
Abstract
Single nucleotide substitutions are the most common type of somatic mutations in cancer genome. The goal of this study was to use publicly available somatic mutation data to quantify negative and positive selection in individual lung tumors and test how strength of directional and absolute selection is associated with clinical features. The analysis found a significant variation in strength of selection (both negative and positive) among tumors, with median selection tending to be negative even though tumors with strong positive selection also exist. Strength of selection estimated as the density of missense mutations relative to the density of silent mutations showed only a weak correlation with tumor mutation burden. In the “all histology together” analysis we found that absolute strength of selection was strongly correlated with all clinically relevant features analyzed. In histology-stratified analysis selection was strongest in small cell lung cancer. Selection in adenocarcinoma was somewhat higher compared to squamous cell carcinoma. The study suggests that somatic mutation- based quantifying of directional and absolute selection in individual tumors can be a useful biomarker of tumor aggressiveness.
Similar content being viewed by others


Introduction
Single nucleotide substitutions (SNSs) are the major type of somatic variation in tumors1,2. Even though the absolute majority of the point mutations are neutral3, there are many examples of positive and negative selection of point mutations in carcinogenesis4,5. The absolute majority of the analyses of selection in tumors has been done at the level of individual genes5,6,7, while a quantitative assessment of the direction and strength of selection at the tumor level has never been addressed according to our best knowledge.
Quantifying of the strength of selection at the tumor level can be used for a better understanding of tumor biology and can reflect tumor aggressiveness because quickly evolving tumors can better adapt to the host immune response and chemotherapy, and as a result, survive better and proliferate more quickly8.
The most commonly used global somatic mutation-based biomarker is tumor mutational burden (TMB). TMB is a tumor feature that predicts survival9, risk of metastasis, progression10,11, and response to treatment, especially to immunotherapy12,13,14. We hypothesized that aside from TMB, strength of selection in a tumor may also reflect the speed of tumor evolution and therefore can be associated with clinically relevant features.
We hypothesized that assessment of the global selection based on somatic mutations in a tumor is associated with clinical features and potentially could be used as a biomarker of cancer aggressiveness. Tumor development is an evolutionary process comprising differential survival and proliferation of genetically different cell lineages (clones)15,16. Cancer cells that survive better and proliferate faster have a selective advantage and over time become a predominant clone of genetically heterogeneous tumor17. This evolution happens even before any treatment is applied, though treatment itself is a very strong selective factor that drives tumor evolution18.
Individual tumors as well as clones inside a tumor differ by their intrinsic propensity to produce somatic mutations, which depends on their DNA repair capacity as well as environmental exposures which is especially relevant to lung cancer19,20. These factors contribute to the tumor’s ability to evolve through Darwinian selection. Fast-evolving tumors tend to be more aggressive since they better adapt to the host immune response and proliferate more quickly compared to slowly evolving tumors21.
The direction and strength of selection in coding regions of the human genome can be quantified by the ratio of substitution rates at non-synonymous and synonymous sites, dN/dS. Even though it is not perfect22, this metric is widely used across different research fields23. The measure was first based on the comparison of homologous sequences to estimate selection strength in species divergence, and recently the approach became a popular tool to quantify selection strength in tumor4,24,25. Nonsynonymous to synonymous mutation ratio was used to estimate strength of selection in individual genes across cancer types26,27,28. A study by Persi et al.29 used dN/dS ratio for a pan-cancer analysis of selection in 6,721 tumors representing 23 cancer types. They found that strength of selection in tumor is associated with tumor fitness and found “likely clinical implications” of dN/dS.
The goal of our study is to quantify global selection in individual lung tumors and to test if the strength of selection is clinically relevant. Clinical relevance is assessed by the analysis of correlation of the strength of positive, negative and absolute selection in individual lung tumors with clinical features.
Methods
Description of the approach
We have estimated the direction (negative or positive) and strength of selection by a comparison of the densities of nonsynonymous (missense) to synonymous (silent) mutations. In this respect our approach is similar to the commonly used ratio on nonsynonymous to synonymous substitutions dN/dS22,30,31. Our approach, however, differs from dN/dS method in the manner of how the normalization of the mutation numbers is done. Our goal was to quantify the strength of positive and negative selection at genome level while dN/dS estimates are designed for an assessment of selection in individual genes. At the genome level, exactly the same single nucleotide substitution may produce nonsynonymous or synonymous mutation depending on what transcript is considered. This ambiguity stems from the fact that the absolute majority of the genes in the human genome undergo alternative splicing32. Together with the common (up to a quarter of all genes) cases of overlapping genes33 this leads to a quite common situation when the same nucleotide substitution results in either a nonsynonymous or a synonymous substitution depending on what transcript is analyzed. The key parameter in our analysis is the number of potential sites for missense and silent mutations in the human genome. To estimate the number of potential sites for silent and missense mutations in the human genome we computationally “mutated” each nucleotide in coding regions into 3 possible single nucleotide substitutions and ran the “mutated” sequence against all known transcripts to see if it produced a silent or a missense mutation34. Therefore, we counted the total number of missense and silent mutations that can be produced by all possible single nucleotide substitutions in the human genome in the context of all existing transcripts. This approach fits well with how somatic mutations are reported in the Catalog Of Somatic Mutations In Cancer (COSMIC) database which we have used as the data source for the study. In COSMIC the same point mutation may be reported as missense or silent depending on the transcript. The other difference of our approach from dN/dS method was that we have used the logarithm of the ratio instead of the simple ratio of nonsynonymous to synonymous mutations. This was done to make the distribution more symmetrical and therefore more suitable for statistical comparisons.
Estimation of the number of potential sites in the human genome for missense and silent mutations
We used the latest build of the human genome project—GRCh38 to estimate the number of potential sites for missense and silent mutations. We first identified all nucleotide positions in the consensus protein coding sequence (CCDS) database35. Then we computationally mutated each nucleotide into the three possible single nucleotide substitutions (SNSs) and checked if a given SNS led to a missense or a silent mutation. If the SNS produced both missense and silent mutations it was counted both ways: as a potential site for both missense and silent mutations. This way we have estimated the total number of potential sites for missense and nonsense mutations in the human genome to be equal to 74,038,110 and the total number of potential sites for silent mutations to be 22,654,380.
Quantifying of negative and positive selection
The number of missense mutations in a tumor can be used to detect the type of selection (negative or positive) and to quantify the strength of selection. Negative selection against missense mutations will result in their lower number while positive selection will increase their number, and both affect the missense mutation density. However, selection is not the only factor influencing the number of somatic mutations in tumor. Environmental exposures, for example, tobacco smoke, dramatically increase the number of somatic mutation in lung tumors36. One needs to take into account the overall mutability when estimating direction and strength of selection of missense mutations. Silent mutations can be used to adjust for tumor-specific mutability. Despite anecdotal examples of functionality37,38, silent mutations are generally selectively neutral39 and, therefore, silent mutations can be used as a reference group.
As a measure of selection we used the logarithm of the ratio of the densities of missense to silent mutations, that is, the number of missense mutations per million of potential sites to the number of silent mutations per million of potential sites. Negative log ratio values of relative selection indicate selection against missense mutations (negative selection), and positive log ratios indicate positive selection for missense mutations. To estimate strength of selection regardless of its direction we used the absolute value of the log ratio. We also estimated tumor mutation burden (TMB) for each tumor. TMB was defined as the number of missense mutations detected in a given tumor by whole exome sequencing.
Somatic mutation data
We used somatic mutation data from the Catalog Of Somatic Mutations In Cancer (COSMIC)40. COSMIC is the largest repository of somatic mutations detected in tumor samples. COSMIC is updated quarterly, with the sample size increasing 5–10% with each new version. We used the latest version (V98) of the database. We focused on lung cancer because it has one of the highest numbers of reported somatic mutations compared to other cancers41,42,43. The summary of the data used in this study can be found in Supplementary Table S1.
We tested the association of (1) strength of positive selection, (2) strength of absolute selection regardless of the direction, and (3) tumor mutation burden with three clinically relevant tumor features: tumor stage, patient age at diagnosis and a comparison between primary and metastatic tumors. We used the Spearman rank-order correlation coefficient (rho) to test the association of selection strength with age at diagnosis. To test the association between selection and tumor stage we used nonparametric Spearman's rank correlation coefficient, and t-test to compare strength of selection between primary and metastatic tumors. The clinical characteristics were downloaded from COSMIC website. Stage information was available for 26% of tumors, the age at diagnosis for 85% of all patients, and primary (82%) versus metastatic (18%) for 60% of COSMIC samples .
Analysis of global selection in lung tumors stratified by the presence of driver mutations in EGFR or KRAS
We stratified tumor samples by the presence/absence of driver mutations in EGFR and KRAS genes. We used these genes because the largest number of samples harbored driver mutations in them: 69 samples with an EGFR driver mutation and 160 samples with a KRAS driver mutation. For EGFR we considered as a driver any of the following COSMIC reported mutations: p.L858R, p.L813R, p.T790M, p.T745M, p.R521K, and p.R476K44. For KRAS the following COSMIC reported mutations were considered as drivers: p.G12C, p.G12V, p.G12D, p.G12A, p.G13C, p.G12S, and p.G13D45.
Results
Quantifying selection in individual tumors: joint analysis of all cell types
Figure 1 shows the distribution of log ratios of the density of missense over the density of silent mutations in individual lung tumors. Denote MmD as the missense mutation density estimated as the number of missense mutation per million of potential sites for missense mutations, and SmD, the silent mutation density, as the number of silent mutations per million of potential sites for silent mutations. We found that the mean log(MmD/SmD) in all cell types analyzed together was equal to -0.034 ± 0.007. Single sample t-test against mean log(MmD/SmD) = 0 (no selection) was -5.1, which is highly statistically significant with p = 7 × 10−7. The result indicates that global selection on missense mutations in lung tumors is negative.

The distribution of the log ratio of the density of missense to the density of silent mutations. The vertical red line marks the relative density expected when the global selection is zero, that is, the density of missense mutations equals the density of silent mutations. The median log ratio is shown as a vertical line on the box plot. The standard deviation SD = 0.268 is shown as a horizontal box. Vertical bars show the 95% confidence interval.
The association of global tumor selection with clinically relevant features.
Table 1 shows the results of statistical analyses of the association of directional and absolute selection with clinically relevant features available. The table also shows the association between clinically relevant features and tumor mutation burden. The total number of missense mutations detected in a given tumor was used as TMB. Directional selection was significantly associated with age at diagnosis and primary versus metastatic tumors. Absolute global selection was significantly associated with all clinically relevant features while TMB does not show any significant association with clinically relevant features.
The shape of the association of directional selection with clinically relevant features
To study the shape of the associations between clinically relevant features and selection we have stratified all tumors into five categories based on the selection strength: (1) strong negative selection—log ratio < -0.2 (total 189 tumors), (2) weak negative selection, -0.02 ≤ log ratio < -0.05 (total 661 tumors), (3) no obvious selection, -0.05 ≤ log ratio < 0.05 (total 461 tumors), (4) weak positive selection, 0.05 ≤ log ratio < 0.2 (total 157 tumors), and (5) strong positive selection, log ratio ≥ 0.2 (total 97 tumors). The categorization was based on the following considerations: (i) to facilitate the interpretation, categories needed to be distributed symmetrically relative to zero (as zero means no selection); (ii) the categories were made maximally similar in size, to make the comparisons more robust. That was not a simple task because the whole distribution is shifted to the left relative to zero.
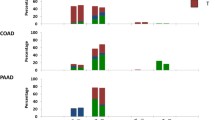
The upper panel of the Fig. 2 shows the positions of the five categories (colored boxes) relative to the distribution of the strength of directional selection. The four lower panels of the Fig. 2 show the results of the analysis. For the study of the shape of association between directional selection and stage (second row, left panel), stage was treated as ordered numbers reflecting tumor progression, with Stage I being least and Stage IV most advanced. For all analyzed clinically relevant traits we observed a U-shaped or an inverse U-shaped association between directional selection and the analyzed features. The results indicate that the absolute strength of the selection rather than the direction of selection is clinically relevant.

Upper panel shows stratification of tumors into five categories of strength of directional selection. Vertical red line marks the point of zero global selection. The middle and low rows show distributions of values of clinically relevant features in tumors categorized by strength of the directional selection. Note U-shaped or reverse U-shaped associations with the clinically relevant features.
Histology-specific analysis of the global selection in lung tumors
Figure 3 shows the distributions of log(MmD/SmD) in three major lung cancer cell types: adenocarcinoma—685 tumors, squamous cell carcinoma—713 tumors, and small cell lung cancer—167 tumors. In all cell types combined the mean log ratio was lower than zero, indicating global negative selection. The mean log ratio for adenocarcinoma was -0.06 ± 0.01 which is significantly lower than zero: t = 5.9, p = 6.1 × 10−9. For squamous cell carcinoma the mean log ratio was -0.070 ± 0.005, t = 15.1, p < 10−24, and for small cell lung cancer the mean ratio was positive: 0.23 ± 0.04, t-test = 6.0, p = 10−8. The positive mean global selection in small cell lung cancer is due to the presence of a cluster of tumors with strong positive selection (see the far right part of the distribution). However, the median value of the log ratio for small cell lung cancer was negative − 0.02, along with the median values for adenocarcinoma and squamous cell carcinoma, -0.06 and -0.07, correspondingly.

The distribution of the ratio of the density of missense to the density of silent mutations in adenocarcinoma (top panel), squamous cell carcinoma (middle panel), and small cell lung cancer (lower panel). The vertical red line marks the relative density expected in the absence of selection. Median log ratio is shown as the vertical line on the box plot.
Association of global tumor selection with three clinically relevant features: histology specific analysis
Table 2 shows the results of the statistical analysis of the associations between selection and clinically relevant features in analyses stratified by histology. The absolute selection shows four significant associations across histology. TMB has three and directional selection—two significant associations with clinically relevant characteristics.
Analysis of the strength of global selection in lung tumors stratified by the presence of common driver mutations
Table 3 describes the results of the analysis of the strength of selection in lung tumors stratified by presence/absence of driver mutations in EGFR and KRAS. Log(dN/dS) in tumor samples with EGFR driver mutations was -0.13 ± 0.01 which is significantly lower compared to the strength of global selection in samples without EGFR driver mutations − 0.06 ± 0.01; t-test = 4.54, p = 3.1 × 10−6. For KRAS we observed the opposite difference: log(dN/dS) for samples with KRAS driver mutations was 0.01 ± 0.01 and for samples without a KRAS driver mutation − 0.06 ± 0.01 : t-test = 5.81, p = 2.5 × 10−11. The differences in the direction of the effect can be related to the fact that EGFR is an oncogene46 and wild type KRAS is a wild type tumor suppressor47 (see Discussion section for details).
Discussion
Somatic mutations play an important role in cancer development48,49,50. Missense and silent mutations are the two most common types of somatic mutations. Though most missense mutations are neutral48,49,50,51, some of them are functional and play an important role in tumorigenesis52,53. As for silent mutations, the absolute majority of them are neutral, with only rare examples of functionality54, and for this reason they can be used as a reference group to quantify strength and direction of selection on missense mutations. If the density of missense mutations in a tumor is lower compared to the density of silent mutations, the global selection is negative. Conversely, if the density of missense mutations is higher than the density of silent mutations, the global selection is positive.
Strength of selection is an indicator of how quickly a tumor evolves: tumors with signs of strong selection evolve more quickly compared to the tumors that do not show signs of strong selection4,55,56. Quickly evolving tumors better survive and propagate faster and generally tend to be more aggressive compared to slower evolving tumors15,57. It is important, therefore, to quantify strength of global selection in individual tumors as a potential biomarker of tumor aggressiveness. The goal of this study is to introduce somatic mutation-based quantitative measure of selection in individual tumors and provide its initial validation as a biomarker of tumor aggressiveness. We also compared strength of selection (both directional and absolute) with tumor mutation burden by studying their associations with select clinically relevant characteristics.
We found that lung tumors are very diverse in terms of strength and direction of selection. In the “all histology together” analysis we found that the average global selection is negative; however, some tumors bear strong signs of positive selection. We used three clinically relevant features available from COSMIC database: clinical stage, age at diagnosis, and source of the tumor tissue (primary versus metastatic) which can be useful to assess the role of selection in metastasizing. We found that in the “all histology together” analysis all clinically relevant features show U-shaped or inverse U-shaped associations with the directional selection. This observation suggests that absolute selection will be a better predictor of clinically relevant features than directional selection. This is exactly what we have found (Table 1).
Many studies have been published on the utility of somatic mutations as predictors of tumor progression, recurrence, metastasizing and response to treatment58,59,60,61,62. Tumor mutation burden is the most commonly used somatic mutation-derived biomarker63. TMB is associated with survival and response to treatment in many cancer types including lung cancer64,65,66. The goal of our study was to define global absolute selection in individual tumors and introduce it as a potential biomarker that is different from TMB. One of the drawbacks of TMB is that it depends not only on strength of selection but also on the overall mutability of the tumor. We take into account the overall mutability by using the ratio of the density of missense to the density of silent mutations. We believe that the absolute logarithm of the ratio of mutation densities better reflects strength of selection than TMB does. The results of this analysis indicate that the absolute selection may be a complementary biomarker of cancer aggressiveness to TMB. Even though we found a significant positive correlation between TMB and directional selection, the correlation was relatively small: rho = 0.12, n = 1.565, p = 0.00002. The correlation between TMB and absolute (non-directional) strength of selection was also significant but negative: rho = -0.06, n = 1.565, p = 0.01. These results suggest that global selection in tumor can be used as an independent predictor of cancer aggressiveness. The relative utility of TMB and strength of selection as biomarkers is a topic of future studies.
Histology-stratified analysis of selection demonstrated significant differences in selection among the three major lung cancer cell types. The cell types differ by absolute strength of selection, with squamous cell carcinoma showing the weakest, adenocarcinoma showing intermediate, and small cell carcinoma—the strongest absolute selection. Interestingly, the variation in absolute strength of selection followed aggressiveness, with squamous cell carcinoma considered to be slow growing and the least aggressive form of lung cancer67, small cell lung cancer considered most aggressive68, and adenocarcinoma showing intermediate aggressiveness69. This supports the idea that absolute strength of selection in tumor can be an indicator of tumor aggressiveness.
One of the possible reasons why absolute strength of global selection in tumor can be a better biomarker compared to tumor mutational burden is its dependency of copy number variation (CNV). CNVs, especially those involving whole chromosomes and large chromosomal regions, directly influence the total number of somatic mutations and, as a result, directly influence TMB. Since estimates of global selection in tumor are based on the ratio of non-synonymous to synonymous substitutions, the measure is less sensitive to the copy number variation than TMB and therefore may be more reliable.
We found that the presence of driver mutations in lung tumors was associated with significant changes in the strength of the global selection, which is not surprising taking into account the profound effect of driver mutations on clonal evolution and tumor growth rate70,71. Interestingly, oncogenic driver mutations in EGFR are associated with more negative while oncogenic driver mutations in KRAS are associated with more positive selection. This can be explained by different effects of EGFR and KRAS driver mutations on DNA repair. Driver mutations in EGFR are associated with decreased DNA repair capacity in non‐small cell lung cancer72. KRAS driver mutations, on the other hand, are associated with more efficient DNA repair in lung tumors73 which may contribute to poor response of KRAS driver mutation-positive tumors to radiotherapy74. Since the absolute majority of de novo mutations tend to have negative effect on fitness at both the population75 and cellular levels76, one can expect that higher mutability associated with EGFR drivers will result in stronger negative selection, while improved DNA repair associated with KRAS drivers will have an opposite effect: reduced negative selection as it was observed in this study.
Conclusion
To conclude, we propose to use the absolute value of the logarithm of relative densities of missense to silent mutations as a quantitative measure of selection in tumor. We hypothesize that the strength of absolute selection reflects tumor aggressiveness and may be used as a biomarker of tumor aggressiveness.
Data availability
All data generated or analyzed during this study are included in this published article (and its supplementary information files). The corresponding author will share any additional relevant data upon request (ivan.gorlov@bcm.edu).
References
Gerstung, M. et al. The evolutionary history of 2,658 cancers. Nature 578, 122–128. https://doi.org/10.1038/s41586-019-1907-7 (2020).
Ghareyazi, A. et al. Whole-genome analysis of de novo somatic point mutations reveals novel mutational biomarkers in pancreatic cancer. Cancers Basel https://doi.org/10.3390/cancers13174376 (2021).
Williams, M. J., Werner, B., Barnes, C. P., Graham, T. A. & Sottoriva, A. Identification of neutral tumor evolution across cancer types. Nat. Genet. 48, 238–244. https://doi.org/10.1038/ng.3489 (2016).
Martincorena, I. et al. Universal patterns of selection in cancer and somatic tissues. Cell 171(1029–1041), e1021. https://doi.org/10.1016/j.cell.2017.09.042 (2017).
Zapata, L. et al. Signatures of positive selection reveal a universal role of chromatin modifiers as cancer driver genes. Sci. Rep. 7, 13124. https://doi.org/10.1038/s41598-017-12888-1 (2017).
Banyai, L., Trexler, M., Kerekes, K., Csuka, O. & Patthy, L. Use of signals of positive and negative selection to distinguish cancer genes and passenger genes. Elife https://doi.org/10.7554/eLife.59629 (2021).
Zapata, L. et al. Negative selection in tumor genome evolution acts on essential cellular functions and the immunopeptidome. Genome Biol. 19, 67. https://doi.org/10.1186/s13059-018-1434-0 (2018).
Margaryan, N. V. et al. The stem cell phenotype of aggressive breast cancer cells. Cancers Basel https://doi.org/10.3390/cancers11030340 (2019).
Valero, C. et al. The association between tumor mutational burden and prognosis is dependent on treatment context. Nat. Genet. 53, 11–15. https://doi.org/10.1038/s41588-020-00752-4 (2021).
Schnidrig, D., Turajlic, S. & Litchfield, K. Tumour mutational burden: Primary versus metastatic tissue creates systematic bias. Immunooncol. Technol. 4, 8–14. https://doi.org/10.1016/j.iotech.2019.11.003 (2019).
Stein, M. K. et al. Tumor mutational burden is site specific in non-small-cell lung cancer and is highest in lung adenocarcinoma brain metastases. JCO Precis. Oncol. 3, 1–13. https://doi.org/10.1200/PO.18.00376 (2019).
Aggarwal, C. et al. Assessment of tumor mutational burden and outcomes in patients with diverse advanced cancers treated with immunotherapy. JAMA Netw. Open 6, e2311181. https://doi.org/10.1001/jamanetworkopen.2023.11181 (2023).
Jardim, D. L., Goodman, A., de Melo Gagliato, D. & Kurzrock, R. The challenges of tumor mutational burden as an immunotherapy biomarker. Cancer Cell 39, 154–173. https://doi.org/10.1016/j.ccell.2020.10.001 (2021).
Strickler, J. H., Hanks, B. A. & Khasraw, M. Tumor mutational burden as a predictor of immunotherapy response: Is more always better?. Clin. Cancer Res. 27, 1236–1241. https://doi.org/10.1158/1078-0432.CCR-20-3054 (2021).
Casas-Selves, M. & Degregori, J. How cancer shapes evolution, and how evolution shapes cancer. Evol. NY 4, 624–634. https://doi.org/10.1007/s12052-011-0373-y (2011).
Merlo, L. M., Pepper, J. W., Reid, B. J. & Maley, C. C. Cancer as an evolutionary and ecological process. Nat. Rev. Cancer 6, 924–935. https://doi.org/10.1038/nrc2013 (2006).
Zhu, L. et al. A narrative review of tumor heterogeneity and challenges to tumor drug therapy. Ann. Trans. Med. 9, 1351. https://doi.org/10.21037/atm-21-1948 (2021).
Thol, K., Pawlik, P. & McGranahan, N. Therapy sculpts the complex interplay between cancer and the immune system during tumour evolution. Genome Med. 14, 137. https://doi.org/10.1186/s13073-022-01138-3 (2022).
Balmain, A. The critical roles of somatic mutations and environmental tumor-promoting agents in cancer risk. Nat. Genet. 52, 1139–1143. https://doi.org/10.1038/s41588-020-00727-5 (2020).
Orlow, I. et al. DNA damage and repair capacity in patients with lung cancer: Prediction of multiple primary tumors. J. Clin. Oncol. 26, 3560–3566. https://doi.org/10.1200/JCO.2007.13.2654 (2008).
McGranahan, N. & Swanton, C. Clonal heterogeneity and tumor evolution: past, present, and the future. Cell 168, 613–628. https://doi.org/10.1016/j.cell.2017.01.018 (2017).
Kryazhimskiy, S. & Plotkin, J. B. The population genetics of dN/dS. PLoS Genet 4, e1000304. https://doi.org/10.1371/journal.pgen.1000304 (2008).
Jeffares, D. C., Tomiczek, B., Sojo, V. & dos Reis, M. A beginners guide to estimating the non-synonymous to synonymous rate ratio of all protein-coding genes in a genome. Methods Mol. Biol. 1201, 65–90. https://doi.org/10.1007/978-1-4939-1438-8_4 (2015).
Gu, X. d(N)/d(S)-H, a new test to distinguish different selection modes in protein evolution and cancer evolution. J. Mol. Evol. 90, 342–351. https://doi.org/10.1007/s00239-022-10064-2 (2022).
Pérez-Figueroa, A. & Posada, D. Interpreting dN/dS under different selective regimes in cancer evolution. bioRxiv 2021.2011.2030.470556 https://doi.org/10.1101/2021.11.30.470556 (2021).
Chandrashekar, P. et al. Somatic selection distinguishes oncogenes and tumor suppressor genes. Bioinformatics 36, 1712–1717. https://doi.org/10.1093/bioinformatics/btz851 (2020).
Temko, D., Tomlinson, I. P. M., Severini, S., Schuster-Bockler, B. & Graham, T. A. The effects of mutational processes and selection on driver mutations across cancer types. Nat. Commun. 9, 1857. https://doi.org/10.1038/s41467-018-04208-6 (2018).
Zhao, S. et al. Detailed modeling of positive selection improves detection of cancer driver genes. Nat. Commun. 10, 3399. https://doi.org/10.1038/s41467-019-11284-9 (2019).
Persi, E., Wolf, Y. I., Leiserson, M. D. M., Koonin, E. V. & Ruppin, E. Criticality in tumor evolution and clinical outcome. Proc. Natl. Acad. Sci. USA. 115, E11101–E11110. https://doi.org/10.1073/pnas.1807256115 (2018).
Nielsen, R. Molecular signatures of natural selection. Ann. Rev. Genet. 39, 197–218. https://doi.org/10.1146/annurev.genet.39.073003.112420 (2005).
Spielman, S. J. & Wilke, C. O. The relationship between dN/dS and scaled selection coefficients. Mol. Biol. Evol. 32, 1097–1108. https://doi.org/10.1093/molbev/msv003 (2015).
Liu, Q., Fang, L. & Wu, C. Alternative splicing and isoforms: from mechanisms to diseases. Genes Basel https://doi.org/10.3390/genes13030401 (2022).
Nakayama, T., Asai, S., Takahashi, Y., Maekawa, O. & Kasama, Y. Overlapping of genes in the human genome. Int. J. Biomed. Sci. 3, 14–19 (2007).
Gorlova Olga, K. M., Spiridon, T., Christopher, A. & Ivan, G. Identification of lung cancer drivers by comparison of the observed and the expected numbers of missense and nonsense mutations in individual human genes. Oncotarget 14, 17–29 (2022).
Pruitt, K. D. et al. The consensus coding sequence (CCDS) project: Identifying a common protein-coding gene set for the human and mouse genomes. Genome Res. 19, 1316–1323. https://doi.org/10.1101/gr.080531.108 (2009).
Alexandrov, L. B. et al. Mutational signatures associated with tobacco smoking in human cancer. Science 354, 618–622. https://doi.org/10.1126/science.aag0299 (2016).
Faheem, M., Zhang, C. J., Morris, M. N., Pleiss, J. & Oelschlaeger, P. Role of synonymous mutations in the evolution of TEM beta-lactamase genes. Antimicrob. Agents Chemother. https://doi.org/10.1128/AAC.00018-21 (2021).
Zheng, S., Kim, H. & Verhaak, R. G. W. Silent mutations make some noise. Cell 156, 1129–1131. https://doi.org/10.1016/j.cell.2014.02.037 (2014).
Gorlov, I. P., Kimmel, M. & Amos, C. I. Strength of the purifying selection against different categories of the point mutations in the coding regions of the human genome. Hum. Mol. Genet. 15, 1143–1150. https://doi.org/10.1093/hmg/ddl029 (2006).
Sondka, Z. et al. The COSMIC cancer gene census: Describing genetic dysfunction across all human cancers. Nat. Rev. Cancer 18, 696–705. https://doi.org/10.1038/s41568-018-0060-1 (2018).
Campbell, B. B. et al. Comprehensive analysis of hypermutation in human cancer. Cell 171(1042–1056), e1010. https://doi.org/10.1016/j.cell.2017.09.048 (2017).
Izumi, M. et al. Mutational landscape of multiple primary lung cancers and its correlation with non-intrinsic risk factors. Sci. Rep. 11, 5680. https://doi.org/10.1038/s41598-021-83609-y (2021).
Lusk, C. M. et al. Profiling the mutational landscape in known driver genes and novel genes in African American non-small cell lung cancer patients. Clin. Cancer Res. 25, 4300–4308. https://doi.org/10.1158/1078-0432.CCR-18-2439 (2019).
Chevallier, M., Borgeaud, M., Addeo, A. & Friedlaender, A. Oncogenic driver mutations in non-small cell lung cancer: Past, present and future. World J. Clin. Oncol. 12, 217–237. https://doi.org/10.5306/wjco.v12.i4.217 (2021).
Huang, L., Guo, Z., Wang, F. & Fu, L. KRAS mutation: From undruggable to druggable in cancer. Signal Transduct. Target Ther. 6, 386. https://doi.org/10.1038/s41392-021-00780-4 (2021).
Arteaga, C. L. The epidermal growth factor receptor: from mutant oncogene in nonhuman cancers to therapeutic target in human neoplasia. J. Clin. Oncol. 19, 32S-40S (2001).
Jancik, S., Drabek, J., Radzioch, D. & Hajduch, M. Clinical relevance of KRAS in human cancers. J. Biomed. Biotechnol. 2010, 150960. https://doi.org/10.1155/2010/150960 (2010).
Araujo, L. H. et al. Somatic mutation spectrum of non-small-cell lung cancer in African Americans: A pooled analysis. J. Thorac. Oncol. 10, 1430–1436. https://doi.org/10.1097/JTO.0000000000000650 (2015).
Ding, L. et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature 455, 1069–1075. https://doi.org/10.1038/nature07423 (2008).
Shen, H. B. et al. Impact of somatic mutations in non-small-cell lung cancer: A retrospective study of a Chinese cohort. Cancer Manag. Res. 12, 7427–7437. https://doi.org/10.2147/CMAR.S254139 (2020).
Tan, K. P., Kanitkar, T. R., Kwoh, C. K. & Madhusudhan, M. S. Packpred: Predicting the functional effect of missense mutations. Front. Mol. Biosci. 8, 646288. https://doi.org/10.3389/fmolb.2021.646288 (2021).
Malhotra, S. et al. Understanding the impacts of missense mutations on structures and functions of human cancer-related genes: A preliminary computational analysis of the COSMIC cancer gene census. PLoS One 14, e0219935. https://doi.org/10.1371/journal.pone.0219935 (2019).
Nishi, H. et al. Cancer missense mutations alter binding properties of proteins and their interaction networks. PLoS One 8, e66273. https://doi.org/10.1371/journal.pone.0066273 (2013).
Sharma, Y. et al. A pan-cancer analysis of synonymous mutations. Nat. Commun. 10, 2569. https://doi.org/10.1038/s41467-019-10489-2 (2019).
Fortunato, A. et al. Natural selection in cancer biology: From molecular snowflakes to trait hallmarks. Cold Spring. Harb. Perspect. Med. https://doi.org/10.1101/cshperspect.a029652 (2017).
Khong, H. T. & Restifo, N. P. Natural selection of tumor variants in the generation of tumor escape phenotypes. Nat. Immunol. 3, 999–1005. https://doi.org/10.1038/ni1102-999 (2002).
Northcott, J. M., Dean, I. S., Mouw, J. K. & Weaver, V. M. Feeling stress: The mechanics of cancer progression and aggression. Front. Cell Dev. Biol. 6, 17. https://doi.org/10.3389/fcell.2018.00017 (2018).
Gussow, A. B., Koonin, E. V. & Auslander, N. Identification of combinations of somatic mutations that predict cancer survival and immunotherapy benefit. NAR Cancer 3, zcab017. https://doi.org/10.1093/narcan/zcab017 (2021).
Horlings, H. M., Shah, S. P. & Huntsman, D. G. Using somatic mutations to guide treatment decisions: Context matters. JAMA Oncol. 1, 275–276. https://doi.org/10.1001/jamaoncol.2015.35 (2015).
Lipsyc, M. & Yaeger, R. Impact of somatic mutations on patterns of metastasis in colorectal cancer. J. Gastrointest. Oncol. 6, 645–649. https://doi.org/10.3978/j.issn.2078-6891.2015.045 (2015).
O’Malley, A. J., Frank, R. G. & Normand, S. L. Estimating cost-offsets of new medications: Use of new antipsychotics and mental health costs for schizophrenia. Stat. Med. 30, 1971–1988. https://doi.org/10.1002/sim.4245 (2011).
Peng, J., Xiao, L., Zou, D. & Han, L. A somatic mutation signature predicts the best overall response to anti-programmed cell death protein-1 treatment in epidermal growth factor receptor/anaplastic lymphoma kinase-negative non-squamous non-small cell lung cancer. Front. Med. Lausanne 9, 808378. https://doi.org/10.3389/fmed.2022.808378 (2022).
Fusco, M. J., West, H. J. & Walko, C. M. Tumor mutation burden and cancer treatment. JAMA Oncol. 7, 316. https://doi.org/10.1001/jamaoncol.2020.6371 (2021).
McFarland, D. C. et al. Tumor mutation burden and depression in lung cancer: Association with inflammation. J. Natl. Compr. Canc. Netw. 18, 434–442. https://doi.org/10.6004/jnccn.2019.7374 (2020).
Ricciuti, B. et al. Association of high tumor mutation burden in non-small cell lung cancers with increased immune infiltration and improved clinical outcomes of PD-L1 blockade across PD-L1 expression levels. JAMA Oncol. 8, 1160–1168. https://doi.org/10.1001/jamaoncol.2022.1981 (2022).
Wang, Z. et al. Assessment of blood tumor mutational burden as a potential biomarker for immunotherapy in patients with non-small cell lung cancer with use of a next-generation sequencing cancer gene panel. JAMA Oncol 5, 696–702. https://doi.org/10.1001/jamaoncol.2018.7098 (2019).
Howell, J.Y., and Ramsey, M.L. Squamous cell skin cancer. In StatPearls (2023).
Rudin, C. M., Brambilla, E., Faivre-Finn, C. & Sage, J. Small-cell lung cancer. Nat. Rev. Dis. Primers 7, 3. https://doi.org/10.1038/s41572-020-00235-0 (2021).
Myers, D.J., and Wallen, J.M. Lung Adenocarcinoma. In StatPearls (2023).
Gomez, K. et al. Somatic evolutionary timings of driver mutations. BMC Cancer 18, 85. https://doi.org/10.1186/s12885-017-3977-y (2018).
Salichos, L., Meyerson, W., Warrell, J. & Gerstein, M. Estimating growth patterns and driver effects in tumor evolution from individual samples. Nat. Commun. 11, 732. https://doi.org/10.1038/s41467-020-14407-9 (2020).
Zhang, L., Pradhan, B., Guo, L., Meng, F. & Zhong, D. EGFR exon 19-deletion aberrantly regulate ERCC1 expression that may partly impaired DNA damage repair ability in non-small cell lung cancer. Thorac. Cancer 11, 277–285. https://doi.org/10.1111/1759-7714.13253 (2020).
Caiola, E. et al. Base excision repair-mediated resistance to cisplatin in KRAS(G12C) mutant NSCLC cells. Oncotarget 6, 30072–30087. https://doi.org/10.18632/oncotarget.5019 (2015).
Yang, L. et al. Oncogenic KRAS drives radioresistance through upregulation of NRF2-53BP1-mediated non-homologous end-joining repair. Nucl. Acids Res. 49, 11067–11082. https://doi.org/10.1093/nar/gkab871 (2021).
Acuna-Hidalgo, R., Veltman, J. A. & Hoischen, A. New insights into the generation and role of de novo mutations in health and disease. Genome Biol. 17, 241. https://doi.org/10.1186/s13059-016-1110-1 (2016).
McFarland, C. D. et al. The damaging effect of passenger mutations on cancer progression. Cancer Res. 77, 4763–4772. https://doi.org/10.1158/0008-5472.CAN-15-3283-T (2017).
Author information
Authors and Affiliations
Contributions
I.P.G. had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis. Study concept and design: I.P.G., O.Y.G., and C.I.A. Acquisition of data: S.T. Analysis: I.P.G., O.Y.G., and S.T. Methodology: I.P.G. and C.I.A. Investigation: all authors. Interpretation of data: I.P.G., O.Y.G., and C.I.A. Writing first the draft of the manuscript: I.P.G. Critical revision of the manuscript for important intellectual content and approval: O.Y.G. and C.I.A.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gorlov, I.P., Gorlova, O.Y., Tsavachidis, S. et al. Strength of selection in lung tumors correlates with clinical features better than tumor mutation burden. Sci Rep 14, 12732 (2024). https://doi.org/10.1038/s41598-024-63468-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-63468-z
- Springer Nature Limited