
Abstract
Twelve polyoxygenated cyclohex(a/e)ne diterpene esters, named albiflorenes A–L (1–12), were isolated from the whole plants of Kaempferia albiflora, known as “Prao Mang Mum.” Their structures and relative stereochemistry were determined by extensive spectroscopic analysis. Furthermore, the comparison of experimental electronic circular dichroism (ECD) curves with the curves predicted by TDDFT was used to determine the absolute configurations. Albiflorenes contain polyoxygenated cyclohexane (or cyclohexene) derivatives, which are linked to either isopimarane or abietane diterpene acid units. The discovery marks the first occurrence of a conjugate between polyoxygenated cyclohexane (or cyclohexene) rings and diterpenoids. Among the isolates, albiflorene C specifically exhibited antibacterial activity against Bacillus cereus with MIC and MBC values of 3.13 and 6.25 μg/mL, respectively.
Similar content being viewed by others
Introduction
The genus Kaempferia, which is part of the Zingiberaceae family, encompasses around 60 species globally. This genus is predominantly found in Southeast Asia, with a notable concentration in Thailand1,2. Several species of this genus hold economic value for their medicinal or culinary purposes, for example, K. parviflora, which has gained significant interest in the last twenty years as a supplement for enhancing vitality; and K. galanga, which is known for its rhizome that serves as a flavor spice of various cooking. In recent years, there has been growing interest in Kaempferia plants due to their diverse structural variations with interesting biological properties3. Several studies on this genus have led to the discovery of flavonoids4, diterpenoids5,6,7, and polyoxygenated cyclohexenes8,9, many of which possess antimalarial4,5, antimicrobial5,10, anti-inflammatory11,12,13,14, and cytotoxic properties15.
The Kaempferia genus can be categorized into two subgenera16. One is the Kaempferia subgen. Kaempferia17 which generates inflorescence on the pseudostems, while the other is the Kaempferia subgen. Protanthium (Horan.) Baker18 which produces inflorescence directly from the rhizome before emerging of the leafy shoot. The Kaempferia subgen. Protanthium denotes a minority group, and currently only about 15 species have been acknowledged18. To date, there has been no documented research on the chemical profiles of this subgenus.
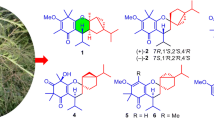
With the significance of discovering novel plant resources that could potentially be employed in the development of medicines, our research group has a sustained interest in exploring bioactive compounds derived from plants within the Kaempferia genus. Recently, we have investigated K. albiflora (syn. K. uttaraditensis)19, a plant belonging to the subgenus Protanthium of the Kaempferia genus, which is endemic to Thailand. This study provided a detailed structural elucidation of twelve unprecedented compounds (1–12), along with nine previously reported compounds (13–21), isolated from the combined CH2Cl2–MeOH (1:1) extract and the EtOAc extract of the whole plants of K. albiflora. Bioassay probing the potential antimicrobial activity of the isolates was performed, and the results showed that albiflorene C exhibited good antibacterial activity against Bacillus cereus (Fig. 1).

The morphological characteristics of K. albiflora and the interesting structure named albiflorene C exhibit notable antibacterial activity against B. cereus.
Results and discussion
By employing successive column chromatography on silica gel, along with gel filtration and reversed-phase HPLC, a total of twelve unprecedented polyoxygenated cyclohex(a/e)ne diterpene esters were effectively isolated, together with nine recognized compounds, from the whole K. albiflora plants.
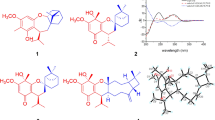
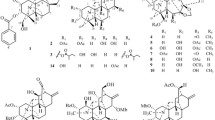
Albiflorene A (1) (Fig. 2), a yellow powder, had a molecular formula of C38H48O9 inferred from the HRESIMS [M+Na]+ ion peak at m/z 671.3195 (calcd 671.3191), and supported by 13C and DEPT135 NMR data, implying fifteen unsaturation indices. UV absorption bands λmax at 229 and 203 nm indicated a characteristic of an aromatic system, and the IR spectrum showed the νmax for a hydroxy (3432 cm–1) group and a conjugated carbonyl (1722 cm–1) functionality. Extensive NMR data were used to clarify the planar structure of 1, which unveiled its composition comprising two subunits designated as unit A and unit B. In unit A, the 1H-NMR (Table 1) showed signals for three methyl groups at δH 1.03 (s, H3-17), 1.21 (s, H3-18), and 0.82 (s, H3-20), a tri-substituted olefin at δH 5.20 (br s, H-14), and a vinyl group with the δH value of 5.75 (dd, J = 17.5, 10.6 Hz), 4.89 (dd, J = 17.5, 1.4 Hz), and 4.87 (dd, J = 10.6, 1.4 Hz). The 13C NMR (Table 1), combined with HSQC and DEPT spectra revealed 20 carbon resonances consisting of three methyls, eight methylenes including one alkene (δC 110.3), four methines including two olefinic (δC, 129.3, 149.1), and five quaternary carbons including one olefinic (δC 136.7) and one carbonyl (δC 178.8). The above spectroscopic data aligned with a pimarane diterpenoid skeleton. In addition, the 1H- and 13C-NMR data for unit A closely resembled those of isopimaric acid20, implying that the isopimaric acid served as unit A and constituted part of compound 1. The HMBC correlations confirmed the planar structure of the isopimaric acid portion, and this was substantiated by NOESY correlations of H-5/H-9 and H3-18/H3-20/H3-17 (Fig. S7). The isopimaric acid structure in nature exists in two forms: the normal form, known as sandaracopimaric acid20, and the ent form, referred to as ent-isopimar-8(14),15-dien-19-oic acid (also known as ent-isopimaric acid)21. These two compounds exhibited identical NMR data and very similar specific optical rotation values (\({\left[\alpha \right]}_{\text{D}}^{26}\) –17.7 for sandaracopimaric acid and \({\left[\alpha \right]}_{\text{D}}^{25}\) –12.5 for ent-isopimaric acid), making it impossible to determine the absolute configuration using optical rotation alone. Due to the absence of circular dichroism (CD) information, we resorted to employ the electronic circular dichroism (ECD) method to determine the absolute configuration of isopimaric acid structure in unit A. In our study, we fortunately isolated the diterpenoid isopimaric acid 13 (Fig. 2). Following biogenesis reasoning, this compound was anticipated to act as a precursor for polyoxygenated cyclohexene isopimaryl esters found in this plant. Therefore, determining the absolute configuration of 13 could contribute to defining the unit A segment in compounds 1 and 2. Through a comparison of the calculated ECD data with the experimental results (Fig. S17), it was determined that 13 shared the absolute configuration with ent-isopimaric acid (4S,5S,9R,10S,13S), as indicated by the strong qualitative agreement between their ECD spectra. This finding confirmed that both unit A of 1 and the following unit A of 2 are ent-isopimaric acid.

Structural compounds (1–21) derived from K. albiflora through isolation.
In unit B, signals corresponding to four oxymethines were observed at the following chemical shifts: δH 5.58 (d, J = 5.2 Hz, H-2ʹ), 4.13 (td, J = 5.2, 2.5 Hz, H-3ʹ), 5.16 (dd, J = 6.0, 2.5 Hz, H-4ʹ), and 5.58 (d, J = 5.2 Hz, H-5ʹ). Furthermore, an oxymethylene signal appeared at δH 4.81 (H-7ʹ), and an olefinic methine signal was observed at δH 5.99 (H-6ʹ). Two acetyl groups were also identified by signals at δH 2.12 and 2.14. The presence of one aromatic group was also evident from the 1H NMR signals in the range of δH 7.47–8.03 (5H) (Table 1). These data suggested the polyoxygenated cyclohexene moiety with aromatic and acetoxy substituents. The occurrence of a polyoxygenated cyclohexene moiety was confirmed through cross peaks in the HMBC correlations from OH-3ʹ to C-2ʹ and C-4ʹ; from H-2ʹ/H-5ʹ to C-3ʹ, C-4ʹ, and C-7ʹ; from H-3′ to C-1′; H-4′ to C-2ʹ, C-5′, and CO; from H-6ʹ to C-4ʹ, C-5ʹ, and C-7ʹ; and from H-7′ to C-1′, C-6′, and CO-7ʹ′. Lastly, the connection of two structural parts was identified as an ester bond at C-19, evidenced by the HMBC cross peak between H-2ʹ (unit B) and C-19 (unit A). The relative configuration of polyoxygenated cyclohexene moiety was identified through an analysis of the coupling constant data and the NOESY correlations (Fig. S7). The small coupling constant value of JH-5ʹ/H-6ʹ, which appeared as a broad singlet, indicated pseudo-axial orientation of H-5ʹ. This orientation permits a dihedral angle of approximately 90 degrees22 between the allylic proton H-5ʹ and the adjacent olefinic proton H-6ʹ within the preferred conformation of cyclohexene ring as shown in Fig. 3. Furthermore, The NOESY correlations observed between H-2ʹ/H-3ʹ, H-3ʹ/H-4ʹ, and H-4ʹ/H-5ʹ (Fig. 3), combined with the coupling constants of JH-2ʹ/H-3ʹ = 5.2 Hz, JH-3ʹ/H-4ʹ = 2.5 Hz (Table 1), indicated a cis relationship among them. The JH-4ʹ/H-5ʹ coupling constants exhibited some differences (6.0 and 5.2 Hz, respectively), but these values still implied a cis relationship. Also, the COSY analysis clearly demonstrated the correlation between these relevant signals. The above observation suggested that H-2ʹ, H-3ʹ, H-4ʹ, and H-5ʹ were axially oriented on the same side. The similar multiplicities and coupling constants in the 1H NMR spectra of 1’s cyclohexene ring (H-2ʹ to H-6ʹ) to those of conduritol D (cyclohex-5-ene-1,2,3,4-tetraol)23, which exclusively adopted an all-cis stereochemistry, strongly suggested a shared relative configuration between unit B of 1 and conduritol D.

Selected NOESY correlations for polyoxygenated cyclohex(a/e)ne rings of 1–9, 11, and 12.
The remaining orientation to be determined is the arrangement of the ester bond linking C-19 to C-2ʹ of the cyclohexene moiety. Since the cyclohexene moiety (unit B) could attach to ent-isopimaric acid (unit A) in two possible orientations, this resulted in two potential absolute configurations for compound 1 as (4S,5S,9R,10S,13S,2ʹS,3ʹS,4ʹS,5ʹR) and (4S,5S,9R,10S,13S,2ʹR,3ʹR,4ʹR,5ʹS). However, due to its conformational flexibility of the ester linkage, NOESY correlations could not be used to establish their relationship. Furthermore, a biphasic pattern observed in the ECD spectrum of 1 made it unfeasible to ascertain the absolute configuration of unit B by comparing with calculated spectrum. As a consequence, on the basis of the aforementioned data, the absolute configuration of 1 could be identified as (4S,5S,9R,10S,13S,2ʹS*,3ʹS*,4ʹS*,5ʹR*).
Albiflorene B (2) (Fig. 2) was obtained as a white powder. The molecular formula of C38H48O9 was deduced from the HRESIMS [M+Na]+ at m/z 671.3190 (calcd 671.3191). Compound 2 displayed almost identical UV absorption bands and IR spectrum, as well as 1D- and 2D-NMR spectra, to compound 1, as illustrated in Fig. S12–S22, suggesting a structural similarity of these two compounds, comprising ent-isopimaric acid (unit A) and the cyclohexene moiety (unit B). The only difference observed was the location of two acetoxy groups at C-3ʹ and C-5ʹ on the cyclohexene ring in 2 instead. Verification of these acetoxy positions was achieved through the HMBC cross-peaks of δH 5.25 (H-3ʹ) and 5.46 (H-5ʹ) with the respective acetyl carbonyls at δC 170.4 and 171.0, and the COSY spin-coupling system from H-2ʹ to H-6ʹ for C-1ʹ to C-6ʹ which indicated their connectivity. The relative configuration of cyclohexene moiety was clearly identifiable through its coupling constants and NOESY correlations. The small coupling constant of JH-2ʹ/H-3ʹ (5.4 Hz), JH-3ʹ/H-4ʹ (2.5 Hz), and JH-4ʹ/H-5ʹ (5.4 Hz) indicated a cis orientation of these protons, with H-5ʹ positioned in a pseudo-axial configuration due to the small coupling constant of olefinic proton H-6ʹ (2.6 Hz) (Table 1). A NOESY experiment (Fig. S18), performed on 2, enabled the assignment of its relative stereochemistry in the same manner as described for 1. Similar to compound 1, the structure of compound 2 consisted of two units linked by an ester bond, resulting in two potential absolute configurations for compound 2 as (4S,5S,9R,10S,13S,2ʹS,3ʹR,4ʹR,5ʹR) and (4S,5S,9R,10S,13S,2ʹR,3ʹS,4ʹS,5ʹS), defined as 2 and 2-dias, respectively, as depicted in Fig. 4A. Following this, the calculated ECD spectra of these two possible structures, 2 and 2-dias, were established. By comparison them with the experimental ECD spectrum, the absolute configuration of 2 was then determined to be (4S,5S,9R,10S,13S,2ʹS,3ʹR,4ʹR,5ʹR).


Comparison of the experimental and calculated ECD curves of compounds 2 (A), 3 (B), 6 (C), and 8 (D).
Albiflorene C (3) (Fig. 2) presented a molecular formula C34H43O7, determined from the HRESIMS ion at m/z 563.3007 [M+Na]+ (calcd for 563.3003) and 1D-NMR data. Its IR spectrum showed absorption bands at 3357 cm–1 for hydroxy, 1717 cm–1 for carbonyl, and 1636 cm–1 for olefinic and aromatic moieties. The NMR data (Table 1) of unit A in compound 3 indicated the presence of three aromatic protons [δH 6.85 (d, J = 1.5 Hz, 1H), 7.00 (s, 1H), and 7.15 (s, 1H)], an isopropyl group [δH 1.21 (s, each of 3H) and 2.80 (m, 1H)], and two additional methyl singlets [δH 0.80, (s, 3H) and 1.21 (s, 3H)]. The 1D- and 2D-NMR spectra revealed 20 carbons, including a carboxylic carbonyl (δC 178.6, C-19) and six aromatic carbons between δC 124.1 and 146.6. These characteristics are typical of an abietane diterpenoid, resembling dehydroabietic acid24,25. Additionally, in the course of our isolation, the diterpenoid dehydroabietic acid (14) was also isolated. Its structure and absolute configuration (4R,5R,10S), were verified by comparing spectroscopic data, including ECD patterns, with previously recorded information. Consequently, in conjunction with biogenetic reasoning suggesting that this compound serves as another precursor for polyoxygenated cyclohexane diterpenoid esters (3–12) found within this plant, the structure and absolute stereochemistry of unit A segment in compound 3 were subsequently affirmed as dehydroabietic acid. The 1H-NMR data of unit B in 3 showed resonances indicative of a benzoyl group (5H) resonating between δH 7.41 and 8.03, along with a polyoxygenated cyclohexene moiety. This includes AB doublet methylene protons [δH 4.73 and 4.80 (2H, d, J = 13.5 Hz, H2-7ʹ)], four oxymethines [δH 5.49 (d, J = 3.6 Hz, H-2ʹ), 4.04 (t, J = 3.2 Hz, H-3ʹ), 3.75 (dd, J = 6.0, 2.6 Hz, H-4ʹ), and 4.39 (d, J = 6.0 Hz, H-5ʹ)], and one olefinic proton [δH 6.04 (dd, J = 1.9 Hz, H-6ʹ)]. These data suggested the similarity of its planar structure to 7-O-benzoyl streptol26. However, the observed small coupling constant values (JH-2ʹ/H-3ʹ = 3.2 Hz and JH-3ʹ/H-4ʹ = 2.6 Hz, and JH-4ʹ/H-5ʹ = 6.0 Hz) indicated co-facial cis relationships of these protons. Their co-directional orientation was further confirmed by NOE experiments. Irradiation at δ 3.75 (H-4ʹ) enhanced signals at δ 4.02 (H-3ʹ) and 4.39 (H-5ʹ), while irradiation at δ 4.02 (H-3ʹ), enhanced signals at δ 5.49 (H-2ʹ) and H-4ʹ. The connection between structural units A and B recognized as the ester bond at C-19, was verified through the cross peak in the HMBC spectrum between H-2ʹ and C-19 (Fig. 5). Finally, by comparing theoretical and experimental ECD curves (Fig. 4B), the structure and absolute configuration (4R,5R,10S,2ʹS,3ʹR,4ʹR,5ʹR) of compound 3 was verified as shown.

Key COSY (red bonds) and HMBC (black arrows) correlations of polyoxygenated cyclohexane ring (Unit B) of selected compounds.
Albiflorene D (4) (Fig. 2), a white powder, had a molecular formula of C36H44O8 inferred from HRESIMS and 1D-NMR data. Analysis of the 1D-NMR data indicated structural similarity between compounds 4 and 3, with the observed 42 Da difference in molecular weights between the two compounds implying that 4 likely contained an additional acetyl substitution compare to 3. The 1H-NMR spectrum of unit B revealed resonances corresponding to methines (four oxygenated) [δH 5.59 (d, J = 3.9 Hz), 5.20 (dd, J = 3.9, 2.5 Hz), 4.35 (br s), and 3.92 (dd, J = 6.8, 3.5 Hz)], an oxymethylene (δH 4.77, br s), an olefinic methine (δH 6.08, d, J = 2.2 Hz), and one acetyl group (δH 2.12, s) (Table 2). The 13C-NMR spectrum subsequently supported these findings, revealing signals for five oxygenated carbons (δC 64.2, 68.4, 69.0, 72.0, and 72.7), one benzoyl group (δC 128.6 × 2, 129.78, 129.9 × 2, 133.4, and 166.1), two alkene carbons (δC 130.6 and 131.4), and an acetyl group (δC 21.0 and 171.0). The connectivity (indicated by red lines) from C-2ʹ to C-6ʹ was verified by analyzing COSY correlations, and was further substantiated through the HMBC cross-peak (Fig. 5). The HMBC correlations between δH 4.77 (H2-7ʹ) and δC 68.4 (C-2ʹ), 130.6 (C-6ʹ), and 131.4 (C-1ʹ), confirmed the linkage of C-2ʹ, C-6ʹ, and C-7ʹ through quaternary center C-1ʹ. The observed cross-peaks of H2-7ʹ and both of H-2ʹʹ/H-6ʹʹ with OCO-7ʹʹ (δC 166.1) determined the location of the benzoyloxy group at C-7ʹ. Additionally, the position of the acetoxy group at C-3ʹ was identified by the correlations from H-3ʹ (δH 5.20) and the acetyl methyl group (δH 2.12) to OCO-3ʹ (δC 171.0) (Fig. 5). Comparing the coupling constants of the methine protons from H-2ʹ to H-5ʹ in 4 with those in 3, the observed coupling constants JH-2ʹ/H-3ʹ = 3.9 Hz and JH-3ʹ/H-4ʹ = 2.5 Hz indicated a cofacial cis relationship, suggesting that unit B of 4 shared the same relative configuration as 3. The molecular formula of albiflorene E (5) and albiflorene F (6) were determined as C36H44O8 by HRESIMS. Examining the NMR data (Table 2) for these compounds revealed their close resemblance to albiflorene D (4), with the only variation being the occurrence of an acetoxy group at different locations; C-4ʹ (δH 4.95/δC 69.1) for compound 5 and C-5ʹ (δH 5.44/δC 71.4) for compound 6. These placements were established through HMBC correlations (Fig. 5) from H-4ʹ (δH 4.95) to the carbonyl carbon at δC 171.1 in 5 and H-5ʹ (δH 5.44) to the carbonyl carbon at δC 171.4 in 6, together with the COSY correlations (Fig. 5) involving H-3ʹ (δH 4.18)/H-4ʹ (δH 4.95)/H-5ʹ (δH 4.52) in 5 and H-4ʹ (δH 3.93)/H-5ʹ (δH 5.44)/H-6ʹ (δH 5.95) in 6. Furthermore, the 1H NMR spectra of the polyoxygenated cyclohexene moiety (unit B) in compounds 5 and 6 showed similar multiplicities and coupling constants to those of albiflorenes (1–4), indicating their consistent configurations. Consequently, the relative configurations of unit B in compounds 5 and 6 were established (Fig. 2).
Given the close resemblance of the structures of compounds 4–6 with 3, which shared the same unit A and differed slightly in the placement of hydroxy and acetoxy substituents on unit B, along with the biogenesis rationale of the compounds being generated from the same plant, the absolute configurations of compounds 4–6 were proposed as (4R,5R,10S,2ʹS,3ʹR,4ʹR,5ʹR), (4R,5R,10S,2ʹS,3ʹS,4ʹR,5ʹR) and (4R,5R,10S,2ʹS,3ʹR,4ʹS,5ʹR), respectively. The reaffirmation of the absolute configuration could be observed in the ECD curve of 6 which displayed the first negative Cotton effect at λmax 203 nm (∆ε = –25.3), corresponding well to the theoretical ECD curve (Fig. 4C).
Based on the HRESIMS ion peak of albiflorene G (7) at m/z 669.3051 [M+Na]+ (calcd, 669.3034) and albiflorene H (8) at m/z 647.3196 [M + H]+ (calcd, 647.3215), a molecular formula of C38H46O9 was suggested for both compounds. Careful analysis of the NMR data (Tables 2 and 3) revealed that signals in 7 and 8 exhibited structural similarities with albiflorenes A (1) and B (2), respectively. However, a notable difference was observed in the core structure unit A, where dehydroabietic acid replaced ent-isopimaric acid (Fig. 2). HMBC correlations of 7 from H-2ʹ (δH 5.63) to C-19 (δC 178.6), and of 8 from H-2ʹ (δH 5.79) to C-19 (δC 177.3), verified that the polyoxygenated cyclohexene and the dehydroabietic moieties were linked through C-2ʹ–O–C-19. The planar structure of polyoxygenated cyclohexene diterpene ester was determined from the COSY and HMBC correlations, as shown in Fig. 5. Additionally, the almost identical multiplicities and coupling constants of the cyclohexene ring proton resonances (H-2ʹ to H-6ʹ) of 7 and 8 in comparison to those found in albiflorenes A (1) and B (2), suggested that they possessed identical relative configurations. Furthermore, in addition to the NOESY correlations observed between H-2ʹ/H-3ʹ, H-3ʹ/H-4ʹ, and H-4ʹ/H-5ʹ in both compounds, compound 7 also exhibited an additional correlation between H-2ʹ and H-4ʹ, while compound 8 showed a correlation between H-3ʹ and H-5ʹ. These correlations provided confirmation of their co-facial alignment within the six-membered ring. Additional support was observed in the NOE experiments, where irradiation at δH 4.18 (H-3ʹ) amplified signals at δH 5.63 (H-2ʹ) and 5.18 (H-4ʹ), while irradiation at δH 5.18 (H-4ʹ) amplified signals at δH 2.39 (H-3ʹ) and 5.58 (H-4ʹ). For the orientation of the ester bond linking C-19 of dehydroabietic acid to C-2ʹ of the cyclohexene moiety, similar to compound 1, where unit B consisted of identical cyclohexene moiety, the absolute configuration of the unit B in compound 7 could not be determined through comparison with calculated spectra. Consequently, the absolute configuration of compound 7 was assigned as (4R,5R,10S,2ʹS*,3ʹS*,4ʹS*,5ʹR*). On the other hand, in the case of compound 8 where the structure of cyclohexene ring is identical to compound 2, the determination of the absolute configuration of 8 could be achieved by comparing the experimental ECD spectra with the calculated ECD spectra of two possible absolute configurations for compound 8, (4R,5R,10S,2ʹS,3ʹR,4ʹR,5ʹR) and (4R,5R,10S,2ʹR,3ʹS,4ʹS,5ʹS) (Fig. 4D). The matching ECD curves, notably at λmax ~ 200 nm, facilitated the determination of the absolute configuration of compound 8 as 4R,5R,10S,2ʹS,3ʹR,4ʹR,5ʹR.
Albiflorene I (9) was isolated as a yellow powder and its HRESIMS spectrum showed a [M+Na]+ peak at m/z 689.3071, consistent with a molecular formula of C41H46O8. Preliminary assessment of the spectroscopic analysis of 9, utilizing 1D- and 2D-NMR data, revealed dehydroabietic acid and a polyoxygenated cyclohexene moiety (Table 3). This compound resembled the core structure of 3 except for the inclusion of an extra benzoyl group and a disubstituted double bond instead of trisubstituted double bond in the polyoxygenated cyclohexene ring (unit B). The 1H-NMR data (Table 3) of unit B showed resonances for two benzoyl groups [δH 7.94 (2H, m), 7.42–7.38 (4H, m), 7.57–7.53 (2H, m), and 8.04 (2H, m)], two olefinic protons [δH 5.93 and 5.97 (each 1H, dd, J = 10.1, 3.1 Hz)], three oxymethine protons [δH 4.20 (d, J = 6.3 Hz), 5.57 (d, J = 3.1 Hz), and 5.74 (dt, J = 6.3, 1.0 Hz)], and two diastereotopic methylene protons [δH 4.39 and 4.95 (each 1H, d, J = 12.2 Hz)]. The 13C-NMR data (Table 3) of unit B exhibited resonances for two benzoyl groups [δC 128.6 × 2, 129.5 × 2, 129.9, 130.0, 133.5, 133.6, and 167.3 × 2], two olefinic carbons (δC 126.5 and 128.7), an oxygenated carbon (δC 75.1), three oxymethine carbons (δC 70.5, 71.7, and 73.9), and an oxymethylene carbon (δC 66.9). These NMR data presented were characteristics of a polyoxygenated cyclohexene ring, resembling those found in (–)-6-acetylzeylenol27,28, with a distinct difference involving the conjugation of the hydroxy group at C-2ʹ, forming an ester linkage (C-2ʹ–OCO-19), with dehydroabietic acid in 9 instead of acetyl group in (–)-6-acetylzeylenol. This ester linkage was confirmed by the HMBC correlation of H-2ʹ (δH 5.57) with the dehydroabietic acid carbonyl carbon (δC 177.8). The positions of benzoyloxy groups at C-1ʹ (δC 75.1) and C-5ʹ (δC 73.9) were established through the HMBC correlations from H-2ʹ to C-7ʹ (δC 66.9) and from H-5ʹ to C-3ʹ (δC 128.7), and the olefinic protons at H-3ʹ (δH 5.97) and H-4ʹ (δH 5.93 /δC 126.5) were identified by their cross-peaks with C-2ʹ (δC 70.5) and C-5ʹ. Furthermore, the correlations of H-2ʹ/H-3ʹ and H-5ʹ/H-6ʹ in the COSY spectrum indicated connectivity of the protons and carbons in cyclohexene ring. The relative stereochemistry of the cyclohexene ring was deduced from the coupling constant between H-2ʹ and H-3ʹ (JH-2ʹ/H-3ʹ = 3.1 Hz) and between H-5ʹ with H-6ʹ (JH-5ʹ/H-6ʹ = 6.3 Hz), which indicated that H-2ʹ adopted a pseudo-equatorial orientation, and existed a trans diaxial relationship between H-5ʹ and H-6ʹ in the favored half-chair conformation (Fig. 3). The methylene protons (H2-7ʹ) of the (benzoyloxy)methyl group showed a cross-peak with H-6ʹ in NOESY spectrum, indicating their positioning on the same face of the cyclohexene ring. The above data confirmed their closed resemblance of their structure and relative stereochemistry of the polyoxygenated cyclohexene moiety in 9 to that of (–)-6-acetylzeylenol. However, their optical rotations displayed opposite directions (\({\left[\alpha \right]}_{\text{D}}^{26}\)= + 38.4 for 9 and \({\left[\alpha \right]}_{\text{D}}^{23}\)= – 63.5 for (–)-6-acetylzeylenol), suggesting that unit B in compound 9 likely had an opposite stereochemistry. Based on the provided data, the structure of compound 9 was proposed to have the (4R,5R,10S,1ʹR,2ʹR,5ʹS,6ʹR) absolute configuration.
Albiflorene J (10) was obtained as a yellow powder. Its HRESIMS spectrum revealed a peak [M+Na]+ at m/z 743.3426 (calcd, 743.3426), corresponding to the molecular formula of C43H51O11. The NMR data (Table 3) of compound 10 revealed its constitution, comprising a dehydroabietic acid (unit A) and a polyoxygenated cyclohexane moiety (unit B) moieties. In unit B, signals corresponding to two benzoyl groups, one acetate group, five oxygenated methines, one oxygenated methylene, and one oxygenated quaternary were identified (Fig. 2). A contiguous spin system formed by H-2ʹ (δH 5.67)/H-3ʹ (δH 4.28)/H-4ʹ (δH 3.96)/H-5ʹ (δH 5.62)/H-6ʹ (δH 5.45) as evidenced by COSY and HSQC correlations (Fig. 5), allowed the identification of the positions of OH-3ʹ and OH-4ʹ on their respective carbons. The two benzoyloxy groups were placed at C-5ʹ (δC 73.8) and C-7ʹ (δC 65.7) by HMBC correlations of H-5ʹ with δC 166.9 (CO) and H2-7ʹ (δH 4.08 and 4.59) with δC 167.3 (CO), respectively. Additionally, the acetoxy group was identified to be located at C-6ʹ (δC 71.7) by the HMBC correlation between H-6ʹ and the acetyl carbonyl at δC 170.1 (Fig. 6). NOESY experiments involving H-2ʹ/H-5ʹ, H-3ʹ/H-5ʹ, H-4ʹ/H-6ʹ, and H-6ʹ/H2-7ʹ, while showing the absence of a cross peak between H2-7ʹ and H-2ʹ, provided evidence for a chair conformation of the cyclohexane ring and indicated α-linkage at C-2ʹ with the diterpene acid ester (Fig. 6). In this conformation, the protons of C-4ʹ, C-6ʹ, and CH2-7ʹ linked to C-1ʹ were aligned on the same face, while the protons of C-2ʹ, C-3ʹ, and C-5ʹ were oriented in the opposite direction. Therefore, the relative stereochemistry of unit B of compound 10 was established. The ECD pattern observed in the spectrum of compound 10 could not provide a conclusive determination of the absolute configuration of unit B by comparing it with the calculated spectrum. As a result, the absolute configuration of compound 10 was assigned as (4R,5R,10S,1ʹS*,2ʹR*,3ʹR*,4ʹR*,5ʹS*,6ʹR*).

Selected HMBC & NOESY correlations for the assignment of 10.
Albiflorene K (11), a yellow powder, had the molecular formula C36H46O10 inferred from HRESIMS. The 1H- and 13C-NMR spectra of 11 (Table 3) revealed a dehydroabietic acid (unit A) and a polyoxygenated cyclohexane moiety (unit B), similar to albiflorene J (10). However, differences associated with the substitutions on the cyclohexane ring in unit B and the linkage of the two units were observed. In unit B, the 1H-NMR resonance assignments (H-2ʹ to H-6ʹ) were identified using sequential correlations in the COSY spectrum (Fig. 5). The chemical shift of H-2ʹ suggested that there was no ester bond formation on the hydroxy group at C-2ʹ, which was now positioned in the β-orientation. The position of acetoxy substitutions were determined through HMBC spectra which showed long-range correlations from the ring methine protons H-3ʹ (δH 5.13) to an acetyl carbonyl at δC 170.1, confirming the location of acetoxy group at C-3ʹ. Furthermore, the HMBC cross-peaks from H-4ʹ (δH 3.15) to C-19 (δC 177.8) verified the connection between the polyoxygenated cyclohexane and the dehydroabietic moieties through C-4ʹ–O–CO-19 rather than the C-2ʹ–O–CO-19 linkage previously identified in compounds 1–10. Another isomer, compound 12, showed a molecular formula of C36H46O10 inferred from HRESIMS analysis. Although, its 1H and 13C NMR spectra displayed similarities to those of 11, the resonance of H-2ʹ of 12 exhibited a downfield shifted instead of that of H-3ʹ, indicating the placement of an acetoxy group at C-2ʹ in 12. This acetoxy group at C-2ʹ was further confirmed by a cross-peak detected in the HMBC spectrum from H-2ʹ (δH 5.45) to the acetyl carbonyl (δC 171.0). Additionally, HMBC correlations between H-4ʹ (δH 5.04) and C-19 (δC 178.5) also verified the linkage between the polyoxygenated cyclohexene and the dehydroabietic moieties via C-4ʹ–O–CO-19. In the NOESY spectra of 11 and 12, the presence of a cross peak between H-7ʹ and H-2ʹ, and between H-7ʹ and H-6ʹ were observed. These cross-peaks clearly indicated an β-orientation of these protons. Furthermore, H-2ʹ, H-3ʹ, H-4ʹ, and H-5ʹ were found to be on the same side of the structure as in β-orientation according to the small coupling constants ranging from 2.0 to 6.0 Hz observed between these protons in both 11 and 12. Therefore, the relative stereochemistry of unit B of both compounds was established. Unfortunately, comparing the quantum chemical calculations of the ECD spectra with the experimental spectra of these two compounds proved difficult in ascertaining the absolute configurations of unit B. Consequently, the absolute stereochemistry of compounds 11 and 12 were identified as (4R,5R,10S,1ʹS*,2ʹR*,3ʹS*,4ʹR*,5ʹR*,6ʹS*), and (4R,5R,10S,1ʹS*,2ʹR*,3ʹR*,4ʹR*,5ʹR*,6ʹS*), respectively.
It is worth mentioning that polyoxygenated cyclohex(a/e)nes, existing as individual molecules, are prevalent in the Annonaceae29 family, and are found in lower abundance in the Zingiberaceae28 and Euphorbiaceae30 families. The polyoxygenated cyclohex(a/e)ne diterpene esters 1–12, represent the first occurrence of a polyoxygenated cyclohex(a/e)ne and diterpenoid conjugate. Their existence should potentially serve as a chemotaxonomic marker for K. albiflora.
The nine, previously recognized, compounds isolated from K. albiflora were identified as ent-isopimar-8(14),15-dien-19-oic acid (13)21, dehydroabietic acid (14)24, dehydroabietinol (15)31, 18-norabieta-8,11,13-trien-4-ol (16)32, pomiferin D (17)33, 7-oxodehydroabietinol (18)31, boesenboxide (19)34, (–)-1,6-desoxytingtanoxide (20)35,36, and stigmasterol stearate (21)37 by comparison of their experimental and reported spectroscopic data.
Except for compounds 2, 4, and 5, which were available in limited quantities, the remaining seventeen compounds (1, 3, and 6–20) were evaluated for their antimicrobial activity against eight bacterial strains, including gram-positive bacteria Staphylococcus aureus, Staphylococcus epidermidis, Enterococcus faecalis, and Bacillus cereus, as well as gram-negative bacteria Pseudomonas aeruginosa, Escherichia coli, Shigella flexneri, and Salmonella Typhimurium. Among the polyoxygenated cyclohexane diterpene esters, compound 3 showed the most potent inhibitory activity against B. cereus strain with MIC value of 3.13 μg/mL and MBC value of 6.25 μg/mL (see Table S1). These values suggested that compound 3 exhibited bactericidal activity against bacterial organisms, as its MIC index (MIC/MBC) is less than four38. The other polyoxygenated cyclohex(a/e)ne diterpene esters (1 and 6–12) did not exhibit noteworthy antibacterial activity and their MIC values exceeded 200 μg/mL. Interestingly, the substituents on the cyclohexene moiety appeared to be crucial as only the presence of free hydroxy substituents (as in 3) showed enhanced activity, whereas the replacement of the OH group with the OAc group at any position(s) resulted in the decrease of activity, as observed in compounds 3–8. For other known compounds isolated from K. albiflora, ent-isopimar-8(14),15-dien-19-oic acid (13) exhibited inhibitory activity against the B. cereus strain with MIC and MBC values of 6.25 and 25.0 μg/mL, respectively (see Table S1). In addition to this, it was previously reported to be active against both pathogens, Streptococcus mutans ATCC 25175 and Actinomyces viscosus ATCC 27044, with MIC values of 125.0 μg/mL21. It should be pointed out that the dehydroabietic acid (14), the diterpenoid core structure of compounds 3–12, showed less potent inhibitory activity compared to compound 13, the diterpenoid core structure of compounds 1–2. Therefore, exploring other polyoxygenated cyclohex(a/e)ne substituents on compound 13 might be a worthwhile avenue for further investigation.
METHODS
General experimental procedures
Sephadex LH-20 (GE Healthcare Bio-Sciences AB) and silica gel 60 (Merck, 0.063–0.200 mm and less than 0.063 mm) were used for column chromatography (CC). TLC was visualized under UV light at 254 and 366 nm to monitor the outcomes, followed by spraying with Godin’s reagent and heating to identify UV-negative compounds and detect color changes in UV-positive spots. Analytical and semipreparative HPLC were performed using Waters Delta 1525 pumps with a Waters 2998 photodiode array detector set at 210 nm to monitor the compounds of interest. NMR spectra were acquired using Bruker Advance 400 and 600 NMR spectrometers. HRESIMS analysis was achieved using Bruker Micro TOFLC spectrometer. Optical rotation was measured on a JASCO P-1020 polarimeter and IR spectra on a Perkin-Elmer Spectrum One Spectrophotometer using the ATR (a universal attenuated total reflectance) technique. ECD spectra were recorded on a JASCO J-810 spectropolarimeter.
Plant materials
The whole plants of K. albiflora Jenjitt. & Ruchis., belonging to the Zingiberaceae, were collected in May 2021 from Mueang Tak district of Tak province, Thailand, at the GPS location (Lat. 16°52′22.7″N, Long, 99°19′27.4″E). Researchers have obtained permission, along with a consent form, to utilize the benefits of Thai medicinal plants from the Department of Agriculture of Thailand. Identification of the plant material was conducted by a taxonomist, Assist. Prof. Dr. Saroj Ruchisansakun. A voucher specimen (accession SR 1511) has been placed in the collection at the Suan Luang Rama IX Botanic Garden, Thailand. All procedures for plant collection were performed in accordance with relevant institutional, national, and international guidelines and legislation.
Extraction and isolation
The 7.7 kg of the shade-dried whole plants of K. albiflora were macerated and soaked in 22 L of 95% EtOH, 13.0 L of 50% CH2Cl2-MeOH, and 4.0 L of EtOAc for 48 h (2 times) at room temperature, respectively. After filtration, the solvent was evaporated and dried under vacuum, resulting in 163.8 g of EtOH extract, 42.5 g of 50% CH2Cl2–MeOH extract, and 5.0 g of EtOAc extract.
A total of 53.52 g of whole plant extracts from K. albiflora, derived by combining 50% CH2Cl2–MeOH and EtOAc crude extracts due to their closely matching NMR spectra, underwent CC using a silica gel column with a gradient of hexane–CH2Cl2 (100:0 → 0:100) followed by CH2Cl2–MeOH (100:0 → 80:20), resulting in 39 fractions (F1–F39). F3 (emerging in 20% CH2Cl2–hexane, 734.1 mg) was separated on a silica gel column eluting with a gradient of hexane–CH2Cl2 (100:0 → 0:100), yielding 10 fractions (F3.1–F3.10). Compound 21 (123.7 mg) was obtained from F3.10. F13 (emerging in 70% CH2Cl2–hexane, 257.9 mg) was separated using preparative HPLC (YMC-Pack ODS-A, 20 × 250 mm) using a gradient of MeCN–H2O (80:20 → 100:0) over 30 min followed by 100% MeCN for 20 min, at a flow rate of 10 mL/min, yielding compounds 16 (tR 15.84 min, 5.6 mg) and 17 (tR 19.55 min, 8.4 mg). Additionally, compound 20 (2.3 mg) was purified by analytical HPLC, transitioning from tR 11.28 min to tR 21.81 min), using a gradient of MeCN–H2O (80:20 → 100:0) over 30 min. F15 (emerging in 80% CH2Cl2–hexane, 324.2 mg) was separated using preparative HPLC (C18, Hichrome, 10 × 250 mm) using a gradient of MeCN–H2O (40:60 → 100:0) over 100 min followed by 100% MeCN for 15 min, at a flow rate of 10 mL/min, to obtain compounds 14 (tR 56.73 min, 45.7 mg) and 13 (tR 73.0 min, 41.2 mg). Additionally, a peak at tR 80.27 min was further purified using analytical HPLC (C18, YMC, 4.6 × 250 mm, gradient, 90:10 to 100:0 MeCN–H2O in 30 min, flow rate 1 mL/min] yielding compounds 3 (tR 11.0 min, 8.5 mg) and 6 (tR 16.48 min, 2.2 mg). F16 (emerging in 85% CH2Cl2–hexane, 326.3 mg) was separated using preparative HPLC (C18, YMC, 20 × 250 mm) using a gradient of MeCN–H2O (90:10 → 100:0) over 30 min followed by 100% MeCN for 20 min, at a flow rate of 10 mL/min, yielding compounds 14 (tR 11.0 min, 94.8 mg), 15 (tR 19.63 min, 0.6 mg), and 13 (tR 21.53 min, 21.2 mg), respectively. F20 (emerging in 1% MeOH–CH2Cl2, 175.9 mg) was separated using preparative HPLC (C18, YMC, 20 × 250 mm) using a gradient of MeCN–H2O (40:60 → 100:0) over 100 min followed by 100% MeCN for 15 min, at a flow rate of 10 mL/min, yielding compounds 19 (tR 38.25 min, 35.8 mg) and 14 (tR 60.7 min, 52.3 mg). A portion of F22 (emerging in 3% MeOH–CH2Cl2, 11.77 g), (1000 mg × 3 times) were chromatographed over Sephadex LH-20, using a MeOH–CH2Cl2 (80:20) solvent system, to afford 5–7 subfractions F22.1 (7 subfractions, F22.1.1–F22.1.7); F22.2 (5 subfractions, F22.2.1–F22.2.5); F22.3 (5 subfractions, F22.3.1–F22.3.5). Then, subfraction F22.1.5 (641 mg) was purified through preparative HPLC (C18, YMC, 20 × 250 mm) using a gradient of MeCN–H2O (30:70 → 100:0) over 120 min at a flow rate of 10 mL/min, yielding compounds 18 (tR 55.02 min, 2.0 mg), 8 (tR 85.84 min, 36.7 mg), and 7 (tR 86.84 min, 30.0 mg). Subfraction F22.2.4 (596.2 mg) was chromatographed over Sephadex LH-20, using a MeOH–CH2Cl2 (80:20) solvent system, to provide four subfractions (F22.2.4.1–F22.2.4.4). Subfraction F22.2.4.3 (406 mg) was further separated using preparative HPLC (C18, YMC, 21.2 × 250 mm) using a gradient of MeCN–H2O (40:60 → 100:0) over 100 min, followed by 100% MeCN for 15 min at a flow rate of 10 mL/min, yielding compounds 12 (tR 68.09 min, 4.5 mg), 11 (tR 70.60 min, 4.3 mg), 10 (tR 76.17 min, 4.0 mg), and 1 (tR 98.18 min, 5.2 mg), respectively. Last, subfraction F22.3.3 (902 mg) was repeated subjection to CC over Sephadex LH-20, eluting with MeOH–CH2Cl2 (80:20) to afford five subfractions (F22.3.3.1–F22.3.3.5). Subfraction F22.3.3.3 (389.6 mg) was purified by analytical HPLC (C18, YMC, 0.4 × 250 mm, gradient 30:70 to 100:0 MeCN–H2O in 100 min, followed by 100% MeCN for 15 min, flow rate 1 mL/min) to give 12 subfractions (F22.3.3.3.1–F22.3.3.3.12). Subfraction F22.3.3.3.9 (110.3 mg) was further purified by HPLC (C18, YMC, 0.4 × 250 mm) using a gradient of MeCN–H2O (90:10 → 100:0) 60 min followed by 100% MeCN for 20 min at a flow rate 1 mL/min, to give compounds 4 (tR 49.75 min, 0.9 mg) and 5 (tR 52.56 min, 3.4 mg). Additionally, a peak at tR 101.5 min was further purified by analytical HPLC (C18, YMC, 4.6 × 250 mm, gradient 90:10 to 100:0 MeCN–H2O, in 60 min, flow rate 1 mL/min) to provide compounds 2 (tR 31.3 min, 1.1 mg) and 1 (tR 32.3 min, 1.3 mg). Subfraction F22.3.3.4 (419.1 mg) was separated by analytical HPLC (C18, YMC, 4.6 × 250 mm) using a gradient of 40:60 to 100:0 MeCN–H2O over 60 min at a flow rate of 1 mL/min, to obtain compound 9 (tR 75.1 min, 1.0 mg).
Albiflorene A or 2ʹ-(ent-isopimar-8(14),15-dien-19-carbonyl)oxy-4ʹ,5ʹ-diacetoxy-3ʹ-hydroxycyclohex-1ʹ-en-1ʹ-yl methyl benzoate (1): A yellow powder; \({\left[\alpha \right]}_{\text{D}}^{23.6}\)–25.4 (c 0.16, MeOH); UV (MeOH) λmax (log ε) 229 (0.18) nm; CD (c 1.20 mM, MeOH) λmax (mdeg) 204 (–33.59), 200 (+ 35.49) nm; IR (ATR) νmax 3432, 2927, 1722, 1451, 1370, 1315, 1266, 1228, 1175, 1109, 1057, 1027, 979, 712 cm−1; 1H- and 13C-NMR data, see Table 1; HRESIMS m/z 671.3195 [M+Na]+ (calcd for C38H48O9Na, 671.3191, ∆ = 0.60 ppm).
Albiflorene B or 2ʹ-(ent-isopimar-8(14),15-dien-19-carbonyl)oxy-3ʹ,5ʹ-diacetoxy-4ʹ-hydroxycyclohex-1ʹ-en-1ʹ-yl methyl benzoate (2): A white powder; \({\left[\alpha \right]}_{\text{D}}^{24}\)+19.5 (c 0.085, MeOH); UV (MeOH) λmax (log ε) 228 (0.71), 204 (1.01) nm; CD (c 0.026 mM, MeOH) λmax (mdeg) 238 (–25.39), 199 (+ 61.63) nm; IR (ATR) νmax 3360, 3193, 2921, 2851, 1727, 1659, 1633, 1470, 1450, 1370, 1268, 1231, 1175, 1110, 1026, 712 cm−1; 1H- and 13C-NMR data, see Table 1; HRESIMS m/z 671.3190 [M+Na]+ (calcd for C38H48O9Na, 671.3191, ∆ = –0.13 ppm).
Albiflorene C or 2ʹ-(abieta-8,11,13-trien-19-carbonyl)oxy-3ʹ,4ʹ,5ʹ-trihydroxycyclohex-1ʹ-en-1ʹ-yl methyl benzoate (3): A white powder; \({\left[\alpha \right]}_{\text{D}}^{24}\)+20.6 (c 0.47, MeOH); UV (MeOH) λmax (log ε) 221 (0.83), 203 (1.15) nm; CD (c 0.74 mM, MeOH) λmax (mdeg) 240 (+ 17.28), 229 (–9.94), 203 (+ 17.17) nm; IR (ATR) νmax 3357, 2928, 1717, 1636, 1497, 1451, 1315, 1269, 1243, 1173, 1106, 1070, 968, 822, 710 cm−1; 1H- and 13C-NMR data, see Table 2; HRESIMS m/z 563.3007 [M+Na]+ (calcd for C34H43O7Na, 563.3003, ∆ = 0.69 ppm).
Albiflorene D or 2ʹ-(abieta-8,11,13-trien-19-carbonyl)oxy-3ʹ-acetoxy-4ʹ,5ʹ-dihydroxycyclohex-1ʹ-en-1ʹ-yl methyl benzoate (4): A white powder; \({\left[\alpha \right]}_{\text{D}}^{24}\)+108.4 (c 0.05, MeOH); UV (MeOH) λmax (log ε) 219 (0.93) nm; CD (c 0.83 mM, MeOH) λmax (mdeg) 203 (–12.67), 199 (+ 50.13) nm; IR (ATR) νmax 3421, 2929, 1723, 1452, 1371, 1268, 1241, 1174, 1108, 1037, 712 cm−1; 1H- and 13C-NMR data, see Table 2; HRESIMS m/z 627.2916 [M+Na]+ (calcd for C36H44O8Na, 627.2928, ∆ = –1.93 ppm).
Albiflorene E or 2ʹ-(abieta-8,11,13-trien-19-carbonyl)oxy-4ʹ-acetoxy-3ʹ,5ʹ-dihydroxycyclohex-1ʹ-en-1ʹ-yl methyl benzoate (5): A white powder; \({\left[\alpha \right]}_{\text{D}}^{25}\)+10.1 (c 0.17, MeOH); UV (MeOH) λmax (log ε) 220 (0.75), 203 (1.05) nm; CD (c 1.41 mM, MeOH) λmax (mdeg) 203 (–17.05), 199 (+ 40.89) nm; IR (ATR) νmax 3360, 3192, 2920, 2851, 1723, 1659, 1633, 1470, 1424, 1377, 1269, 1243, 1173, 1108, 1036, 970, 821, 711 cm−1; 1H- and 13C-NMR data, see Table 2; HRESIMS m/z 627.2931 [M+Na]+ (calcd for C36H44O8Na, 627.2928, ∆ = 0.43 ppm).
Albiflorene F or 2ʹ-(abieta-8,11,13-trien-19-carbonyl)oxy-5ʹ-acetoxy-3ʹ,4ʹ-dihydroxycyclohex-1ʹ-en-1ʹ-yl methyl benzoate (6): A white powder; \({\left[\alpha \right]}_{\text{D}}^{24}\)+2.4 (c 0.14, MeOH); UV (MeOH) λmax (log ε) 221 (0.61), 203 (0.89) nm; CD (c 0.83 mM, MeOH) λmax (mdeg) 203 (–25.33) nm; IR (ATR) νmax 3364, 2927, 2101, 1725, 1636, 1456, 1370, 1269, 1233, 1171, 1107, 1026, 971, 822, 712 cm−1; 1H- and 13C-NMR data, see Table 2; HRESIMS m/z 627.2935 [M+Na]+ (calcd for C36H44O8Na, 627.2928, ∆ = 0.98 ppm).
Albiflorene G or 2ʹ-(abieta-8,11,13-trien-19-carbonyl)oxy-4ʹ,5ʹ-diacetoxy-3ʹ-hydroxycyclohex-1ʹ-en-1ʹ-yl methyl benzoate (7): A yellow powder; \({\left[\alpha \right]}_{\text{D}}^{24}\) –7.6 (c 2.85, MeOH); UV (MeOH) λmax (log ε) 221 (0.43), 203 (0.72) nm; CD (c 0.62 mM, MeOH) λmax (mdeg) 249 (+ 18.75), 200 (+ 33.67) nm; IR (ATR) νmax 3444, 2928, 1723, 1451, 1370, 1269, 1237, 1173, 1107, 1070, 1026, 970, 823, 712 cm−1; 1H- and 13C-NMR data, see Table 2; HRESIMS m/z 669.3051 [M+Na]+ (calcd for C38H46O9Na, 669.3034, ∆ = 2.46 ppm).
Albiflorene H or 2ʹ-(abieta-8,11,13-trien-19-carbonyl)oxy-3ʹ,5ʹ-diacetoxy-4ʹ-hydroxycyclohex-1ʹ-en-1ʹ-yl methyl benzoate (8): A yellow powder; \({\left[\alpha \right]}_{\text{D}}^{24}\) –3.9 (c 0.15, MeOH); UV (MeOH) λmax (log ε) 221 (0.67), 203 (0.94) nm; CD (c 2.32 mM, MeOH) λmax (mdeg) 201 (+ 105.87) nm; IR (ATR) νmax 3405, 2937, 1724, 1451, 1370, 1266, 1228, 1170, 1119, 1107, 1026, 712 cm−1; 1H- and 13C-NMR data, see Table 3; HRESIMS m/z 647.3196 [M + H]+ (calcd for C38H47O9, 647.3215, ∆ = –2.81 ppm).
Albiflorene I or 2ʹ-(abieta-8,11,13-trien-19-carbonyl)oxy-5ʹ-benzoyl-1ʹ,6ʹ-dihydroxycyclohex-3ʹ-en-1ʹ-yl methyl benzoate (9): A yellow powder; \({\left[\alpha \right]}_{\text{D}}^{26}\)+38.4 (c 0.24, MeOH); UV (MeOH) λmax (log ε) 226 (0.24) nm; CD (c 0.90 mM, MeOH) λmax (mdeg) 231 (–5.08), 205 (+ 30.76), 198 (+ 14.47) nm; IR (ATR) νmax 3462, 2925, 2854, 1720, 1602, 1451, 1380, 1316, 1269, 1176, 1110, 1070, 1027, 970, 710 cm−1; 1H- and 13C-NMR data, see Table 3; HRESIMS m/z 689.3071 [M+Na]+ (calcd for C41H46O8Na, 689.3085, ∆ = –2.06 ppm).
Albiflorene J or 2ʹ-(abieta-8,11,13-trien-19-carbonyl)oxy-6ʹ-acetoxy-5ʹ-benzoyl-1ʹ,3ʹ,4ʹ-trihydroxycyclohexane-1ʹ-yl methyl benzoate (10): A yellow powder; \({\left[\alpha \right]}_{\text{D}}^{24}\)+21.6 (c 0.13, MeOH); UV (MeOH) λmax (log ε) 225 (0.37) nm; CD (c 0.69 mM, MeOH) λmax (mdeg) 246 (–20.41), 199 (+ 16.48) nm; IR (ATR) νmax 3443, 2953, 2927, 1723, 1602, 1496, 1451, 1377, 1268, 1225, 1177, 1105, 1070, 1027, 971, 905, 822, 710 cm−1; 1H- and 13C-NMR data, see Table 3; HRESIMS m/z 743.3426 [M+Na]+ (calcd for C43H51O11Na, 743.3426, ∆ = 0.0 ppm).
Albiflorene K or 4ʹ-(abieta-8,11,13-trien-19-carbonyl)oxy-3ʹ-acetoxy-1ʹ,2ʹ,5ʹ,6ʹ-tetrahydroxycyclohexane-1ʹ-yl methyl benzoate (11): A yellow powder; \({\left[\alpha \right]}_{\text{D}}^{26}\)+13.3 (c 0.22, MeOH); UV (MeOH) λmax (log ε) 220 (0.29), 203 (0.47) nm; CD (c 0.78 mM, MeOH) λmax (mdeg) 203 (+ 16.51), 199 (–15.92), 193 (+ 10.91) nm; IR (ATR) νmax 3452, 2956, 2931, 1724, 1451, 1369, 1267, 1241, 1175, 1119, 1051, 1037, 972, 822, 712 cm−1; 1H- and 13C-NMR data, see Table 3; HRESIMS m/z 661.2999 [M+Na]+ (calcd for C36H46O10Na, 661.2983, ∆ = 2.34 ppm).
Albiflorene L or 4ʹ-(abieta-8,11,13-trien-19-carbonyl)oxy-2ʹ-acetoxy-1ʹ,3ʹ,5ʹ,6ʹ-tetrahydroxycyclohexane-1ʹ-yl methyl benzoate (12): A yellow powder; \({\left[\alpha \right]}_{\text{D}}^{24}\)–12.7 (c 0.07, MeOH); UV (MeOH) λmax (log ε) 221 (0.43), 203 (0.64) nm; CD (c 1.09 mM, MeOH) λmax (mdeg) 201 (+ 47.95) nm; IR (ATR) νmax 3422, 2926, 1723, 1665, 1602, 1451, 1379, 1314, 1268, 1245, 1175, 1110, 1069, 1026, 971, 821, 710 cm−1; 1H- and 13C-NMR data, see Table 3; HRESIMS m/z 661.2984 [M+Na]+ (calcd for C36H46O10Na, 661.2983, ∆ = 0.12 ppm).
Calculation method
Conformer–Rotamer Ensemble Sampling Tool (CREST) with iMTD-GC conformational search algorithm and GBSA solvent model of methanol was used to examine a conformational ensemble. Gaussian 16 Rev. C.01 program was used to execute all DFT calculations39,40. The low-energy conformers within an energy window of 5 kcal/mol were further optimized at ωB97XD/cc-PVDZ level of theory. To confirm the true minimum of electronic potential energy of all optimized conformers, the vibrational frequencies at the same level were calculated, and no imaginary frequency was detected. Each conformer with the population over 2% based on Boltzmann distribution with Gibbs free energies were subject to following ECD calculations. TD-DFT at M06-2x/def2-SVP level of theory with IEFPCM of methanol solvent model with 30 excited states for each conformer was used to calculate simulated ECD spectra of compounds 2, 3, 6, 8, and 1341,42,43. The simulated ECD curves of Boltzmann average for all conformers were plotted with overlapping Gaussian function with an exponential half-width (σ = 0.35) by using SpecDis44,45. The direct inversion of simulated ECD spectra were plotted for the related enantiomers. All structures were virtualized by using CYLview46.
Antimicrobial measurements
Eight strains from the American Type Culture Collection (ATCC) were used in susceptibility tests for this study. These were four species of Gram-positive (Staphylococcus aureus ATCC6538, Enterococcus faecalis ATCC19433, Staphylococcus epidermidis ATCC12228, and Bacillus cereus ATCC11778) and four species of Gram-negative bacteria (Salmonella Typhimurium ATCC13311, Escherichia coli ATCC8739, Shigella flexneri ATCC12022, and Pseudomonas aeruginosa ATCC27853). The susceptibility test of compounds was performed in adherence to the CLIS (Clinical and Laboratory Standards Institute) M100 protocol47. The tested bacteria were cultured overnight in an incubator for 18–24 h at 37 ºC on Mueller–Hinton agar (MHA). The harvested bacterial colonies were then placed in sterile normal saline and adjusted to 1 × 104 CFU/mL in Mueller–Hinton broth (MHB). The compounds were present in concentrations ranging from 0.097 to 200 µg/mL. The MICs (minimum inhibitory concentrations) and MBCs (minimum bactericidal concentration) values were determined in triplicate.
Conclusions
In summary, twelve previously undescribed polyoxygenated cyclohexane diterpene esters have been isolated from K. Albiflora. This report represents the first documentation of polyoxygenated cyclohexene diterpene esters in K. albiflora, signifying its chemotaxonomic significance. The present phytochemical findings could contribute to the categorization of the genus within Kaempferia plants and other related genera in Zingiberaceae family. Albiflorene C demonstrated notable antibacterial effectiveness in inhibiting B. cereus. The results from this study provide valuable insights into the chemical constituents present in K. albiflora and their antimicrobial characteristics, offering potential for the research and development into practical applications.
Data availability
The data that support the findings of this study are available in the Supplementary Information. These included 1D- and 2D-NMR, HRESIMS, CD, and IR spectra of compounds 1–14, 1H- and 13C-NMR, and HRESIMS of compounds 15–21, the results of antimicrobial activities of crude extracts and isolated compounds, and Simulated and calculated ECD spectra of compound 13.
References
Techaprasan, J., Klinbunga, S., Ngamriabsakul, C. & Jenjittikul, T. Genetic variation of Kaempferia (Zingiberaceae) in Thailand based on chloroplast DNA (psbA-trnH and petA-psbJ) Sequences. Genet. Mol. Res. 9, 1957–1973. https://doi.org/10.4238/vol9-4gmr873 (2010).
Singh, A. et al. The industrially important genus Kaempferia: An ethnopharmacological review. Front. Pharmacol. https://doi.org/10.3389/fphar.2023.1099523 (2023).
Van Anh, C., Duc, D. X. & Son, N. T. Kaempferia diterpenoids and flavonoids: An overview on phytochemistry, biosynthesis, synthesis, pharmacology, and pharmacokinetics. Med. Chem. Res. https://doi.org/10.1007/s00044-023-03169-w (2023).
Yenjai, C., Prasanphen, K., Daodee, S., Wongpanich, V. & Kittakoop, P. Bioactive flavonoids from Kaempferia parviflora. Fitoterapia 75, 89–92. https://doi.org/10.1016/j.fitote.2003.08.017 (2004).
Thongnest, S., Mahidol, C., Sutthivaiyakit, S. & Ruchirawat, S. Oxygenated pimarane diterpenoids from Kaempferia marginata. J. Nat. Prod. 68, 1632–1636. https://doi.org/10.1021/np050186l (2005).
Boonsombat, J. et al. Roscotanes and roscoranes: Oxygenated abietane and pimarane diterpenoids from Kaempferia roscoeana. Phytochemistry 14, 36–44. https://doi.org/10.1016/j.phytochem.2017.07.008 (2017).
Booranaseensuntorn, P. et al. Diterpenoids an p-methoxycinnamic acid diol ester from Kaempferia saraburiensis Picheans. (Zingiberaceae): Structural assignment of saraburol and their biological activities. Phytochemistry https://doi.org/10.1016/j.phytochem.2022.113181 (2022).
Pancharoen, O., Tuntiwachwuttikul, P. & Taylor, W. C. Cyclohexene oxide derivatives from Kaempferia angustifolia and Kaempferia species. Phytochemistry 28, 1143–1148. https://doi.org/10.1016/0031-9422(89)80198-0 (1989).
Lallo, S., Lee, S., Dibwe, D. F., Tezuka, Y. & Morita, H. A new polyoxygenated cyclohexane and other constituents from Kaempferia rotunda and their cytotoxic activity. Nat. Prod. Res. 28, 1754–1759. https://doi.org/10.1080/14786419.2014.945175 (2014).
Wisetsai, A. et al. Isopimarane-type diterpenoids from the rhizomes of Kaempferia galanga L. and their biological activities. Nat. Prod. Res. 37, 1106–1115. https://doi.org/10.1080/14786419.2021.1989681 (2023).
Win, N. N., Hardianti, B., Ngwe, H., Hayakawa, Y. & Morita, H. Anti-inflammatory activities of isopimara-8(9),15-diene diterpenoids and mode of action of kaempulchraols B-D from Kaempferia pulchra rhizomes. J. Nat Med. https://doi.org/10.1007/s11418-020-01389-7 (2020).
Win, N. N. et al. Anti-inflammatory activities of isopimara-8(14),-15-diene diterpenoids and mode of action of kaempulchraols P and Q from Kaempferia pulchra rhizomes. Bioorg. Med. Chem. Lett. 30, 126841. https://doi.org/10.1016/j.bmcl.2019.126841 (2020).
Chokchaisiri, R. et al. Marginaols G-M, anti-inflammatory isopimarane diterpenoids, from the rhizomes of Kaempferia marginata. Phytochemistry 200, 113225. https://doi.org/10.1016/j.phytochem.2022.113225 (2022).
Jongsomjainuk, O. et al. Kaemtakols A-D, highly oxidized pimarane diterpenoids with potent anti-inflammatory activity from Kaemferia takensis. Nat. Prod. Bioprospect. https://doi.org/10.1007/s13659-023-00420-0 (2023).
Kongwaen, P. et al. Cytotoxic isopimarane diterpenoids from Kaempferia koratensis rhizomes. Rev. Bras. Farmacogn. 33, 415–421. https://doi.org/10.1007/s43450-023-00359-w (2023).
Leong-Škorničková, J. & Newman, M. Kaempferia L. In Gingers of Cambodia (eds Leong-Škorničková, J. & Newman, M.) 203–207 (Singapore Botanic Gardens, 2015).
Kam, Y. K. Taxonomic studies in the genus Kaempferia (Zingiberaceae). Notes RBG Edinb. 38, 1–12 (1980).
Boonma, T., Saensouk, S. & Saensouk, P. Kaempferia sipraiana (Zingiberaceae), a new species from Thailand and a new record of Kaempferia pseudoparviflora for Myanmar. Biodiversitas. 23, 2203–2211. https://doi.org/10.13057/biodiv/d230456 (2022).
Jenjittikul, T. & Ruchisansakun, S. Kaempferia albiflora (Zingiberaceae), a new species from Thailand. Kew. Bull. 75, 13. https://doi.org/10.1007/s12225-020-9868-4 (2020).
Sugimoto, N. et al. Identification of the main constituents in sandarac resin, a natural gum base. J. Food Hygiene Soc. Jpn. 47, 76–79. https://doi.org/10.3358/shokueishi.47.76 (2006).
Liu, X. T. et al. Antibacterial diterpenoids from Sagittaria pygmaea. Planta Med. 73, 84–90. https://doi.org/10.1055/s-2006-951773 (2007).
Singh, J., Dhar, K. L. & Atal, C. K. Studies on the genus Piper-X. Structure of pipoxide. A new cyclohexene epoxide from P. hookeri Linn. Tetrahedron 26, 4403–4406. https://doi.org/10.1016/S0040-4020(01)93087-X (1997).
Carless, H. A. J., Busia, K., Dove, Y. & Malik, S. S. Syntheses of conduritol D derivatives from aromatic compounds. J. Chem. Soc. Perkin Trans. 1, 2505–2506. https://doi.org/10.1002/chin.199406239 (1993).
De Carvalho, M. S., Baptistella, L. H. B. & Imamura, P. M. 13C and 1H NMR signal assignments of some new synthetic dehydroabietic acid derivatives. Magn. Reson. Chem. 46, 381–386. https://doi.org/10.1002/mrc.2187 (2008).
Imhoff, J. E., Sun, M., Wiese, J., Tank, M. & Zeeck, A. First evidence of dehydroabietic acid production by a marine phototrophic gammaproteobacterium, the purple sulfur bacterium Allochromatium vinosum MT86. Mar. Drugs 16, 1–8. https://doi.org/10.3390/md16080270 (2018).
Kroutil, W., Hagmann, L., Schuez, T. C., Jungmann, V. & Pachlatko, J. P. Esterification of streptol—A cyclitol derivative–by Candida rugosa lipase: Influence of the acyl donor on regioselectivity. J. Mol. Catal. B. 32, 247–252. https://doi.org/10.1016/j.molcatb.2004.12.012 (2005).
Kijjoa, A. et al. Polyoxygenated cyclohexene derivatives from Ellipeiopsis cherrevensis. Phytochemistry 59, 543–549. https://doi.org/10.1016/s0031-9422(01)00465-4 (2002).
Stevenson, P., Veitch, N. C. & Simmonds, M. S. Polyoxygenated cyclohexane derivatives and other constituents from Kaempferia rotunda L. Phytochemistry 68, 1579–1586. https://doi.org/10.1016/j.phytochem.2007.03.015 (2007).
Nyandoro, S. S. et al. Polyoxygenated cyclohexenes and other constituents of Cleistochlamys kirkii leaves. J. Nat. Prod. 80, 114–125. https://doi.org/10.1021/acs.jnatprod.6b00759 (2017).
Kupchan, S. M., Hemingway, R. J. & Smith, R. M. Tumor inhibitors. XLV. Crotepoxide, a novel cyclohexane diepoxide tumor inhibitor from Croton macrostachys. J. Org. Chem. 34, 3898–3902. https://doi.org/10.1021/jo01264a033 (1969).
González, M. A., Pérez-Guaita, D., Correa-Royero, J. & Zapata, B. Synthesis and biological evaluation of dehydroabietic acid derivatives. Eur. J. Med. Chem. 45, 811–816. https://doi.org/10.1016/j.ejmech.2009.10.010 (2010).
Lee, C. K., Fang, J. M. & Cheng, Y. S. Norditerpenes from Juniperus chinensis. Phytochemistry 39, 391–394. https://doi.org/10.1016/0031-9422(94)00868-T (1995).
Ulubelen, A. & Topcu, G. Abietane diterpenoids from Salvia pomifera. Phytochemistry 31, 3949–3951. https://doi.org/10.1016/S0031-9422(00)97560-5 (1992).
Tuntiwachwuttikul, P. et al. Constituents of the Zingiberaceae. XI Structues of (+)-(1R,2S,3R,4S)-2-benzoyloxy-methylcyclohex-5-ene-1,2,3,4-tetrol 4-benzoate [(+)-zeylenol] and (+)-(1R,2R,4R,5S,6R,7R)-4-benzoyloxymethyl-3,8-dioxatricyclo-[5.1.0.02,4]octane-5,6-diol 5-acetate 6-benzoate (Boesenboxide) isolated from a new Boesenbergia species. Aust. J. Chem. 40, 2049–2061. https://doi.org/10.1071/CH9872049 (1987).
Kodpinid, M., Sadavongvivad, C., Thebtaranonth, C. & Thebtaranonth, Y. Structures of β-senepoxide, tingtanoxide, and their diene precursors. Constituents of Uvaria ferruginea. Tetrahedron. Lett. 24, 2019–2022. https://doi.org/10.1016/S0040-4039(00)81832-8 (1983).
Nguyen, N. L. et al. Bioassay-guided isolation and HPLC quantification of antiproliferative metabolites from Stahlianthus thorelii. Molecules 25, 1–14. https://doi.org/10.3390/molecules25030551 (2020).
Dupont, M. P., Llabrés, G., Delaude, C., Tchissambou, L. & Gastmans, J. P. Sterolic and triterpenoidic constituents of stem bark of Drypetes gossweileri. Planta Med. 63, 282–284. https://doi.org/10.1055/s-2006-957678 (1997).
Radhakrishnan, N., Gnanamani, A. & Mandal, A. B. A potential antibacterial agent Embelin, a natural benzoquinone extracted from Embelia ribes. Biol. Med. 3, 1–7 (2011).
Frisch, M. J. et al. Gaussian 16, Revision C.01, (Gaussian, Inc., 2016).
Dennington, R., Keith Todd, A. & Millam John, M. GaussView, Version 6.1. (Semichem Inc., 2016).
Zhao, Y. & Truhlar, D. G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 120, 215–241. https://doi.org/10.1007/s00214-007-0310-x (2008).
Weigend, F. Accurate Coulomb-fitting basis sets for H to Rn. Phys. Chem. Chem. Phys. 8, 1057–1065. https://doi.org/10.1039/B515623H (2006).
Weigend, F. & Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 7, 3297–3305. https://doi.org/10.1039/B508541A (2005).
Bruhn, T., Schaumlöffel, A., Hemberger, Y. & Bringmann, G. SpecDis: Quantifying the comparison of calculated and experimental electronic circular dichroisom spectra. Chirality 25, 243–249 (2013).
Bruhn, T., Schaumlöffel, A., Hemberger, Y. & Pescitelli, G. SpecDis—A tool to compare calculated and experimental (chir) optical spectra, http://specdis-software.jimdo.com (2017).
Legault, C. Y. CYLview20 (Université de Sherbrooke, 2020). http://www.cylview.org.
M100 Performance Standards for Antimicrobial Susceptibility Testing, 32th Edition (Clinical and Laboratory Standards Institute, 2022).
Acknowledgements
This work was in part supported by the Thailand Science Research and Innovation (TSRI), Chulabhorn Research Institute (Grant No. 48296/4691995), and Center of Excellence on Environmental Health and Toxicology (EHT), OPS, Ministry of Higher Education, Science, Research and Innovation. Pornpuk Booranaseensuntorn acknowledges a grant from the Chulabhorn Graduate Scholarship Commemorating the 84th Birthday Anniversary of His Majesty King Bhumibol Adulyadej the Great.
Author information
Authors and Affiliations
Contributions
P.Booranaseensuntorn: compound isolation and purification; J.B.: project planning, coordination, data analysis, and manuscript preparation; S.Thongnest: project planning, guidance, compound identification, data analysis, and manuscript preparation; N.R. and P.Khlaychan: antimicrobial activity assessment; P.B.: ECD work; S.Ruchisansakul: plant collection and identification; P.K. and S.T.: review and editing; C.M. and S.R.: project supervision. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interest.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Booranaseensuntorn, P., Boonsombat, J., Thongnest, S. et al. Albiflorenes A–L, polyoxygenated cyclohex(a/e)ne diterpene esters from Kaempferia albiflora. Sci Rep 14, 13967 (2024). https://doi.org/10.1038/s41598-024-64889-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-64889-6
- Springer Nature Limited