 1164 software to construct a maximum likelihood (ML) phylogenetic tree (bootstraps:1000), and construction of BI phylogenetic tree using MrBayes 3.2.765 based on PhyloSuite v1.2.366 software looking for the best model GTR + F + I + G4.
Ethics approval and consent to participate
The authors declare that the present study of A. crispa is not indexed by IUCN. The experimental research work on the plants described in this paper conforms to institutional, national and international guidelines. The voucher specimens of the plants are kept in the Herbarium of Guizhou University of Traditional Chinese Medicine, Guiyang, Guizhou Province, China.
Data availability
The complete chloroplast genomes and annotations are available at the NCBI database (Ardisia crispa: OP762693).
Abbreviations
- cpDNA :
-
Chloroplast DNA
- SSR :
-
Simple sequence repeat
- SNP :
-
Single nucleotide polymorphism
- ITS :
-
Internal transcribed spacer
- IRs :
-
Inverted repeats
- LSC :
-
Large single copy
- SSC :
-
Small single copy
- rRNA :
-
Ribosomal RNA
- tRNA :
-
Transfer RNAs
References
Guizhou Medical Products Administration. Quality standards for traditional Chinese medicinal materials and ethnic medicinal materials in Guizhou Province Guiyang (Guizhou Science and Technology Press, 2003).
Zhang, N. L. et al. Chemical constituents of Ardisia crispa (Thunb.) A. DC. Nat. Prod. Res. Dev. 22, 587–589. https://doi.org/10.16333/j.1001-6880.2010.04.039 (2010).
Li, M. et al. Investigation and application evaluation of Ardisia resources in Guizhou. Guizhou Agric. Sci. 47, 140–144 (2019).
Chen, C. Flora Reipublicae Popularis Sinica, Tomus Vol. 58, 1–147 (Science Press, 1979).
Takhtajan A L. Song Thuc Vat. Leningad. 5, 106–108(1981).
The Angiosperm Phylogeny Group. An update of the Angiosperm Phylogeny Group classification for the orders and families of fl owering plants: APG III. Bot. J. Linn Soc. 161, 105–121. https://doi.org/10.1111/j.1095-8339.2009.00996.x (2009).
The Angiosperm Phylogeny Group. An update of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants: APG IV. Bot. J. Linn Soc. 181, 1–20. https://doi.org/10.1111/boj.12385 (2016).
Editorial Committee of Flora of China. Chinese Academy of Sciences. Flora of China (Science Press, 1979).
Tong, J. Y. et al. Investigation of genus Ardisia in Bencao literature. Chin. J. Chin. Materia Medica. 42, 396–404. https://doi.org/10.19540/j.cnki.cjcmm.20161222.021 (2017).
Xue, S. et al. Comparative analysis of the complete chloroplast genome among Prunus mume, P. armeniaca, and P. salicina. Hort. Res. https://doi.org/10.1038/s41438-019-0171-1 (2019).
Li, X. et al. Complete chloroplast genome sequence of Magnolia grandiflora and comparative analysis with related species. Sci. Chin. Life Sci. 56, 189–198. https://doi.org/10.1007/s11427-012-4430-8 (2013).
Chen, Q., Hu, H. & Zhang, D. DNA Barcoding and phylogenomic analysis of the genus Fritillaria in China based on complete chloroplast genomes. Front. Plant Sci. 13, 764255. https://doi.org/10.3389/fpls.2022.764255 (2022).
Chen, Q. et al. Complete chloroplast genomes of 11 Sabia samples: Genomic features, comparative analysis, and phylogenetic relationship. Front. Plant Sci. 13, 1052920. https://doi.org/10.3389/fpls.2022.1052920 (2022).
Shi, W. et al. Comparative chloroplast genome analyses of diverse Phoebe (Lauraceae) species endemic to China provide insight into their phylogeographical origin. PeerJ. 11, e14573. https://doi.org/10.7717/peerj.14573 (2023).
Annette, M. et al. Chloroplast genome assemblies and comparative analyses of commercially important Vaccinium berry crops. Sci. Rep. 12, 21600. https://doi.org/10.1038/s41598-022-25434-5 (2022).
Abdullah, et al. Chloroplast genome of Hibiscus rosa-sinensis (Malvaceae): Comparative analyses and identification of mutational hotspots. Genomics 112, 581–591. https://doi.org/10.1016/j.ygeno.2019.04.010 (2020).
Deng, C. Y. et al. Characterization of the complete chloroplast genome of Dalbergia hainanensis (Leguminosae), a vulnerably endangered legume endemic to China. Conserv. Genet. Resour. 11, 105–108. https://doi.org/10.1007/s12686-017-0967-y (2019).
Song, W. et al. Comparative analysis of complete chloroplast genomes of nine species of Litsea (Lauraceae): Hypervariable regions, positive selection, and phylogenetic relationships. Genes 13, 1550. https://doi.org/10.3390/genes13091550 (2022).
Jiang, D. Z. et al. Complete chloroplast genomes provide insights into evolution and phylogeny of Zingiber (Zingiberaceae). BMC Genom. 24, 30. https://doi.org/10.1186/s12864-023-09115-9 (2023).
Jiang, R. et al. Complete chloroplast genome of Paris mairei: Characterization and phylogeny. Chin. Herb. Med. 52, 4014–4022. https://doi.org/10.7501/j.issn.0253-2670.2021.13.024 (2021).
Zeng, X. F. et al. Chloroplast genome resolution and phylogenetic analysis of Ardisia crispa var. amplifolia and Ardisia crispa var. dielsii. Acta Pharmaceutica Sin. 58, 217–228. https://doi.org/10.16438/j.0513-4870.2022-0874 (2023).
Liu, X. W. et al. Comparative and phylogenetic analyses of complete chloroplast genomes in Ardisia crenata. Biotechnol. Bulle. 39, 232–242. https://doi.org/10.13560/j.cnki.biotech.bull.1985.2022-0471 (2023).
Yuan, Q. et al. Impacts of recent cultivation on genetic diversity pattern of a medicinal plant, Scutellaria baicalensis (Lamiaceae). BMC Genet. 11, 1–13. https://doi.org/10.1186/1471-2156-11-29 (2010).
Chmielewski, M. et al. Chloroplast microsatellites as a tool for phylogeographic studies: The case of white oaks in Poland. IFOREST 8, 765. https://doi.org/10.3832/ifor1597-008 (2015).
Asaf, S. et al. Complete chloroplast genome of Nicotiana otophora and its comparison with related species. Front. Plant Sci. 7, 843. https://doi.org/10.3389/fpls.2016.00843 (2016).
Zhuo, L. et al. Advances in the application of SSR markers in the identification of plant germplasm resources. Contemp. Hortic. 44, 9–11. https://doi.org/10.14051/j.cnki.xdyy.2021.15.005 (2021).
Shang, M. Y. et al. Analysis of chloroplast genome structure and phylogeny of endangered Dendrobium devonianum. Chin. Tradit. Herb. Drugs 54, 6424–6433. https://doi.org/10.7501/j.issn.0253-2670.2023.19.023 (2023).
Wang, Y. Z. et al. Comparative analysis of codon usage patterns in chloroplast genomes of ten Epimedium species. BMC Genom. Data 24, 3. https://doi.org/10.1186/s12863-023-01104-x (2023).
Wang, Z. J. et al. Comparative analysis of codon usage patterns in chloroplast genomes of six Euphorbiaceae species. Peer J. 8, e8251. https://doi.org/10.7717/peerj.8251 (2020).
Gao, C. et al. Codon bias analysis of chloroplast genome of Artocarpus heterophyllus. J. Fujian Agric. For. Univ. (Natural Science Edition). 52, 776–784. https://doi.org/10.13323/j.cnki.j.fafu(nat.sci.).2023.06.008 (2023).
Long, T. et al. Codon usage bias analysis in the Acer amplum subsp. catalpifolium genome. J. Northwest For. Univ. https://doi.org/10.3969/j.issn.1001-7461.2023.06.08 (2023).
Hong, S. R. et al. Analysis of the complete chloroplast genome sequence characteristics and its code usage bias of sorghum bicolor. Acta Agrestia Sin. 31(12), 3636 (2023).
Shang, M. Y. et al. Complete chloroplast genome of endangered Dendrobium devonianum Paxt.: characterization and phylogeny. Chinese Traditional and Herbal Drugs. http://kns.cnki.net/kcms/detail/12.1108.R.20230828.1757.004. (2023).
Zhang, J. et al. Characteristics of the chloroplast genome of Glycyrrhiza eurycarpa P.C.Li from Xinjiang with comparison and phylogenetic analysis of the chloroplast genomes of the medicinal plants of Glycyrrhiza. Acta Pharmaceutica Sin. 57, 1516–1525. https://doi.org/10.1643/j.0513-4870.2021-1661 (2022).
Wang, R. J. et al. Dynamics and evolution of the inverted repeat-large single copy junctions in the chloroplast genomes of monocots. BMC Evol. Biol. 8, 1–14. https://doi.org/10.1186/1471-2148-8-36 (2008).
Jiang, M. et al. Assembly and sequence analysis of Tetrastigma hemsleyanum chloroplast genome. Chin. Tradit. Herb. Drugs. 51, 461–468. https://doi.org/10.7501/j.issn.0253-2670.2020.02.024 (2020).
Yang, X. et al. PBR1 selectively controls biogenesis of photosynthetic complexes by modulating translation of the large chloroplast gene Ycf1 in Arabidopsis. Cell Discov. 2, 1–19. https://doi.org/10.1038/celldisc.2016.3 (2016).
Vitti, J. J., Grossman, S. R. & Sabeti, P. C. Detecting natural selection in genomic data. Annu. Rev. Genet. 47, 97–120. https://doi.org/10.1146/annurev-genet-111212-133526 (2013).
Chen, X. Y. et al. Complete chloroplast genome and phylogenetic analysis of Rubia cordifolia. Acta Bot. Boreali-Occidentalia Sin. 43, 1855–1865. https://doi.org/10.7606/j.issn.1000-4025.2023.11.1855 (2023).
Luo, Y. et al. Chloroplast genome sequence characteristics and phylogenetic analysis of Polygala sibirica. Chin. Tradit. Herb. Drugs. 54, 6065–6073. https://doi.org/10.7501/j.issn.0253-2670.2023.18.024 (2023).
Li, Y. H. Bioinformatics analysis of Triticum species chloroplast genomes. Shanxi Univ. https://doi.org/10.2728/d.cnki.gsxiu.2021.000053 (2021).
Li, H. et al. Chloroplast genomic comparison of two sister species Allium macranthum and A. fasciculatum provides valuable insights into adaptive evolution. Genes Genom. 42, 507–517. https://doi.org/10.1007/s13258-020-00920-0 (2020).
Suo, Z. et al. A new nuclear dna marker revealing both microsatellite variations and single nucleotide polymorphic loci: A case study on classification of cultivars in Lagerstroemia indica L. J.Microbial. Biochem. Technol. 8, 266–271. https://doi.org/10.4172/1948-5948.1000296 (2016).
Li, E. Z. et al. Insights into the phylogeny and chloroplast genome evolution of Eriocaulon (Eriocaulaceae). BMC Plant Biol. 23, 32. https://doi.org/10.1186/s12870-023-04034-z (2023).
Yang, L. et al. Comparative chloroplast genomics of 34 species in subtribe Swertiinae (Gentianaceae) with implications for its phylogeny. BMC Plant Biol. 23, 164. https://doi.org/10.1186/s12870-023-04034-z (2023).
Pan, J. et al. Screening and identification on ITS Sequences of original plants from Ardisia crispa. Mol. Plant Breed. 18, 8187–8195. https://doi.org/10.13271/j.mpb.018.008187 (2020).
Wen, Q. Q. et al. ITS2 sequence identification of Miao medicine Ardisia crispa medicinal materials and their related mixed counterfeits. J. Chin. Med. Mater. 45, 830–835 (2022).
Xie, C. et al. Comparative genomic study on the complete plastomes of four officinal Ardisia species in China. Sci. Rep. 11, 22239. https://doi.org/10.1038/s41598-021-01561-3 (2021).
Doyle, J. DNA Protocols for Plants: CTAB Total DNA Isolation. In Molecular techniques in taxonomy (eds Hewitt, G. M. & Johnston, A.) (Springer, 1991).
Jin, J. J. GetOrganelle: A simple and fast pipeline for de novo assembly of a complete circular cp genome using genome skimming data. BioRxiv. https://doi.org/10.1101/256479 (2018).
Bankevich, A. et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. https://doi.org/10.1089/cmb.2012.0021 (2012).
Qu, X. J. et al. PGA: A software package for rapid, accurate, and flexible batch annotation of plastomes. Plant Methods. 15, 50. https://doi.org/10.1186/s13007-019-0435-7 (2019).
Kearse, M. et al. Geneious basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649. https://doi.org/10.1093/bioinformatics/bts199 (2012).
Lowe, T. M. & Chan, P. P. tRNAscan-SE On-line: Integrating search and context for analysis of transfer RNA genes. Nucl. Acids Res. 44, W54–W57. https://doi.org/10.1093/nar/gkw413 (2016).
Greiner, S., Lehwark, P. & Bock, R. OrganellarGenomeDRAW (OGDRAW) version 1.3.1: Expanded toolkit for the graphical visualization of organellar genomes. Nucl. Acids Res. 47, W59–W64. https://doi.org/10.1093/nar/gkz238 (2019).
Kurtz, S. et al. REPuter: The manifold applications of repeat analysis on a genomic scale. Nucl. Acids Res. 29, 4633–4642. https://doi.org/10.1093/nar/29.22.4633 (2001).
Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucl. Acids Res. 15, 573–580. https://doi.org/10.1093/nar/27.2.573 (1999).
Beier, S. et al. MISA-web: A web server for microsatellite prediction. Bioinformatics 33, 2583–2585. https://doi.org/10.1093/bioinformatics/btx198 (2017).
Rice, P., Longden, L. & Bleasby, A. EMBOSS: The European molecular biology open software suite. Trends Genet. 16, 276–277. https://doi.org/10.1016/s0168-9525(00)02024-2 (2000).
Amiryousefi, A., Hyvönen, J. & Poczai, P. IRscope: An online program to visualize the junction sites of chloroplast genomes. Bioinformatics. 34, 3030–3031. https://doi.org/10.1093/bioinformatics/bty220 (2018).
Ma, J. Y. et al. The complete chloroplast genome characteristics of Polygala crotalarioides Buch.-Ham. ex DC. (Polygalaceae) from Yunnan China. Mitochondrial DNA B Res. 6, 2838–2840. https://doi.org/10.1080/23802359.2021.1964396() (2021).
Librado, P. & Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 25, 1451–1452. https://doi.org/10.1093/bioinformatics/btp187 (2009).
Katoh, K. et al. MAFFT: Anovel method for rapid multiple sequence alignment basedon fast Fourier transform. Nucl. Acids Res. 30, 3059–3066. https://doi.org/10.1093/nar/gkf436 (2002).
Tamura, K., Stecher, G. & Kumar, S. MEGA11: Molecular evolutionary genetics analysis version 11. Mol. Biol. Evol. 38, 3022–3027. https://doi.org/10.1093/molbev/msab120 (2021).
Huelsenbeck, J. P. & Ronquist, F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics. 17, 754–755. https://doi.org/10.1093/bioinformatics/17.8.754 (2001).
Zhang, D. et al. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 20, 348–355. https://doi.org/10.1111/1755-0998.13096 (2020).
Acknowledgements
This study was supported by the National Key Research and Development Program Project (2018YFC1708101), Guizhou Provincial Science and Technology Program Project (No. Qianke Zhongcuidi [2022] 4016), Guizhou Modern Industrial Technology System Construction of Chinese Herbal Medicines (No. GZCYTX2019-2024), and State Administration of Traditional Chinese Medicine Seedling Breeding of Chinese Herbal Medicines Seeds Required for National Essential Drugs (Guizhou) Base Construction Project (2014-2017).
Author information
Authors and Affiliations
Contributions
F.L.Yan designed the research. Z.K.Wu, Y.Q.J., S.H.Wei, F.L.Yan and L.D. collected the samples, J.Ye, F.L.Yan, Q.Luo, and Y.H.Lang conceived the experiments, J.Ye, F.L.Yan, Q.Luo, and Y.H.Lang did computational analysis and deposited sequences. J.Ye, F.L.Yan, and Y.H.Lang wrote the manuscript. All authors have read and approved the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ye, J., Luo, Q., Lang, Y. et al. Analysis of chloroplast genome structure and phylogeny of the traditional medicinal of Ardisia crispa (Myrsinaceae). Sci Rep 14, 19045 (2024). https://doi.org/10.1038/s41598-024-66563-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-66563-3
- Springer Nature Limited