
Abstract
Chronic obstructive pulmonary disease (COPD) is often associated with lung squamous cell carcinoma (LUSC), which has the same etiology (smoking, inflammation, oxidative stress, microenvironmental changes, and genetics). Smoking, inflammation, and airway remodeling are the most important and classical mechanisms of COPD comorbidity in LUSC patients. Cancer can occur during repeated airway damage and repair (airway remodeling). Changes in the inflammatory and immune microenvironments, which can cause malignant transformation of some cells, are currently being revealed in both LUSC and COPD patients. We obtained the GSE76925 dataset from the Gene Expression Omnibus database. Screening for possible COPD biomarkers was performed using the LASSO regression model and a random forest classifier. The compositional patterns of the immune cell fraction in COPD patients were determined using CIBERSORT. HTR2B expression was analyzed using validation datasets (GSE47460, GSE106986, and GSE1650). HTR2B expression in COPD cell models was determined via real-time quantitative PCR. Epithelial–mesenchymal transition (EMT) marker expression levels were determined after knocking down or overexpressing HTR2B. HTR2B function and mechanism in LUSC were analyzed with the Kaplan‒Meier plotter database. HTR2B expression was inhibited to detect changes in LUSC cell proliferation. A total of 1082 differentially expressed genes (DEGs) were identified in the GSE76925 dataset (371 genes were significantly upregulated, and 711 genes were significantly downregulated). Kyoto Encyclopedia of Genes and Genomes pathway enrichment analysis indicated that the DEGs were mainly enriched in the p53 signaling and β-alanine metabolism pathways. Gene Ontology enrichment analysis indicated that the DEGs were largely related to transcription initiation from the RNA polymerase I promoter and to the regulation of mononuclear cell proliferation. The LASSO regression model and random forest classifier results revealed that HTR2B, DPYS, FRY, and CD19 were key COPD genes. Immune cell infiltration analysis indicated that these genes were closely associated with immune cells. Analysis of the validation sets suggested that HTR2B was upregulated in COPD patients. HTR2B was significantly upregulated in COPD cell models, and its upregulation was associated with increased EMT marker expression. Compared with that in bronchial epithelial cells, HTR2B expression was upregulated in LUSC cells, and inhibiting HTR2B expression led to the inhibition of LUSC cell proliferation. In conclusions, HTR2B might be a new biomarker and therapeutic target in COPD patients with LUSC.
Similar content being viewed by others


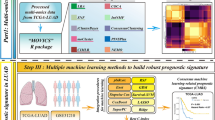
Introduction
Chronic obstructive pulmonary disease (COPD) is an inflammatory lung disease that is mainly caused by chronic smoking and is characterized by irreversible airflow restriction1. COPD is the most disabling and fatal noncommunicable disease and has become one of the top three causes of mortality, with approximately 3 million people dying from COPD each year worldwide2. The current gold standard for diagnosing COPD is testing pulmonary function. However, COPD may be latent in heavy smokers who may even have normal lung function; by the time symptoms, such as difficulty breathing, develop, lung function is often reduced to 50%3. Therefore, it is highly important to search for COPD biomarkers.
Lung cancer is a common malignant tumor, and non-small cell lung cancer (NSCLC) accounts for > 80% of cases4. The incidence of lung cancer is the highest among all malignant tumors, and it has a very high mortality rate5. As early-stage lung cancer has few specific symptoms, it is often diagnosed late, which results in a very serious public health burden6. Therefore, it is highly important to explore NSCLC pathogenesis and identify new diagnostic and therapeutic biomarkers7,8. Lung squamous cell carcinoma (LUSC) accounts for 40–51% of primary lung cancers9. LUSC treatment is less effective than that of lung adenocarcinoma, and there are fewer treatment options available for LUSC10. LUSC has unique epidemiological and clinicopathological features, such as a close association with smoking, a low EGFR mutation rate and a low ALK rearrangement rate, which leads to poor effects of targeted therapy for LUSC11.
COPD is often associated with LUSC, and they share common risk factors and pathogenesis12. The risk factors include a long history of smoking, age > 45 years, and high-risk occupations13. In terms of pathogenesis, a variety of mechanisms may combine to cause disease occurrence and development, including activated cell proliferation pathways, DNA repair gene dysfunction, a chronic inflammatory microenvironment, and an impaired immune response14,15.
In this study, we identified HTR2B as a novel key gene in COPD with LUSC through bioinformatics analysis and experimental validation. HTR2B is a promising therapeutic target and prognostic biomarker for COPD patients with LUSC (Fig. 1A).

(A) Flowchart. (B) Heatmap of DEGs in the GSE76925 dataset. (C) Volcano plot of DEGs in the GSE76925 dataset.
Methods
Data acquisition and processing
The National Center for Biotechnology Information Gene Expression Omnibus (GEO) database (http://www.ncbi.nlm.nih.gov/geo) is an open-access database. In this study, we used the GSE76925, GSE47460, GSE106986, and GSE1650 GEO datasets. The GSE76925 dataset contained lung tissue data from 111 COPD patients and 40 healthy participants. The GSE47460 dataset contained data from lung homogenate samples from 75 COPD patients and 17 healthy participants. The GSE106986 dataset contained lung tissue data from 14 COPD patients and 5 healthy participants. The GSE1650 dataset contained lung tissue data from 18 patients with severe emphysema and 12 patients with mild or no emphysema. The Kaplan‒Meier plotter (http://kmplot.com/analysis/index.php?p=background) is a commonly used online database for differential gene expression and survival analyses. The database included expression data for all genes in > 30,000 samples from 21 tumors and complete patient clinical information. The Human Protein Atlas (HPA) (https://www.proteinatlas.org) provides 24,000 kinds of information on human protein distribution in different tissues and cells and displays immunohistochemical (IHC) staining results for more than 20 types of cancer. Information on HTR2B expression in LUSC patients and complete patient clinical data were downloaded from The Cancer Genome Atlas (TCGA) (https://portal.gdc.cancer.gov/) database.
Screening for differentially expressed genes (DEGs)
We screened for DEGs between patients and controls in the GSE76925 dataset using the R limma package. P < 0.05 and a |log2 fold change (FC)|≥ 0.5 were set as the threshold values for DEG identification. The heatmap and the volcano plot of the DEGs were visualized with the “ggplot2” and “pheatmap” packages of RStudio software (3.1.3) (https://posit.co/download/rstudio-desktop/).
Kyoto encyclopedia of genes and genomes (KEGG) and gene ontology (GO) enrichment analyses
KEGG pathway enrichment analysis was used to identify genes involved in multiple disease pathways and annotate gene functions (https://www.kegg.jp/kegg/kegg1.html)16. GO enrichment analysis was used to reveal the regulatory relationships of target genes related to the biological process, cellular component, and molecular function categories. The KEGG and GO enrichment analyses were conducted using the “org.Hs.eg.db” R package.
Selection of candidate diagnostic markers
LASSO is a regression analytical arithmetic method that performs regularization to ameliorate forecast accuracy. LASSO regression analysis was conducted using the R glmnet package to investigate genes that were significantly associated with the discriminative power between COPD and healthy specimens. We then added the DEGs to the random forest classifier. We performed recurrent random forest classification for all possible numbers of variables and calculated the average error rate of the model.
CIBERSORT analysis
The CIBERSORT (http://cibersort.stanford.edu/) computational approach involves a deconvolutional arithmetic based on gene expression and is used to assess the variations of a gene group relative to the remaining genes within a specimen. CIBERSORT was used to identify the immune cell responses and determine the associations between immune cells and the expression of critical genes in normal and COPD samples. Our primary objective was to identify the associations among these immune cells.
Validation of the roles of key genes in the diagnosis of COPD
We used the “ggpubr” R package to construct receiver operating characteristic (ROC) curves for the key genes in the GSE76925, GSE47460, GSE106986, and GSE1650 datasets.
Cell culture
BEAS-2B, H226, and H1703 cells were purchased from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China). The cells were cultured in RPMI 1640 medium (Gibco, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (Gibco) at 37 °C with 5% CO2.
COPD cell model construction
The COPD cell model was constructed using cigarette smoke extract (CSE; AAPR551-2, GuangZhou Peiyu Biotechnology Co., Ltd., Guangzhou, China). The experiment was conducted according to the manufacturer’s instructions.
Cell transfection
Cells were transfected with a short interfering RNA against HTR2B (si-HTR2B, 5'-GCUGCAGUAUGCUACUAAUTT-3′) (GenePharma, Shanghai, China). Transfection was performed according to the jetPRIME instructions (PolyPlus Transfection, San Marcos, CA, USA).
Construction of an HTR2B-overexpressing cell line
A fragment of the human HTR2B gene sequence was cloned into the pLVX-Puro vector. The BEAS-2B cell line was transfected with the control pLVX-Puro vector or with the pLVX-Puro-HTR2B plasmid (Cas9X, Soochow, China).
Quantitative real-time PCR (RT‒qPCR)
Total RNA was extracted with the RNA-Easy isolation reagent (Vazyme, Nanjing, China) and reverse-transcribed into complementary DNA using a reverse transcription kit (Vazyme) according to the manufacturer’s protocol. RT‒qPCR was performed according to the RT‒qPCR reagent instructions (Vazyme). The primers used were synthesized by Tsingke (Beijing, China) (Table 1).
Cell counting Kit-8 (CCK-8) assay
Cells were seeded in a 96-well plate (3.0 × 103 cells per well), and each group had three replicate wells. The CCK-8 reagent (10 µL; Beyotime, Shanghai, China) was added to the respective wells at 24, 48, and 72 h. After 4 h of incubation, the absorbance of each well was measured at 450 nm in a microplate reader.
Statistical analysis
The public gene expression data were analyzed with RStudio 3.1.3. Statistical analysis was conducted using GraphPad Prism 8.0. A t test was performed to compare data between two groups. Continuous variables are presented as the mean ± standard deviation. Differences were considered statistically significant at *p < 0.05, **p < 0.01, and ***p < 0.001.
Results
Identification of COPD-associated DEGs
We downloaded and analyzed the GSE76925 dataset from the GEO database. The dataset contained a total of 1082 DEGs, among which 371 were upregulated and 711 were downregulated. The heatmap and the volcano plot of the DEGs were shown in Fig. 1(B and C).
KEGG and GO enrichment analyses
KEGG analysis indicated that the DEGs were mainly enriched in the p53 signaling and β-alanine metabolism pathways (Fig. 2A, C). GO analysis indicated that the DEGs were mainly involved in transcription initiation from the RNA polymerase I promoter and in the regulation of mononuclear cell proliferation (Fig. 2B, D).

(A) KEGG pathway enrichment analysis results for the upregulated DEGs. (B) GO enrichment analysis results for the upregulated DEGs. (C) KEGG pathway enrichment analysis results for the downregulated DEGs. (D) GO enrichment analysis results for the downregulated DEGs.
Determination and verification of COPD markers
The underlying biological markers of COPD were selected with two arithmetic algorithms. Each prognostic indicator coefficient track was analyzed using LASSO coefficient profiles by changing log (lambda) in the LASSO algorithm (Fig. 3A). The DEGs were identified by LASSO regression analysis, and eight variables were identified as COPD diagnostic markers (Fig. 3B). The number of variables was small, and the out-of-band error was low. Based on the relationship plot between the model error and the number of decision trees, 400 trees were selected as a parameter for the final model, which revealed a stable error in the model (Fig. 3C). Next, we identified eight DEGs with a significance value > 2 as candidate genes for subsequent analyses (Fig. 3D). Combining the results of the two algorithms yielded four key genes, namely, HTR2B, DPYS, FRY, and CD19.

(A) LASSO coefficient profiles of the risk factors. (B) Eight risk factors selected by LASSO regression analysis. (C) Influence of the number of decision trees on the error rate. X-axis: Number of decision trees; Y-axis: Error rate. The error rate was relatively stable when the number of decision trees was approximately 400. (D) Results of the Gini coefficient method in the random forest classifier. X-axis: Genetic variables; Y-axis: Importance index.
Key genes related to immunocyte infiltration
We studied the coefficients of the four key genes (HTR2B, DPYS, FRY, and CD19) and the immunocyte infiltration status of COPD and normal samples to determine the correlation between immunocyte infiltration status and key gene expression. The immunocyte features were investigated with CIBERSORT, and the relationships between key gene expression and immunocyte infiltration levels were examined. We determined that HTR2B was correlated with CD8+ T cells (Fig. 4A), DPYS was correlated with M0 macrophages (Fig. 4B), FRY was correlated with resting memory CD4+ T cells (Fig. 4C), and CD19 was correlated with plasma cells (Fig. 4D). Our findings suggested that these genes might be involved in COPD progression by regulating the abovementioned immune cells.

Correlations between key genes and infiltrating immune cells in COPD and normal samples.
Validation of differential expression of HTR2B in COPD
We analyzed the GSE76925, GSE47460, GSE106986, and GSE1650 datasets to verify HTR2B expression in COPD patients. Compared with that in the control group, HTR2B expression was significantly upregulated in the COPD group (GSE76925, GSE47460, and GSE106986) (Fig. 5A–C). HTR2B expression in severe emphysema patients was significantly greater than that in mild or no emphysema patients (GSE1650) (Fig. 5D). ROC curves for the key genes in the GSE76925, GSE47460, GSE106986, and GSE1650 datasets were plotted using the RStudio software (Fig. 5E–H).

(A) HTR2B expression in the GSE76925 dataset. (B) HTR2B expression in the GSE47460 dataset. (C) HTR2B expression in the GSE106986 dataset. (D) HTR2B expression in the GSE1650 dataset. (E) ROC curves for key genes in the GSE76925 dataset. (F) ROC curves for key genes in the GSE47460 dataset. (G) ROC curves for key genes in the GSE106986 dataset. (H) ROC curves for key genes in the GSE1650 dataset. (I) HTR2B expression in CSE-stimulated BEAS-2B cells. (J) HTR2B expression in BEAS-2B cells stimulated with 3% CSE.
HTR2B was upregulated in the COPD cell model and promoted epithelial–mesenchymal transition (EMT)
According to the log2 FC (GSE76925), we selected HTR2B for preliminary experimental validation. We stimulated BEAS-2B cells with CSE to simulate the COPD microenvironment and determined that HTR2B expression was the highest after 48 h of stimulation with 3% CSE (Fig. 5I, J). Following GSE76925 dataset analysis, the genes coexpressed with HTR2B (cor > 0.3, P < 0.05) were input into FunRich 3.1.3 for signaling pathway analysis. The results demonstrated that HTR2B might be closely related to EMT (Fig. 6A).

(A) Prediction of downstream signaling pathways regulated by HTR2B in COPD. (B) Inhibition of HTR2B expression in BEAS-2B cells. (C–G) Changes in the mRNA expression levels of EMT markers in BEAS-2B cells with different HTR2B expression levels.
We explored the molecular biological mechanisms of HTR2B in COPD by knocking down and overexpressing HTR2B in BEAS-2B cells (Fig. 6B and Supplementary Figure S1A). HTR2B knockdown led to significant downregulation of the EMT-related markers CDH2 (Fig. 6C), MMP2 (Fig. 6D), VIM (Fig. 6F), and SNAI1 (Fig. 6G), while ZEB1 expression did not significantly change (Fig. 6E). The addition of CSE to simulate a COPD cell microenvironment partially restored the CDH2 (Fig. 6C), MMP2 (Fig. 6D), VIM (Fig. 6F), and SNAI1 (Fig. 6G) expression levels by inhibiting HTR2B expression. HTR2B overexpression led to significant upregulation of CDH2 (Supplementary Figure S1B), MMP2 (Supplementary Figure S1C), ZEB1 (Supplementary Figure S1D), and SNAI1 (Supplementary Figure S1F) expression, while VIM expression (Supplementary Figure S1E) did not significantly change. CSE further upregulated CDH2 (Supplementary Figure S1B), MMP2 (Supplementary Figure S1C), and SNAI1 (Supplementary Figure S1F) expression via HTR2B overexpression.
Bioinformatics analysis of HTR2B expression in LUSC
The Kaplan‒Meier plotter data indicated that HTR2B expression in LUSC tissue was significantly higher than that in normal lung tissue (Fig. 7A, B) and that lower HTR2B expression was linked to longer overall survival (OS) (Fig. 7C) and relapse-free survival (RFS) (Fig. 7D) in patients with LUSC. The IHC staining data in the HPA database further confirmed the upregulation of HTR2B at the protein level in LUSC (Fig. 7E–H). The TCGA LUSC data showed that a high expression level of HTR2B was significantly related to a poor patient prognosis (Fig. 7I). Following Kaplan‒Meier plotter database analysis, the genes coexpressed with HTR2B (cor > 0.3, P < 0.05) were input into FunRich 3.1.3 for signaling pathway analysis. The results demonstrated that HTR2B might be closely related to pathways in cancer, cytokine–cytokine receptor interaction, and the PI3K–Akt signaling pathway (Fig. 7J).

(A, B) Prediction of differences in the HTR2B mRNA expression levels between LUSC and normal tissue samples using the Kaplan‒Meier plotter database. (C) Results of survival analysis of LUSC patients stratified by HTR2B expression by using the Kaplan‒Meier plotter. (D) Results of RFS analysis of patients with LUSC stratified by HTR2B expression by using the Kaplan‒Meier plotter. (E–H) Results of IHC staining analysis of HTR2B in the HPA database. (I) Results of survival analysis of patients based on HTR2B expression in the TCGA database. (J) Prediction of downstream signaling pathways regulated by HTR2B in LUSC.
HTR2B was upregulated in LUSC cells and promoted cell proliferation
RT‒qPCR demonstrated that compared with that in BEAS-2B cells, HTR2B expression was significantly increased in H226 and H1703 cells (Fig. 8A). Furthermore, RT‒qPCR revealed that the HTR2B expression level significantly decreased following si-HTR2B transfection (Fig. 8B, C). The CCK-8 assay demonstrated that si-HTR2B transfection significantly reduced cell proliferation (Fig. 8D, E). In H226 cells, CCND1 expression did not significantly change following HTR2B knockdown (Fig. 8F), while ITGAV and ITGB1 expression significantly decreased (Fig. 8G, H). In H1703 cells, CCND1 expression significantly decreased following HTR2B knockdown (Fig. 8I), while ITGAV and ITGB1 expression did not significantly change (Fig. 8J, K).

(A) HTR2B expression in BEAS-2B, H226, and H1703 cells. (B, C) Inhibition of HTR2B in H226 and H1703 cells. (D, E) LUSC cell proliferation, as determined by the CCK-8 assay. (F, I) CCND1 expression after short interfering RNA (siRNA)-mediated knockdown of HTR2B, as determined by RT‒qPCR. (G, J) ITGAV expression after siRNA-mediated knockdown of HTR2B, as determined by RT‒qPCR. (H, K) ITGB1 expression after siRNA-mediated knockdown of HTR2B, as determined by RT‒qPCR.
Pattern diagram
The pattern diagram demonstrated that HTR2B might be a new biomarker and therapeutic target in COPD patients with LUSC (Fig. 9).

Pattern diagram.
Discussion
COPD is a respiratory disease, and its diagnosis currently depends mainly on lung function17. Researchers have concluded that existing spirometry-based diagnostic methods cannot sufficiently predict COPD progression and related deaths18. Presently, the role of genetic factors in COPD development is not well understood. Some genetic factors may increase COPD risk19. Many candidate genes, including those that encode α1-antitrypsin, secretory inclusion cell protease suppressor, and TNF-α, may increase COPD risk20,21. In the present study, we determined that HTR2B, DPYS, FRY, and CD19 might be promising biomarkers and potential therapeutic targets for COPD.
Lung cancer is the leading cause of cancer-related death worldwide22, and the 5-year mean survival rate is 20%, which decreases with the tumor stage23. This improvement can be attributed to advances in the diagnosis and treatment of patients with early and advanced lung cancer. Despite such advances in lung cancer treatment, increasing the efficiency of early detection is the most promising strategy for improving long-term survival of patients with lung cancer24. In recent years, the rapid development of high-throughput technology has generated large amounts of high-throughput sequencing data and expression profile data in oncology25. The combination of big data and different data types presents an entirely new perspective on the analysis of such data26. In the present study, we determined that HTR2B might be a diagnostic biomarker and potential therapeutic target in LUSC.
HTR2B belongs to the serotonin receptor family. HTR2B has been described as an oncogene in many cancers27,28. However, HTR2B has not been reported in lung cancer. Our results suggested that HTR2B might promote LUSC development. An animal study demonstrated that HTR2A and HTR2B partially mediated smoking-induced airway inflammation and small airway remodeling and that they might be new therapeutic targets for COPD airway remodeling29. Herein, we determined that HTR2B might contribute to COPD progression.
The chronic inflammatory response is considered to be one of the most important factors in the pathogenesis of COPD30. In COPD patients, respiratory tract inflammation can persist after smoking cessation, and there is persistent local infiltration of macrophages, neutrophils, and CD4+ and CD8+ T lymphocytes, which indicates the involvement of autoimmunity in COPD progression31. The main inflammatory reaction in COPD is an increase in the number of CD8+ T cells, while the number of CD4+ T cells is basically unchanged or slightly decreased, and the CD4+/CD8+ T-cell ratio decreases, which leads to the interaction between host T and B lymphocytes and results in an immune imbalance32. By analyzing the relationships between key genes and immune cell infiltration, we confirmed that the key genes might promote COPD progression by regulating immune cells.
During EMT, epithelial cells lose polarity and cell adhesion characteristics, gain the migration ability and aggressiveness, and become mesenchymal cells33. COPD can increase lung cancer risk by 40–50%, and 70% of lung cancers are accompanied by mild to moderate COPD, with EMT being one of the shared pathophysiological mechanisms34. Many studies have reported that the airways of COPD patients exhibit signs of increased EMT marker expression, reticulum basement membrane rupture, and decreased epithelial junction molecule expression, which suggests that EMT might promote small airway remodeling and fibrosis in COPD patients35,36. Our results confirmed that HTR2B might contribute to COPD progression by influencing EMT.
The occurrence and development of lung cancer involve the activation of various signal transduction pathways37. Studies on the PI3K/AKT signaling pathway have made remarkable progress in the field of cancer38. Our findings confirmed that HTR2B might promote LUSC cell proliferation by regulating the PI3K/AKT signaling pathway.
Cells are in constant contact with their surroundings, mainly through cell adhesion, and these contacts are essential for cell differentiation and growth39. Cell adhesion includes intercellular adhesion (direct contact between cells) and cell–matrix adhesion (the interaction between cells and the extracellular matrix)40. ITGAV and ITGB1 are downstream signaling genes of cell adhesion41. Our results confirmed that HTR2B might promote LUSC cell proliferation by regulating the cell adhesion signaling pathway.
Several limitations of the study should be noted. First, there is a lack of relevant in vivo experiments. Animal experiments are required for better clinical application in further studies. Second, the key genes need to be validated with a larger sample size. In the future, we plan to conduct clinical trials to verify the diagnostic efficacy of the key genes. Finally, the specific molecular mechanism by which HTR2B promotes the proliferation of LUSC cells is still unclear and needs to be explored via detailed molecular biology experiments.
In summary, we validated the differential expression and predictive ability of HTR2B in COPD patients across multiple datasets. We confirmed that HTR2B expression was upregulated in LUSC cell lines and that HTR2B knockdown significantly inhibited the proliferation of LUSC cells. Inhibiting HTR2B expression could inhibit the expression of EMT-related genes in COPD cell models and LUSC cells. HTR2B might be a new biomarker and therapeutic target in COPD patients with LUSC.
Data availability
All data involved in this study are available from the corresponding author upon request. The original GEO datasets used for analysis in this study (GSE76925, GSE47460, GSE106986, and GSE1650) are available from the GEO database (https://www.ncbi.nlm.nih.gov/geo/). Kaplan‒Meier plotter can be accessed at http://kmplot.com/analysis/index.php?p=background. The HPA can be accessed at https://www.proteinatlas.org. TCGA can be accessed at https://portal.gdc.cancer.gov/. The KEGG can be accessed at https://www.kegg.jp/kegg/kegg1.html.
References
Kate, M. J. & Kevin, I. D. The growing burden of COPD in the United States. Chest https://doi.org/10.1016/j.chest.2024.01.003 (2024).
Daniel, J. T., Wouter, H. V. G. & Haydn, E. W. Impact of triple therapy vs dual bronchodilator therapy on mortality rates in COPD. Chest https://doi.org/10.1016/j.chest.2023.12.033 (2024).
Agustí, A., Vogelmeier, C. & Faner, R. COPD 2020: changes and challenges. American journal of physiology. Lung Cell. Mol. Physiol. 319, 879–883. https://doi.org/10.1152/ajplung.00429.2020 (2020).
Siqi, N. et al. Prognostic models for immunotherapy in non-small cell lung cancer: A comprehensive review. Heliyon https://doi.org/10.1016/j.heliyon.2024.e29840 (2024).
Reck, M., Carbone, D., Garassino, M. & Barlesi, F. Targeting KRAS in non-small-cell lung cancer: recent progress and new approaches. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 32, 1101–1110. https://doi.org/10.1016/j.annonc.2021.06.001 (2021).
Saurav Kumar, J. et al. Cellular senescence in lung cancer: Molecular mechanisms and therapeutic interventions. Ageing Res. Rev. https://doi.org/10.1016/j.arr.2024.102315 (2024).
Lahiri, A. et al. Lung cancer immunotherapy: progress, pitfalls, and promises. Mol. Cancer 22, 40. https://doi.org/10.1186/s12943-023-01740-y (2023).
Li, Y., Yan, B. & He, S. Advances and challenges in the treatment of lung cancer. Biomed. Pharmacother. 169, 115891. https://doi.org/10.1016/j.biopha.2023.115891 (2023).
Pan, Y., Han, H., Labbe, K., Zhang, H. & Wong, K. Recent advances in preclinical models for lung squamous cell carcinoma. Oncogene 40, 2817–2829. https://doi.org/10.1038/s41388-021-01723-7 (2021).
Kuan-Li W et al. 2024 PROM2 upregulation promotes cancer cell migration and confers a poor prognosis in lung squamous cell carcinoma. Am. J. Cancer Res. https://doi.org/10.62347/eqfy12190
Parth, M., Ruma, R., Raghu, S., Vishal Haribhai, P. & Tapan Kumar, M. Understanding the feasibility of chemotherapeutic and immunotherapeutic targets against non-small cell lung cancers: an update of resistant responses and recent combinatorial therapies. Explor. Target Antitumor. Ther. https://doi.org/10.37349/etat.2023.00171 (2023).
Silvia, R. et al. Effectiveness of immunotherapy in non-small cell lung cancer patients with a diagnosis of COPD: is this a hidden prognosticator for survival and a risk factor for immune-related adverse events?. Cancers (Basel) https://doi.org/10.3390/cancers16071251 (2024).
de Vries, R. et al. Prospective detection of early lung cancer in COPD patients in regular care by electronic nose analysis of exhaled breath. Chest https://doi.org/10.1016/j.chest.2023.04.050 (2023).
Kim, N., Kang, E., Ha, E., Lee, J. & Lee, J. Association of type 2 diabetes mellitus with lung cancer in patients with chronic obstructive pulmonary disease. Frontiers in medicine 10, 1118863. https://doi.org/10.3389/fmed.2023.1118863 (2023).
Forder, A. et al. Mechanisms contributing to the comorbidity of COPD and lung cancer. Int. J. Mol. Sci. https://doi.org/10.3390/ijms24032859 (2023).
Minoru, K., Miho, F., Yoko, S., Masayuki, K. & Mari, I.-W. KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res https://doi.org/10.1093/nar/gkac963 (2022).
Priya, V. GOLD COPD report: 2024 update. Lancet Respir. Med. https://doi.org/10.1016/s2213-2600(23)00461-7 (2023).
Daniel, J. T. et al. Can we use lung function thresholds and respiratory symptoms to identify Pre-COPD? A prospective, population-based cohort study. Am J Respir Crit Care Med https://doi.org/10.1164/rccm.202212-2330OC (2024).
Sayers, I., John, C., Chen, J. & Hall, I. Genetics of chronic respiratory disease. Nat. Rev. Genet. https://doi.org/10.1038/s41576-024-00695-0 (2024).
Davide, P. et al. Comparison among populations with severe and intermediate alpha1-antitrypsin deficiency and chronic obstructive pulmonary disease. Minerva Med. https://doi.org/10.23736/s0026-4806.22.08266-0 (2023).
Schleich, F., Bougard, N., Moermans, C., Sabbe, M. & Louis, R. Cytokine-targeted therapies for asthma and COPD. European respiratory review : an official journal of the European Respiratory Society https://doi.org/10.1183/16000617.0193-2022 (2023).
Berg, C. et al. AIR POLLUTION AND LUNG CANCER A Review by International Association for the Study of Lung Cancer Early Detection and Screening Committee. Journal of thoracic oncology : official publication of the International Association for the Study of Lung Cancer https://doi.org/10.1016/j.jtho.2023.05.024 (2023).
Tyler, B. K. et al. Lung cancer statistics, 2023. Cancer https://doi.org/10.1002/cncr.35128 (2024).
Duffy, M. Circulating tumor DNA (ctDNA) as a biomarker for lung cancer: Early detection, monitoring and therapy prediction. Tumour biology : the journal of the International Society for Oncodevelopmental Biology and Medicine 46, S283–S295. https://doi.org/10.3233/tub-220044 (2024).
Brockley, L. et al. Sequence-Based Platforms for Discovering Biomarkers in Liquid Biopsy of Non-Small-Cell Lung Cancer. Cancers (Basel) https://doi.org/10.3390/cancers15082275 (2023).
Villaruz, L., Socinski, M. & Weiss, J. Guidance for clinicians and patients with non-small cell lung cancer in the time of precision medicine. Front. Oncol. 13, 1124167. https://doi.org/10.3389/fonc.2023.1124167 (2023).
Li, T. et al. Serotonin receptor HTR2B facilitates colorectal cancer metastasis via CREB1-ZEB1 axis mediated epithelial-mesenchymal transition. Molecular cancer research : MCR https://doi.org/10.1158/1541-7786.Mcr-23-0513 (2024).
Tu, R. et al. Neurotransmitter receptor HTR2B regulates lipid metabolism to inhibit ferroptosis in gastric cancer. Cancer Res. 83, 3868–3885. https://doi.org/10.1158/0008-5472.Can-23-1012 (2023).
Yang, T. et al. Serotonin receptors 5-HTR2A and 5-HTR2B are involved in cigarette smoke-induced airway inflammation, mucus hypersecretion and airway remodeling in mice. Int. Immunopharmacol. 81, 106036. https://doi.org/10.1016/j.intimp.2019.106036 (2020).
Xiang, Y. & Luo, X. Extrapulmonary comorbidities associated with chronic obstructive pulmonary disease: A review. Int. J. Chron. Obstruct. Pulmon. Dis. 19, 567–578. https://doi.org/10.2147/copd.S447739 (2024).
Pei, Y. et al. Combining single-cell RNA sequencing of peripheral blood mononuclear cells and exosomal transcriptome to reveal the cellular and genetic profiles in COPD. Respir. Res. 23, 260. https://doi.org/10.1186/s12931-022-02182-8 (2022).
Xue, W., Ma, J., Li, Y. & Xie, C. Role of CD T and CD T lymphocytes-mediated cellular immunity in pathogenesis of chronic obstructive pulmonary disease. J. Immunol. Res. 2022, 1429213. https://doi.org/10.1155/2022/1429213 (2022).
Zhao, Z. & Zhu, Y. FAP, CD10, and GPR77-labeled CAFs cause neoadjuvant chemotherapy resistance by inducing EMT and CSC in gastric cancer. BMC Cancer 23, 507. https://doi.org/10.1186/s12885-023-11011-0 (2023).
Qi, C., Sun, S. & Xiong, X. From COPD to lung cancer: mechanisms linking, diagnosis, treatment, and prognosis. Int. J. Chron. Obstruct. Pulmon. Dis. 17, 2603–2621. https://doi.org/10.2147/copd.S380732 (2022).
Mei-Yu, L. et al. Abhd2, a candidate gene regulating airway remodeling in COPD via TGF-β. Int. J. Chron. Obstruct. Pulmon. Dis. https://doi.org/10.2147/copd.S440200 (2024).
Hou, W. et al. Cigarette smoke induced lung barrier dysfunction, EMT, and tissue remodeling: A possible link between COPD and lung cancer. BioMed Res. Int. 2019, 2025636. https://doi.org/10.1155/2019/2025636 (2019).
Combes, A., Samad, B. & Krummel, M. Defining and using immune archetypes to classify and treat cancer. Nature reviews. Cancer https://doi.org/10.1038/s41568-023-00578-2 (2023).
Premila, D. L. & Chandrakanth, A. PI3K/Akt/mTOR Signaling Pathway as a Target for Colorectal Cancer Treatment. Int. J. Mol. Sci. https://doi.org/10.3390/ijms25063178 (2024).
Siamak, A. K., Bhavna, R. & Staffan, J. Cell Cycle Regulation by Integrin-Mediated Adhesion. Cells https://doi.org/10.3390/cells11162521 (2022).
Fan, C. et al. Role of adhesion molecules in cancer and targeted therapy Science China. Life Sci. 67, 940–957. https://doi.org/10.1007/s11427-023-2417-3 (2024).
Lor Huai, C. et al. The role of cell-matrix adhesion and cell migration in breast tumor growth and progression. Front Cell Dev Biol https://doi.org/10.3389/fcell.2024.1339251 (2024).
Acknowledgements
The authors gratefully acknowledge the Henan Key Laboratory of Pharmacology for Liver Diseases for providing experimental platform support.
Funding
This study was supported by the Medical Science and Technology Research Project of Henan Province (LHGJ20230169).
Author information
Authors and Affiliations
Contributions
Zhe Cheng and Pengfei Li designed the experiments. Yue Li and Yu Wang analyzed the data and wrote the manuscript. Ruhao Wu provided helpful discussion and reviewed the manuscript. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, Y., Wang, Y., Wu, R. et al. HTR2B as a novel biomarker of chronic obstructive pulmonary disease with lung squamous cell carcinoma. Sci Rep 14, 13206 (2024). https://doi.org/10.1038/s41598-024-63896-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-63896-x
- Springer Nature Limited