
Abstract
Although there is an association between abdominal aortic aneurysm (AAA) and circulating immune cell phenotypes, the exact causal relationship remains unclear. This study aimed to explore the causal relationships between immune cell phenotypes and AAA risk using a bidirectional two-sample Mendelian randomization approach. Data from genome-wide association studies pertaining to 731 immune cell traits and AAA were systematically analyzed. Using strict selection criteria, we identified 339 immune traits that are associated with at least 3 single nucleotide polymorphisms. A comprehensive MR analysis was conducted using several methods including Inverse Variance Weighted, Weighted Median Estimator, MR-Egger regression, Weighted Mode, and Simple Median methods. CD24 on switched memory cells (OR = 0.922, 95% CI 0.914–0.929, P = 2.62e−79) at the median fluorescence intensities level, and SSC-A on HLA-DR + natural killer cells (OR = 0.873, 95% CI 0.861–0.885, P = 8.96e−81) at the morphological parameter level, exhibited the strongest causal associations with AAA. In the reverse analysis, no significant causal effects of AAA on immune traits were found. The study elucidates the causal involvement of multiple circulating immune cell phenotypes in AAA development, signifying their potential as diagnostic markers or therapeutic targets. These identified immune traits may be crucial in modulating AAA-related inflammatory pathways.
Similar content being viewed by others


Introduction
Abdominal aortic aneurysms (AAA) are characterized by localized, permanent dilation of the abdominal aorta exceeding 50% of normal diameter or 3 cm in width1,2. This chronic degenerative condition results from progressive structural deterioration and weakening of the aortic wall. Known risk factors for AAA include advanced age, male sex, white race, smoking, dyslipidemia, and atherosclerosis3,4. If AAA dilation remains undetected and untreated, continued expansion can eventually precipitate life-threatening aortic rupture associated with high mortality. AAA affects roughly 2–5% of men and 0.5–1.5% of women over 60 years old in developed nations, representing a major source of cardiovascular morbidity and mortality5.
The pathogenesis of AAA is driven by chronic inflammation, extracellular matrix proteolysis, and smooth muscle cell apoptosis within the aortic wall2,3. AAA tissues exhibit infiltration of various leukocytes including macrophages, T cells, B cells, and mast cells6,7,8,9. Based on this evidence, the circulating immune cells are suspected to play a central role in AAA pathogenesis. However, the causal influence of systemic immune cell populations on AAA risk remains uncertain. While numerous observational studies have reported associations between circulating immune cells and presence, progression, or complications of AAA, residual confounding and reverse causation limit robust causal inference10,11,12,13.
Mendelian randomization (MR) leverages genetic variants as instrumental variables to strengthen causal conclusions from observational data. Since alleles are randomly allocated during meiosis independently of confounders, MR studies are less prone to confounding biases14,15. Additionally, the unidirectional flow of information from germline DNA sequence to downstream molecular traits minimizes reverse causation. Here, we performed bidirectional MR analyses to definitively investigate for causal relationships between circulating immune cell phenotypes and AAA risk. Elucidating the causal role of peripheral immune cells and inflammatory pathways in AAA may uncover potential drug targets or biomarkers to guide therapeutic monitoring and early intervention.
Methods
Study design and the assumption of MR
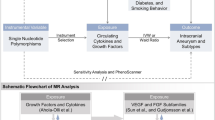
We utilized publicly available large-scale Genome-Wide Association Study (GWAS) data to conduct a two-sample MR analysis. This enabled us to explore the genetic associations circulating immune cell phenotypes and AAA. Concurrently, we reversed the exposure and outcome variables to perform a complementary inverse MR analysis, adding robustness to our findings. The schematic flow of our study methodology is graphically depicted in Fig. 1.

Schematic Representation of the Mendelian Randomization (MR) Analysis. The flowchart of the study based on three assumptions: (1) the instrumental variables (IVs) selected from datasets were associated with exposure; (2) the IVs were not associated with confounders; and (3) the IVs influences the outcome only through exposure. AAA, Abdominal Aortic Aneurysm, SNP single nucleotide polymorphism.
Data sources
To provide a more robust and comprehensive understanding of the causal links between immune indicators and AAA, we leveraged the largest GWAS available for immunophenotyping of peripheral blood. This GWAS comprehensively characterized 731 distinct immune cell phenotypes in a cohort of 3757 individuals from Sardinia16. The study captured a wide array of immune cell types, providing detailed measurements that included 118 absolute cell (AC) counts, 389 median fluorescence intensities (MFIs) reflecting surface antigen expression levels, 32 morphological parameters (MPs) (captured through forward scatter (FSC) and side scatter analyses (SSC), proportional to cell size and complexity), and 192 relative cell (RC) counts. Approximately 22 million Single Nucleotide Polymorphisms (SNPs) were genotyped using high-density arrays or were imputed based on a Sardinian-specific reference panel. All analyses were rigorously adjusted for sex and age confounding variables. The summary statistics for each of the 731 phenotypic GWAS outcomes are publicly accessible through the GWAS Catalog (accession numbers GCST0001391 to GCST0002121).
GWAS summary statistics for AAA were sourced from a comprehensive meta-analysis of 17 individual GWAS spanning 14 distinct discovery cohorts, as reported in Roychowdhury et al.17. This all-encompassing meta-analysis, the most extensive of its kind to date, incorporated data from 39,221 AAA cases—37,214 of European descent and 2007 of African descent—and 1,086,107 controls derived from diverse population-based studies and biobanks. Prominent contributors to the dataset include illustrious studies like the Million Veteran Program, UK Biobank, and deCODE genetics, among others (a detailed breakdown is provided in Supplemental Table 1). Genotyping in the discovery cohorts was performed using various high-density SNP arrays, and genotype imputation was carried out using population-specific reference panels. After quality control and meta-analysis, summary statistics for 33.4 million SNPs were generated. These data have been made publicly available (https://csg.sph.umich.edu/willer/public/AAAgen2023/).
Instrumental variable selection
Consistent with prior MR analyses, we adopted the same cut-off value of P < 5 × 10–8 to identify significant SNPs for each immune trait with the same criteria in our MR analysis18. When conducting a reverse MR analysis with AAA as the exposure, we set the same threshold for selecting SNPs.
The following three assumptions were satisfied for the two sample MR: (1) the instrumental variables (IVs) selected from datasets were associated with exposure. (2) the IVs were not associated with confounders. (3) the IVs influences the outcome only through exposure14. Second, the clump program in PLINK software was adopted to exclude the dependent instrumental variable of r2 < 0.001 (clumping window size = 10,000 kb), which was obtained using the 1000 Genome Projects reference panel in Europe19. Third, SNPs with minor allele frequency (MAF) < 0.01 were removed. Fourth, an important step in MR is to ensure that the effects of the SNPs on the exposure correspond to the same allele as the effects on the outcome. To avoid distortion of strand orientation or allele coding, we deleted palindromic SNPs (e.g. with A/T or G/C alleles).
The F-statistic of the IVs was calculated to detect whether or not there was a weak IV bias, an F-value less than 10 indicated a weak instrument and was excluded20. Through the PhenoScanner website (http://www.phenoscanner.medschl.cam.ac.uk) and the GWAS catalog (https://www.ebi.ac.uk/gwas/), we removed SNPs associated with "smoke", "high density lipoprotein", "low density lipoprotein", "Triglycerides", "total cholesterol", "apolipoprotein A1", "apolipoprotein B", "Hypertension", "Coronary artery disease", in order to limit the influence of confounding factors21. Then we utilized the remaining IVs for MR analysis. We further only included immune trait with at least three SNPs as IVs in downstream analyses.
Statistical analysis for MR
We conducted bidirectional MR analysis to test both whether immune cell traits causally influence AAA and vice versa. Independent SNPs associated with each immune trait and AAA at genome-wide significance were used as instrumental variables. To test for causal effects of immune cells on AAA, we applied inverse variance weighted (IVW)22,23, weighted median (WME)24, weighted mode (WMo)25, MR-Egger26, and Simple Mode (SM) methods. We evaluated reverse direction effects of AAA on immune cells similarly. Multiple testing was adjusted using the FDR approach.
Heterogeneity and pleiotropy analyses
We assessed heterogeneity through the application of Cochran's Q statistics, considering a P < 0.05 as indicative of significant heterogeneity27. To evaluate the direct relationship between the chosen IVs and the outcome, horizontal pleiotropy was tested using MR-Egger regression and MR-PRESSO global tests28,29. Any outliers of significance identified via the MR-PRESSO analysis were excluded to minimize the impact of horizontal pleiotropy. Additionally, we conducted a leave-one-out analysis to validate the robustness of the conclusions27.
Analysis software
All statistical analyses were conducted using R version 4.2.3 (R Foundation for Statistical Computing, Vienna, Austria, https://cran.r-project.org/src/base/R-4/R-4.2.3.tar.gz). For MR analyses, we utilized the TwosampleMR (Version 0.5.7, https://github.com/MRCIEU/TwoSampleMR) and MR-PRESSO (Version 1.0, https://github.com/rondolab/MR-PRESSO)29 R packages.
Results
Exploration of the causal effect of immunophenotypes on AAA
In our two-sample MR study, we explored a comprehensive array of 731 circulating immune cell traits to discern their potential causal relationship with AAA. A stringent series of selection criteria narrowed this to 339 immune traits associated with at least three SNPs, as detailed in Supplemental Table 2.
Through the application of the IVW approach in MR, we identified 46 immune traits significantly associated with AAA risk. Importantly, 34 of these traits had consistent odds ratio (OR) directions across all employed methodologies—including IVW, WME, MR-Egger regression, WMo, and SM methods. After adjusting for multiple comparisons using the False Discovery Rate (FDR) method and performing additional sensitivity analyses, 16 immune trait-AAA pairs remained statistically significant (FDR-adjusted P < 0.05). These pairs included 8 MFIs, 5 ACs, 1 RCs, and 2 MPs, details in Supplemental Table 3 and Figs. 2, 3.

Causal Effects of immunophenotypes on Abdominal Aortic Aneurysm. IVW inverse variance weighted, WMe weighted median, MFI median fluorescence intensitie.

Scatter plots of the effect of the immunophenotypes on Abdominal Aortic Aneurysm.
For traits measured at the Absolute Count level, we found strong evidence for causal relationships with AAA risk. Specifically, CD86 + myeloid DC AC (OR = 1.03, 95% confidence interval (CI) 1.021–1.039, P = 2.04e − 09) and CD20- CD38- AC (OR = 1.072, 95% CI 1.036–1.109, P = 2.28e − 03) demonstrated a significant positive causal effect on AAA risk. On the other hand, Naive DN (CD4-CD8-) AC (OR = 0.983, 95% CI 0.974–0.992, P = 1.05e − 02), CD14- CD16- AC (OR = 0.985, 95% CI 0.976–0.993, P = 1.05e − 02), and CD62L- CD86 + myeloid DC AC (OR = 0.982, 95% CI 0.972–0.992, P = 1.05e − 02) showed a significant negative causal effect on AAA risk.
When scrutinizing traits gauged through Mean Fluorescence Intensities, our MR analyses revealed significant causal relationships with AAA risk. CD24 on sw mem (OR = 0.922, 95% CI 0.914–0.929, P = 2.62e − 79) and CCR2 on monocyte (OR = 0.978, 95% CI 0.971–0.986, P = 1.99e − 06) demonstrated a significant negative causal effect on AAA risk. Similarly, CD16-CD56 on NKT (OR = 0.978, 95% CI 0.969–0.986, P = 2.35e − 05), CD62L on monocyte (OR = 0.981, 95% CI 0.971–0.990, P = 2.67e − 03), CD127 on CD28- CD8br (OR = 0.976, 95% CI 0.962–0.990, P = 1.32e − 02), CD25 on CD39 + activated Treg (OR = 0.973, 95% CI 0.956–0.989, P = 2.59e − 02), and CD25 on activated Treg (OR = 0.991, 95% CI 0.985–0.997, P = 3.45e − 02) also indicated a significant negative causal impact on AAA risk. In contrast, CD45 on CD8br (OR = 1.047, 95% CI 1.037–1.056, P = 1.80e − 21) showed a significant positive causal effect on AAA risk.
In terms of Morphological Parameters, SSC-A on HLA DR + NK (OR = 0.873, 95% CI 0.861–0.885, P = 8.96e − 81) and SSC-A on HLA DR + T cell (OR = 0.982, 95% CI 0.973–0.992, P = 1.07e − 02) indicated a significant negative causal effect on AAA risk.
Regarding traits characterized by Relative Counts, IgD + CD38br %lymphocyte (OR = 0.964, 95% CI 0.944–0.984, P = 1.15e−02) exhibited a significant negative causal impact on AAA risk.
In the MR-Egger regression analysis, four immune cell phenotypes demonstrated horizontal pleiotropy and were consequently excluded. Specifically, these included: Absolute count of CD20- CD38- AC with a P-value of 0.039, Mean Fluorescence Intensity (MFI) of CCR2 on monocytes with a P-value of 0.034, MFI of CD127 on CD28- CD8br with a P-value of 0.025, and MFI of CD25 on activated Treg with a P-value of 0. In addition, evidence of heterogeneity was uncovered by both Cochran's Q statistics and the MR-PRESSO global test. Consequently, we relied on the results from the IVW random effects model as our final outcomes. Notably, these findings remained robust after outlier correction, except for MFI of CCR2 on monocytes (Outlier-Corrected P = 0.138), as detailed in Supplemental Table 4. Moreover, a leave-one-out sensitivity analysis confirmed that no individual SNP disproportionately influenced the causal estimates associated with AAA, as illustrated in Fig. 4. Ultimately, we identified 12 immune cell phenotypes exerting a causal effect on AAA risk. Most notably, CD24 on switched memory cells (OR = 0.922, 95% CI 0.914–0.929, P = 2.62e−79) at the MFI level, and SSC-A on HLA-DR + natural killer cells (OR = 0.873, 95% CI 0.861–0.885, P = 8.96e−81) at the morphological parameter level, exhibited the strongest causal associations with AAA.

Leave-one-out plots of the effect of immunophenotypes on Abdominal Aortic Aneurysm.
Exploration of the causal effect of AAA onset on immunophenotypes
In our comprehensive analysis focused on the causal impact of AAA onset on 731 immunophenotypes, we employed the IVW method. This revealed that AAA was significantly associated with 18 immunophenotypes. Notably, 10 of these immunophenotypes displayed consistent OR directions across all employed analytical techniques. However, after adjusting for multiple comparisons via the FDR methodology and conducting additional sensitivity tests, AAA was found to have no significant causal effect on any of the investigated immunophenotypes (Supplemental Tables 5, 6).
Discussion
Our bidirectional MR study offers a comprehensive analysis of the causal links between circulating immune cell phenotypes and the development of AAA. Notably, 10 immune traits remained statistically significant (FDR-adjusted P < 0.05) after rigorous statistical adjustments, offering potential therapeutic targets. However, our results do not suggest a significant causal effect of AAA onset on any of the investigated immune traits. Herein, we categorize our discussion of the Mechanisms of Association into four parts according to immune phenotype types. It's important to note that the potential mechanisms discussed below are based on hypotheses from previous literature and require further studies for validation.
Absolute count
Higher counts of CD86 + myeloid dendritic cells were associated with increased AAA risk. CD86 is an immune co-stimulatory molecule predominantly expressed on antigen-presenting cells like dendritic cells and macrophages. It serves as a crucial facilitator for both innate and adaptive immune responses30,31. Elevated levels of circulating CD86 + myeloid DCs may potentiate the risk of AAA by amplifying proinflammatory T-cell activities.
CD86 interacts with CD28 on naïve T cells, providing the necessary co-stimulatory signals for T-cell activation32. This interaction effectively lowers the threshold for T-cell receptor-mediated responses. Once activated, T cells may contribute to the inflammatory cascade by releasing metalloproteinases (MMPs) which degrade the aortic wall's extracellular matrix30,33.
Relative count
IgD + CD38br B cells Relative count displayed inverse association with AAA risk. IgD + CD38br B cells represent an activated B cell subset that can rapidly secrete IgM antibodies and participate in early immune responses34,35. Elevated levels of these activated B cells have been reported in some autoimmune diseases36. However, their specific role in AAA pathophysiology remains unclear and warrants further investigation.
Mean fluorescence intensities
Our MR result indicates a negative association between CD24 and AAA risk, suggesting a potential protective role. As a cell adhesion molecule, CD24 is ubiquitously expressed but shows high prevalence in switched memory B cells. This negative correlation could potentially be linked to two primary mechanisms. First, reduced levels of CD24 are likely to limit leukocyte migration to the aortic wall, consequently mitigating the local inflammatory response, a well-established driver in AAA pathogenesis. Second, CD24 serves as an inhibitory modulator of immune responses through its interaction with specific siglec receptors37,38. These phosphatases inhibit downstream signaling pathways that would otherwise facilitate B cell activation and autoantibody production, processes known to exacerbate AAA39. Previous research corroborates CD24's immune inhibitory functions, showing that its deficiency augments humoral immune responses and raises susceptibility to autoimmune diseases40. Thus, elevated CD24 expression, particularly on switched memory B cells, could potentially serve as a protective mechanism by suppressing pro-inflammatory immune activities. These insights collectively contribute to the understanding that AAA development is modulated by immune-mediated mechanisms, and they also underscore the potential role of CD24 as a biomarker or therapeutic target in AAA.
In addition, higher expression of CD45 on CD8 bright T cells was causally associated with increased AAA risk. The CD45 antigen is a protein tyrosine phosphatase involved in regulation of T cell receptor signaling. The tyrosine phosphatase activity of CD45 is integral for the regulation of multiple signaling pathways, not only those associated with the T cell receptor but also with integrin and cytokine receptors41,42. This multi-faceted role suggests that elevated levels of CD45 could potentially have a broader impact, affecting various immune cells that are involved in the inflammatory milieu characteristic of AAA. Elevated CD45 levels may promote autoreactive T cell responses that drive AAA development. Further research is required to elucidate the precise mechanisms by which CD45 influences AAA risk.
Morphological parameter
Our findings demonstrate that genetically predicted elevations in SSC-A on HLA-DR + NK cells and HLA-DR + T cells correspond to a reduced risk of AAA, highlighting the potential protective role of these cells.
HLA-DR is a Major Histocompatibility Complex (MHC) class II molecule that plays an integral role in antigen presentation and activation of CD4 + T cells. HLA-DR is constitutively expressed on professional antigen presenting cells like dendritic cells and B cells. However, HLA-DR can also be inducibly expressed on other cell types including NK cells and CD4 + T cells upon activation43,44,45. Recent studies have shown that a subset of NK cells expand and express HLA-DR during cytomegalovirus (CMV) infection. These HLA-DR + NK cells demonstrate enhanced cytokine production and ability to present antigens to CD4 + T cells compared to conventional NK cells46,47. Additionally, HLA-DR + NK cells have been found to process and present antigens through the HLA-DR pathway, suggesting they may help regulate antigen-specific CD4 + T cell responses47.
This association between HLA-DR expression on NK cells and a reduced AAA risk indicates that a robust presence or activity of this specific NK subset might regulate CD4 + T cell responses against AAA-associated antigens more effectively. This could potentially lead to the amplified activation and proliferation of CD4 + T cells, potentially mitigating the immune-mediated damage seen in AAA progression. Additionally, CD4 + T cells, when activated, can express HLA-DR, intensifying immune responses through direct antigen presentation. The increased expression of HLA-DR on these cells could be instrumental in modulating immune reactions related to AAA.
Our MR study provides evidence that multiple circulating immune cell phenotypes are causally implicated in AAA pathogenesis. These findings have a twofold clinical relevance. Firstly, the immune traits identified here may serve as novel therapeutic targets. Secondly, these causally associated immune traits could be examined as potential biomarkers for AAA.
Study limitations
Our study has some limitations. Firstly, the majority of patients included in the GWAS summary data used in our research were of European descent. This discrepancy could potentially introduce biases in the estimates, thereby affecting the universal applicability of our findings. The results of this study may not be extrapolated to other racial or ethnic groups. Secondly, while our study identifies certain immune cell phenotypes as potential causal factors in AAA, the specific mechanisms by which they influence AAA risk remain unclear. Further experimental studies focusing on individual immune cell phenotypes are required to elucidate these mechanisms. Thirdly, we utilized publicly available GWAS summary statistics, which precluded conducting individual level analyses. We were unable to explore gene-environment interactions or subgroup effects. Finally, although we performed sensitivity analyses to assess and mitigate potential pleiotropy, we cannot completely rule out residual pleiotropy that may bias the causal estimates.
Conclusions
Our MR study identifies multiple circulating immune cell phenotypes as playing causal roles in AAA development. Elucidating causal immune pathways provides vital clues to guide biomarker discovery, risk stratification, and immunomodulatory treatment innovations to improve AAA prevention and management.
Data availability
The Abdominal Aortic Aneurysm summary statistics of GWAS used in current study is publicly available (https://csg.sph.umich.edu/willer/public/AAAgen2023/). The summary statistics for each of the 731 phenotypic GWAS outcomes are publicly accessible through the GWAS Catalog (accession numbers GCST0001391 to GCST0002121).
References
Schanzer, A. & Oderich, G. S. Management of abdominal aortic aneurysms. N. Engl. J. Med. 385, 1690–1698 (2021).
Sakalihasan, N. et al. Abdominal aortic aneurysms. Nat. Rev. Dis. Primers 4, 34 (2018).
Nordon, I. M., Hinchliffe, R. J., Loftus, I. M. & Thompson, M. M. Pathophysiology and epidemiology of abdominal aortic aneurysms. Nat. Rev. Cardiol. 8, 92–102 (2011).
Forsdahl, S. H., Singh, K., Solberg, S. & Jacobsen, B. K. Risk factors for abdominal aortic aneurysms: A 7-year prospective study: The Tromsø study, 1994–2001. Circulation 119, 2202–2208 (2009).
Davis, F. M., Daugherty, A. & Lu, H. S. Updates of recent aortic aneurysm research. Arterioscler. Thromb. Vasc. Biol. 39, e83–e90 (2019).
Gong, W., Tian, Y. & Li, L. T cells in abdominal aortic aneurysm: Immunomodulation and clinical application. Front. Immunol. 14, 1240132 (2023).
Cheng, S. et al. Identification of key monocytes/macrophages related gene set of the early-stage abdominal aortic aneurysm by integrated bioinformatics analysis and experimental validation. Front. Cardiovasc. Med. 9, 950961 (2022).
Sun, P. et al. Immune checkpoint programmed death-1 mediates abdominal aortic aneurysm and pseudoaneurysm progression. Biomed. Pharmacother. 142, 111955 (2021).
Márquez-Sánchez, A. C. & Koltsova, E. K. Immune and inflammatory mechanisms of abdominal aortic aneurysm. Front. Immunol. 13, 989933 (2022).
Tang, W. et al. The association of biomarkers of inflammation and extracellular matrix degradation with the risk of abdominal aortic aneurysm: The ARIC study. Angiology 70, 130–140 (2019).
Nahrendorf, M. et al. Detection of macrophages in aortic aneurysms by nanoparticle positron emission tomography-computed tomography. Arterioscler. Thromb. Vasc. Biol. 31, 750–757 (2011).
Yan, H. et al. Neutrophil proteases promote experimental abdominal aortic aneurysm via extracellular trap release and plasmacytoid dendritic cell activation. Arterioscler. Thromb. Vasc. Biol. 36, 1660–1669 (2016).
Duftner, C. et al. High prevalence of circulating cd4+cd28- t-cells in patients with small abdominal aortic aneurysms. Arterioscler. Thromb. Vasc. Biol. 25, 1347–1352 (2005).
Emdin, C. A., Khera, A. V. & Kathiresan, S. Mendelian randomization. JAMA 318, 1925–1926 (2017).
Smith, G. D. & Ebrahim, S. Mendelian randomization: Can genetic epidemiology contribute to understanding environmental determinants of disease?. Int. J. Epidemiol. 32, 1–22 (2003).
Orrù, V. et al. Complex genetic signatures in immune cells underlie autoimmunity and inform therapy. Nat. Genet. 52, 1036–1045 (2020).
Roychowdhury, T. et al. Genome-wide association meta-analysis identifies risk loci for abdominal aortic aneurysm and highlights pcsk9 as a therapeutic target. Nat. Genet. 55, 1831–42 (2023).
Morris, D. R. et al. Genetic predisposition to diabetes and abdominal aortic aneurysm: A two stage mendelian randomisation study. Eur. J. Vasc. Endovasc. Surg. 63, 512–519 (2022).
Purcell, S. et al. Plink: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575 (2007).
Burgess, S. & Thompson, S. G. Avoiding bias from weak instruments in Mendelian randomization studies. Int. J. Epidemiol. 40, 755–764 (2011).
Kamat, M. A. et al. Phenoscanner v2: An expanded tool for searching human genotype-phenotype associations. Bioinformatics 35, 4851–4853 (2019).
Bowden, J. et al. A Framework for the investigation of pleiotropy in two-sample summary data mendelian randomization. Stat. Med. 36, 1783–1802 (2017).
Burgess, S., Bowden, J., Fall, T., Ingelsson, E. & Thompson, S. G. Sensitivity analyses for robust causal inference from mendelian randomization analyses with multiple genetic variants. Epidemiology 28, 30–42 (2017).
Bowden, J., Davey, S. G., Haycock, P. C. & Burgess, S. Consistent estimation in mendelian randomization with some invalid instruments using a weighted median estimator. Genet. Epidemiol. 40, 304–314 (2016).
Hartwig, F. P., Davey, S. G. & Bowden, J. Robust inference in summary data mendelian randomization via the zero modal pleiotropy assumption. Int. J. Epidemiol. 46, 1985–1998 (2017).
Bowden, J., Davey, S. G. & Burgess, S. Mendelian randomization with invalid instruments: effect estimation and bias detection through egger regression. Int. J. Epidemiol. 44, 512–525 (2015).
Bowden, J. & Holmes, M. V. Meta-analysis and mendelian randomization: A review. Res. Synth. Methods 10, 486–496 (2019).
Morrison, J., Knoblauch, N., Marcus, J. H., Stephens, M. & He, X. Mendelian randomization accounting for correlated and uncorrelated pleiotropic effects using genome-wide summary statistics. Nat. Genet. 52, 740–747 (2020).
Verbanck, M., Chen, C. Y., Neale, B. & Do, R. Detection of widespread horizontal pleiotropy in causal relationships inferred from mendelian randomization between complex traits and diseases. Nat. Genet. 50, 693–698 (2018).
Frohwein, T. A., Sonnenburg, A., Zuberbier, T., Stahlmann, R. & Schreiner, M. Unsaturated compounds induce up-regulation of cd86 on dendritic cells in the in vitro sensitization assay lcsa. Arch. Toxicol. 90, 927–936 (2016).
Zhang, Q. et al. Cd86 is associated with immune infiltration and immunotherapy signatures in aml and promotes its progression. J. Oncol. 2023, 9988405 (2023).
Sakthivel, P., Shively, V., Kakoulidou, M., Pearce, W. & Lefvert, A. K. The soluble forms of cd28, cd86 and ctla-4 constitute possible immunological markers in patients with abdominal aortic aneurysm. J. Intern. Med. 261, 399–407 (2007).
Zhang, X. L. et al. Imbalance between cd205 and cd80/cd86 in dendritic cells in patients with immune thrombocytopenia. Thromb. Res. 135, 352–361 (2015).
Tangye, S. G., Avery, D. T., Deenick, E. K. & Hodgkin, P. D. Intrinsic differences in the proliferation of naive and memory human b cells as a mechanism for enhanced secondary immune responses. J. Immunol. 170, 686–694 (2003).
He, J. et al. Circulating precursor ccr7(lo)pd-1(hi) cxcr5+ cd4+ t cells indicate tfh cell activity and promote antibody responses upon antigen reexposure. Immunity 39, 770–781 (2013).
Chini, C. et al. Cd38 ecto-enzyme in immune cells is induced during aging and regulates nad(+) and nmn levels. Nat. Metab. 2, 1284–1304 (2020).
Wang, X. et al. Cd24-siglec axis is an innate immune checkpoint against metaflammation and metabolic disorder. Cell Metab. 34, 1088–1103 (2022).
Barkal, A. A. et al. Cd24 signalling through macrophage siglec-10 is a target for cancer immunotherapy. Nature 572, 392–396 (2019).
Chen, G. Y., Tang, J., Zheng, P. & Liu, Y. Cd24 and siglec-10 selectively repress tissue damage-induced immune responses. Science 323, 1722–1725 (2009).
Duan, S. & Paulson, J. C. Siglecs as immune cell checkpoints in disease. Annu. Rev. Immunol. 38, 365–395 (2020).
Rheinländer, A., Schraven, B. & Bommhardt, U. Cd45 in human physiology and clinical medicine. Immunol. Lett. 196, 22–32 (2018).
Hermiston, M. L., Xu, Z. & Weiss, A. Cd45: A critical regulator of signaling thresholds in immune cells. Annu. Rev. Immunol. 21, 107–137 (2003).
Baecher-Allan, C., Wolf, E. & Hafler, D. A. Mhc class ii expression identifies functionally distinct human regulatory t cells. J. Immunol. 176, 4622–4631 (2006).
Sedlmayr, P. et al. Differential phenotypic properties of human peripheral blood cd56dim+ and cd56bright+ natural killer cell subpopulations. Int. Arch. Allergy. Immunol. 110, 308–313 (1996).
Fogli, M. et al. Significant nk cell activation associated with decreased cytolytic function in peripheral blood of hiv-1-infected patients. Eur. J. Immunol. 34, 2313–2321 (2004).
Fei, F., Rong, L., Jiang, N., Wayne, A. S. & Xie, J. Targeting hla-dr loss in hematologic malignancies with an inhibitory chimeric antigen receptor. Mol. Ther. 30, 1215–1226 (2022).
Costa-García, M. et al. Human cytomegalovirus antigen presentation by hla-dr+ nkg2c+ adaptive nk cells specifically activates polyfunctional effector memory cd4+ t lymphocytes. Front. Immunol. 10, 687 (2019).
Acknowledgements
We extend our gratitude to the committed participants, the researchers who generously shared their insights, the GWAS data platform, and the prominent research consortia, all of which have paved the way for further explorations and discoveries in subsequent studies.
Author information
Authors and Affiliations
Contributions
K.L., W.R. and X.Z. conceptualised and designed the study. W.R., X.Z., T.W., H.L., G.Z. and J.S. collectioned and analyzed the data.W.R. and X.Z. wrote the first draft of the manuscript. K.L., W.R. and X.Z. critically revised the draft. All authors contributed to data interpretation, critical revisions, and final approval of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ruan, W., Zhou, X., Wang, T. et al. Assessing the causal relationship between circulating immune cells and abdominal aortic aneurysm by bi-directional Mendelian randomization analysis. Sci Rep 14, 13733 (2024). https://doi.org/10.1038/s41598-024-64789-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-64789-9
- Springer Nature Limited