
Abstract
Delayed diagnosis in patients with pulmonary tuberculosis (PTB) often leads to serious public health problems. High throughput sequencing was used to determine the expression levels of lncRNAs, mRNAs, and miRNAs in the lesions and adjacent health lung tissues of patients with PTB. Their differential expression profiles between the two groups were compared, and 146 DElncRs, 447 DEmRs, and 29 DEmiRs were obtained between lesions and adjacent health tissues in patients with PTB. Enrichment analysis for mRNAs showed that they were mainly involved in Th1, Th2, and Th17 cell differentiation. The lncRNAs, mRNAs with target relationship with miRNAs were predicted respectively, and correlation analysis was performed. The ceRNA regulatory network was obtained by comparing with the differentially expressed transcripts (DElncRs, DEmRs, DEmiRs), then 2 lncRNAs mediated ceRNA networks were established. The expression of genes within the network was verified by quantitative real-time PCR (qRT-PCR). Flow cytometric analysis revealed that the proportion of Th1 cells and Th17 cells was lower in PTB than in controls, while the proportion of Th2 cells increased. Our results provide rich transcriptome data for a deeper investigation of PTB. The ceRNA regulatory network we obtained may be instructive for the diagnosis and treatment of PTB.
Similar content being viewed by others


Introduction
Pulmonary tuberculosis (PTB) is an infectious disease caused by invasion of lung tissue by Mycobacterium tuberculosis1. PTB remains a formidable challenge to global health, being the deadliest infectious disease despite advances in vaccination, therapy, and diagnostics2. As the second-largest burden of PTB and multidrug resistance, China faces significant challenges in tackling this disease3. PTB accounts for more deaths worldwide than any other infectious disease, and is essentially curable if detected early and treated effectively4. While etiological testing is the standard for diagnosing PTB, it is hampered by several limitations5.
Recent studies have shed light on the role of immune responses and inflammatory factors in the progression of PTB, opening avenues for potential diagnostic and therapeutic targets6,7. PTB is known to impair immune function and cause systemic wasting8,9, with excessive inflammatory responses exacerbating its severity and complicating treatment10,11.
In the realm of molecular biology, gene regulation plays a crucial role in maintaining cellular function and has been implicated in the pathogenesis of complex diseases like PTB12. Noncoding RNAs (ncRNAs), particularly long noncoding RNAs (lncRNAs) and microRNAs (miRNAs), have emerged as significant players in the immune response to PTB13. LncRNAs, for instance, are key regulators in the inflammatory response to infection, particularly influencing T cell responses14, while miRNAs have been increasingly recognized for modulating host-TB bacilli interactions through immune pathways15. Competing endogenous RNAs (ceRNAs) offer a novel mechanism for regulating miRNA activity, acting as sponges through their miRNA response elements16. These findings suggest the potential of the ceRNA network as a biomarker for PTB diagnosis and treatment.
However, despite these advances, the specific roles and interactions of ncRNAs, lncRNAs, and ceRNAs in PTB remain underexplored. Previous studies have laid a foundation, but gaps persist in understanding the full scope of their regulatory functions and potential as diagnostic and therapeutic targets17. This study aims to fill these gaps by focusing on the ceRNA network in PTB. Through whole transcriptome sequencing of patients with active PTB, we seek to identify lncRNA-associated ceRNA regulatory networks, potentially unveiling novel biomarkers for PTB. Such insights could improve the early diagnosis and treatment of this global health threat.
Materials and methods
Participants
A total of 37 patients with active PTB and 37 healthy controls were recruited from Children’s Hospital of Xinjiang Uygur Autonomous Region (Table 1). Among this, 7 cases underwent biopsies due to acute hemoptysis with non-prominent clinical symptoms. The biopsy needle extracted a small amount of tissue from the patient’s lungs for testing. Additionally, lesions refer to areas of granulomatous disease as identified pathologically, and adherent refers to non-granulomatous areas, specifically the tissue surrounding the granulomas (3 cm away from the granulomatous regions). Three patients were randomly selected, including both their lesions and adjacent lung tissues. Peripheral blood samples were obtained from 37 patients with active PTB and 37 healthy controls.
Human studies were reviewed and approved by the Ethics committee of Children’s Hospital of Xinjiang Uygur Autonomous Region (No. 2019030622), and adhered to the Declaration of Helsinki. Written informed consent was obtained from all patients.
Inclusion Criteria for patients with PTB: Confirmed diagnosis of pulmonary tuberculosis based on clinical, radiological, and microbiological evidence. Exclusion Criteria for patients with PTB: Co-infection with HIV or other immunocompromising conditions; History of anti-tuberculosis treatment in the past six months; Presence of other chronic pulmonary diseases. Selection Criteria for Healthy Controls: No history or clinical signs of tuberculosis or other chronic illnesses; Age and sex-matched to the patients with PTB group.
RNA preparation
Total RNA was extracted from lesions and adjacent healthy lung tissues using Trizol (Invitrogen, MA, USA). RNA concentration was quantified using Nanodrop. The integrity and quantity of the RNA were assessed using an Agilent 2100 Bioanalyzer (Agilent Technologies, CA, USA) and a NanoDrop (Thermo, MA, USA), respectively, ensuring a RNA Integrity Number (RIN) > 7.0 for downstream applications.
Transcriptome sequencing
Sequencing libraries were prepared from the high-quality RNA samples using the NEBNext® Ultra™ RNA Library Prep Kit for Illumina® (New England Biolabs, USA), following the manufacturer’s instructions. Briefly, mRNA was enriched and fragmented, followed by first-strand cDNA synthesis using reverse transcriptase and random hexamer primers. Second-strand cDNA synthesis incorporated dUTP to mark the strand used for sequencing. End repair, A-tailing, adapter ligation, and index addition were performed before PCR enrichment of the final library. Libraries were validated for quality and quantified using the Agilent Bioanalyzer 2100 and qPCR. Size selection was targeted to cDNA fragments of 250–300 bp. Following library preparation, the sequencing was performed on an Illumina PE150 platform using the S4 flow cell and Illumina’s standard sequencing chemistry. This targeting a sequencing depth of approximately 30 million reads per sample.
For small RNA sequencing, libraries were generated using NEBNext® Multiplex Small RNA Library Prep Set for Illumina® (NEB, USA). DNA fragments corresponding to 140–160 bp, representing small RNA ligated to adaptors, were recovered and purified. The quality of these libraries was also assessed using the Agilent Bioanalyzer 2100 system. The libraries were sequenced on the same Illumina PE150 platform, targeting a sequencing depth of around 10 million reads per sample to sufficiently capture the small RNA profile.
Data processing
Raw reads of fastq format were firstly processed through in-house perl scripts. Then, a certain range of length from clean reads with high quality was chosen to do downstream analyses. Reference genome and gene model annotation files were downloaded from genome website directly (https://ftp.ensembl.org/pub/release-97/fasta/homo_sapiens/dna/). Index of the reference genome was built using Hisat2 (v2.0.5)18. On the other hand, the small RNA tags were mapped to reference sequence19 without mismatch to analyze their expression and distribution on the reference.
Quantification of transcript expression levels and differential analysis
The reads numbers mapped to each transcript were counted using feature Counts v1.5.0-p3. Then FPKM (Fragments Per Kilobase of transcript sequence per Millions) of each transcript was calculated based on the length of the transcript and reads count mapped to this transcript. The miRNA expression levels were estimated by TPM (transcript per million). Principal component analysis (PCA) was performed using prcomp R function.
Differential expression analysis between lesions and adjacent lung tissues was performed using the DESeq R package20 for miRNAs, lncRNAs and mRNAs. Benjamini–Hochberg method was used to select false discovery rate (FDR) < 0.05 to assign as differentially expressed lncRNAs (DElncRs) and differentially expressed mRNAs (DEmRs). A P < 0.05 were assigned as differentially expressed miRNAs (DEmiRs).
Construction of the ceRNA network
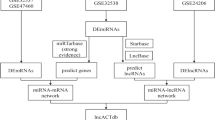
The lncRNAs with target relationship with expressed miRNAs in all samples were predicted according to miRanda software (v22)21. The mRNAs predicted by miRanda and RNAhybrid softwares (v2.1.2)22 to have a target relationship with the expressed miRNAs in all samples and identified the intersection mRNAs. Further, the Pearson’s correlation coefficient was calculated for all lncRNAs or mRNAs that had a targeting relationship with miRNAs, the threshold was set as negative correlation. MiRNAs, mRNAs with a targeted relationship and negatively correlated expression with this miRNA, and lncRNAs with a targeted relationship and negatively correlated expression with this miRNA were screened, respectively. Finally, we constructed lncRNA-miRNA-mRNA ceRNA networks.
Functional annotation analysis
Gene Ontology (GO) analysis was performed using clusterProfiler R package23 for DEmRs. Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways24 were enriched by the Enrichr R package25. We used P < 0.05 to test the significant enrichment results.
Quantitative real-time PCR
Reverse transcription of total RNA extracted from 10 lesions and adjacent healthy lung tissues into cDNA was performed using First-Strand cDNA Synthesis SuperMix (Takara, Dalian, China). Quantitative real-time PCR (qRT-PCR) then was performed with specific primers using SYBR Green PCR kit (Invitrogen), according to the manufacturer’s protocol. Expression of GAPDH was used as internal standard for lncRNAs and mRNAs, U6 was used as internal standard for miRNAs. Results were analyzed using 2−ΔΔCt method. Primer sequences which designed by sagan corporation are shown in Supplemental Table 1. A P < 0.05 was considered significant difference.
Flow cytometry
The whole blood samples of 30 patients with PTB and 30 controls were collected. Cell surface markers were then stained with antibodies against CD4 FITC (Cat 555346), CD196 PE (Cat 559562), CD183 ECD (Cat 551128) from BD (San Diego, USA), according to standard procedures. The gating strategy was CD4 + CD183 + CD196- for Th1, CD4 + CD183− CD196 for Th2, and CD4 + CD183-CD196 + for Th1726. The cells were treated with red blood cell lysate (BD, California, USA) and washed with PBS twice. Flow cytometry of cells was performed with a BD LSRII flow cytometer and analyzed with FlowJo software (v7).
Statistical analyses
R (v4.2.3) and Bioconductor (v3.17) were used for bioinformatic analyses. All data are shown as mean ± standard deviation (SD). All data are under normal distribution. Statistical significance between groups was analyzed by Student’s t-test using the SPSS software (v19.0). All experiments performed in three times.
Ethics approval and consent to participate
The study was conducted strictly in accordance with the Declaration of Helsinki and approved by the Ethics Committee of Children’s Hospital of Xinjiang Uygur Autonomous Region (No.2019030622), all respondents under investigation have signed written informed consent.
Results
Aberrantly expressed transcripts in PTB
PCA showed two relatively different distribution patterns between lesions and adjacent healthy groups (Figure S1). To identify aberrantly expressed transcripts in PTB, we performed differential analysis of transcripts expression in lesions and adjacent health tissues. By threshold screening, we identified 146 differentially expressed lncRNAs, including 64 upregulated and 82 downregulated (Fig. 1A). A total of 447 differentially expressed mRNAs were identified, including 185 upregulated and 262 downregulated (Fig. 1B). And 29 differentially expressed miRNAs were also identified, including 12 upregulated and 17 downregulated (Fig. 1C).

Differentially expressed transcripts between lesions and adjacent healthy lung tissues. (A) Volcano plot of differentially expressed lncRNAs. Red dots represent up-regulated expression, green dots represent down-regulated, and blue dots represent non-differential expression. (B) Volcano plot of differentially expressed mRNAs. Red dots represent up-regulated expression, green dots represent down-regulated, and blue dots represent non-differential expression. (C) Volcano plot of differentially expressed miRNAs. Red dots represent up-regulated expression, green dots represent down-regulated, and blue dots represent non-differential expression.
CeRNA networks for pulmonary tuberculosis
Using the online prediction website, we identified lncRNAs and mRNAs that had targeting relationships with miRNAs. Through correlation analysis, we screened the lncRNAs and mRNAs with negative correlation with miRNAs, respectively. Therefore, we constructed lncRNA-miRNA-mRNA networks. Afterward, to identify ceRNA networks that may have regulatory roles in PTB, we compared the transcripts in constructed lncRNA-miRNA-mRNA networks with the differentially expressed transcripts. Further 3 DElncRs (Fig. 2A), 2 DEmRs (Fig. 2B), 11 DEmiRs (Fig. 2C) were identified in the ceRNA networks. Therefore, we constructed two ceRNA regulator networks (LRRC34/hsa-miR-1185-1-3p/MAPK10; P4HB/hsa-miR-549a-5p/LRBA). Among them, hsa-miR-1185-1-3p and hsa-miR-549a-5p were up-regulated in PTB, and LRRC34, P4HB, MAPK10, and LRBA were down regulated in PTB (Fig. 2D).

Identification of the ceRNA regulator network in pulmonary tuberculosis. (A) Intersection between lncRNAs in ceRNA network and differentially expressed lncRNAs. (B) Intersection between mRNAs in ceRNA network and differentially expressed mRNAs. (C) Intersection between miRNAs in ceRNA network and differentially expressed miRNAs. (D) Heatmap of DElncRs, DEmRs, and DEmiRs of ceRNA regulator network.
Enrichment analysis of DEmRs
To explore the underlying pathological mechanism of PTB, we performed biological function enrichment analysis of DEmRs. GO analysis results (Fig. 3A) showed that metabolic process, cellular component organization, and cellular metabolic process were enriched in biological processes (BP). For cellular component, cytoplasm, membrane-bounded organelle, and intracellular were significantly enriched terms. Major representatives of molecular function included protein binding, binding, and molecular function. In addition, 30 KEGG pathway analysis terms were evaluated, such as adherens junction, PI3K-Akt signaling pathway, JAK-STAT signaling pathway, TGF-beta signaling pathway, B cell receptor signaling pathway, Th1 and Th2 cell differentiation, and Th17 cell differentiation (Fig. 3B, Table S2). Together, we constructed two comprehensive ceRNA regulatory networks (Fig. 3C). As we were more concerned about T cell immunity, LRRC34/hsa-miR-1185-1-3p/MAPK10 contributed to Th1, Th2, and Th17 cell differentiation.

Enrichment of GO and KEGG pathways for DEmRs. (A) Classification of top 20 significant GO terms for DEmRs. (B) Enrichment of significant KEGG pathways for DEmRs. (C) The comprehensive ceRNA regulatory networks in PTB.
Experimental validation of significant genes and immune cells
To further confirm the significant results we obtained, we performed molecular experiments in clinical samples. We examined the expression of genes in the ceRNA regulatory networks in lesions and adjunct health tissues using qRT-PCR experiments (Fig. 4A). Compared with adjacent, hsa-miR-1185-1-3p and hsa-miR-549a-5p showed elevated expression in lesions, and LRRC34, P4HB, MAPK10, and LRBA showed decreased expression in lesions. In addition, we examined the levels of Th1 cells, Th2 cells, and Th17 cells in PTB and healthy controls using flow cytometry (Fig. 4B). Th1 cells and Th17 cells were significantly less abundant in PTB than in controls, while the proportion of Th2 cells was up regulated.

Molecular experiments validate significant analytical results. (A) Expression changes of genes in ceRNA regulatory networks in lesions and adjacent healthy lung tissues of patients with PTB through qRT-PCR detection and analyzed by Student’s t-test. *P < 0.05, **P < 0.01, ***P < 0.001. (B) The proportion change of Th1, Th2, and Th17 cells in patients with PTB and controls detected using flow cytometry.
Discussion
PTB is the leading cause of death worldwide, resulting in increased medical costs and other socioeconomic burdens27. Early and accurate diagnosis is of great importance to effectively control and treat PTB infection, and prevent TB from developing into MDR-TB. Understanding the pathogenesis of pulmonary tuberculosis is beneficial for the prevention and treatment of tuberculosis. The discovery of lncRNAs mediated ceRNA regulatory network reveals new insights into PTB related mechanisms and lays a new theoretical foundation for researchers and physicians to develop effective treatments and diagnoses against this highly prevalent disease28.
This study used high-throughput sequencing analysis to identify differentially expressed lncRNAs, mRNAs, and miRNAs in the lesions and adjacent health tissues of patients with PTB. The results showed significant aberrant expression of lncRNAs, mRNAs, and miRNAs in the lesions. Patients with a history of TB likely exhibit an altered immune landscape compared to TB-naive individuals, which can arise from several factors, including but not limited to, the presence of immunological memory, and the alteration in lung architecture due to previous TB infections. Immunological memory, in particular, could lead to a heightened and more rapid immune response upon re-exposure to Mycobacterium tuberculosis, potentially altering the expression profiles of immune-related genes and pathways identified in our study.
Through miRNAs target prediction and correlation analysis, we identified a ceRNA regulatory network. Two networks were identified according to the differential expression of ceRNA transcripts. The first one consisted of LRRC34/hsa-miR-1185-1-3p/MAPK10 network, hsa-miR-1185-1-3p was up-regulated in PTB, and LRRC34 and MAPK10 were down regulated. In the second network consisting of P4HB/hsa-miR-549a-5p/LRBA, hsa-miR-549a-5p was found to be elevated in PTB and decreased in P4HB and LRBA. These data suggest that lncRNAs mediated ceRNA network, plays a key regulatory role in PTB. These results can enrich our understanding of the pathogenesis of PTB.
LRRC34 has been reported to play an important role in numerous cellular processes such as cell proliferation, differentiation, growth and cell survival29. SNPs at LRRC34 were found to promote interstitial lung abnormalities30. P4HB is more abundantly expressed in pulmonary artery muscle cells31. Previous studies have shown that overexpression of P4HB promotes liver cancer progression32, gastric cancer invasion and metastasis33, non-small cell lung cancer occurrence and growth34. MiR-1185-1-3p is significantly differentially expressed in bladder cancer35, Alzheimer’s disease35 and is involved in disease regulation. miR-549a-5p promotes tumorigenesis and metastasis by promoting angiogenesis through enhancing vascular permeability36. Until now, there have been no reports on LRRC34, P4HB, miR-1185-1-3p, and miR-549a-5p in PTB, although the results of our analysis argue for their significant involvement in the molecular regulation of PTB.
CD4 + T-cell deficiency and NK cell deficiency occur in lipopolysaccharide-responsive beige-like anchor (LRBA) protein deficiency, causing immunosuppression37. LRBA deficiency usually leads to recurrent infections, lymphoproliferative disorders, autoimmune diseases, allergic diseases38. Mitogen activated protein kinase 10 (MAPK10) is a member of the Jun terminal kinase subgroup of mitogen activated protein kinases39. The ceRNA network may be through inhibiting the expression of LRBA and MAPK10, causing immune function decline in patients, and thus promoting the occurrence of PTB. However, our results are inconsistent with those previously reported, Chen et al., found that MAPK10 is upregulated expression in tuberculosis40. This may also require an expanded sample size for validation.
Enrichment analysis showed that the differentially expressed mRNAs were involved in Th1 and Th2 cell differentiation, and Th17 cell differentiation. Interestingly, MAPK10 is also involved in these biological processes. Previous studies have shown that the percentage of Th1 cells is significantly reduced and the ratio of Th1/Th2 cells is imbalanced in patients with PTB, which leads to a decline in the body’s immune function41. Liu et al., also found a significant decrease in serum Th1 cytokine expression and a significant increase in Th2 cytokine expression in patients with PTB compared to controls6. These results suggest that the imbalance of Th1/Th2 cytokines is associated with PTB. Other studies have shown that active TB leads to the generation of mixed Th1/Th242. Transcriptional and clinical analyses revealed that inhibition of Th17 responses is associated with the development of tuberculosis43. Low levels of interleukin-17 in serum have been associated with high mortality in patients with PTB44. However, it has also been shown that the Th17 response is enhanced in patients with TB, and Th17 is involved in the pathological process of TB45. This showed a significant imbalance in Th1, Th2, and Th17 cell differentiation in PTB, which may regulated by ceRNA networks, and contributes to the disease’s immune dysfunction.
This study also has certain limitations. First, the sample size of our study was small, and the follow-up needs to expand the sample size for in-depth exploration. Second, low-level expressed genes may have been missed by our analysis due to insufficient sequencing depth. Then, the potential heterogeneity in immune responses among our study population could contribute to variability in gene expression patterns, making it challenging to delineate the specific effects of current TB infection from the lingering impacts of past infections. Finally, although we performed molecular experiments to validate the significant profiling results, it was not enough to deeply delineate the ceRNA regulatory network’s mechanism of action.
Conclusion
The lncRNAs associated ceRNA network was constructed by integrating lncRNAs, mRNAs, and miRNAs targeted interactions as well as their differential expression in the lesions and adjacent tissues of patients with PTB. They may contribute to PTB through regulatory effects of targeting Th1, Th2, and Th17 pathways for developing novel therapeutic strategies, offering new insights into the molecular mechanisms underlying PTB and paving the way for improved diagnostic and treatment approaches.
Data availability
The datasets generated and analysed in the current study are available in the NCBI BioProject database (Accession PRJNA784123).
Code availability
The codes during the current study are available from GitHub: https://github.com/GithubCDCode/TBceRNA_Python.
References
Chen, J. et al. The characteristics of spatial-temporal distribution and cluster of tuberculosis in Yunnan Province, China, 2005–2018. BMC Public Health 19(1), 1715 (2019).
Peters, J. S. et al. Advances in the understanding of Mycobacterium tuberculosis transmission in HIV-endemic settings. Lancet Infect. Dis. 19(3), e65–e76 (2019).
Wu, Z. et al. Effect of the Xpert MTB/RIF on the detection of pulmonary tuberculosis cases and rifampicin resistance in Shanghai, China. BMC Infect. Dis. 20(1), 153 (2020).
Horne, D. J. et al. Xpert MTB/RIF and Xpert MTB/RIF ultra for pulmonary tuberculosis and rifampicin resistance in adults. Cochrane Database Syst. Rev. 6, CD009593 (2019).
Nahid, P., Pai, M. & Hopewell, P. C. Advances in the diagnosis and treatment of tuberculosis. Proc. Am. Thorac. Soc. 3(1), 103–110 (2006).
Liu, Q. Y. et al. Inflammation responses in patients with pulmonary tuberculosis in an intensive care unit. Exp. Ther. Med. 15(3), 2719–2726 (2018).
Liu, S. et al. Factors associated with differential T cell responses to antigens ESAT-6 and CFP-10 in pulmonary tuberculosis patients. Medicine (Baltimore) 100(8), e24615 (2021).
Peng, X. et al. PPE11 of Mycobacterium tuberculosis can alter host inflammatory response and trigger cell death. Microb. Pathog. 126, 45–55 (2019).
Gupte, A. N. et al. Higher interleukin-6 levels and changes in transforming growth factor-beta are associated with lung impairment in pulmonary tuberculosis. ERJ Open Res. 7(1), 00390 (2021).
Cheng, C., Pan, W., Li, X. & Qu, H. Clinical effect of vitamin D supplementation on patients with pulmonary tuberculosis and its influence on the expression of immune cells and inflammatory factors. Exp. Ther. Med. 20(3), 2236–2244 (2020).
Kumar, N. P., Moideen, K., Banurekha, V. V., Nair, D. & Babu, S. Plasma proinflammatory cytokines are markers of disease severity and bacterial burden in pulmonary tuberculosis. Open Forum Infect. Dis. 6(7), ofz257 (2019).
Silva, C. A. et al. Can miRNA indicate risk of illness after continuous exposure to M. tuberculosis?. Int. J. Mol. Sci. 22(7), 3674 (2021).
Wang, L. et al. MicroRNAs as immune regulators and biomarkers in tuberculosis. Front. Immunol. 13, 1027472 (2022).
Yi, Z., Li, J., Gao, K. & Fu, Y. Identifcation of differentially expressed long non-coding RNAs in CD4+ T cells response to latent tuberculosis infection. J. Infect. 69(6), 558–568 (2014).
Kundu, M. & Basu, J. The role of microRNAs and long non-coding RNAs in the regulation of the immune response to Mycobacterium tuberculosis infection. Front. Immunol. 12, 687962 (2021).
Salmena, L., Poliseno, L., Tay, Y., Kats, L. & Pandolfi, P. P. A ceRNA hypothesis: The rosetta stone of a hidden RNA language?. Cell 146(3), 353–358 (2011).
Song, J. et al. CeRNA network identified hsa-miR-17-5p, hsa-miR-106a-5p and hsa-miR-2355-5p as potential diagnostic biomarkers for tuberculosis. Medicine (Baltimore) 102(11), e33117 (2023).
Kim, D., Paggi, J. M., Park, C., Bennett, C. & Salzberg, S. L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 37(8), 907–915 (2019).
Langmead, B., Trapnell, C., Pop, M. & Salzberg, S. L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10(3), R25 (2009).
Anders, S. & Huber, W. Differential expression analysis for sequence count data. Genome Biol. 11(10), R106 (2010).
Kozomara, A., Birgaoanu, M. & Griffiths-Jones, S. miRBase: From microRNA sequences to function. Nucleic Acids Res. 47(D1), D155–D162 (2019).
Kruger, J. & Rehmsmeier, M. RNAhybrid: microRNA target prediction easy, fast and flexible. Nucleic Acids Res. 34(Web Server issue), W451-454 (2006).
Yu, G., Wang, L. G., Han, Y. & He, Q. Y. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS 16(5), 284–287 (2012).
Kanehisa, M., Furumichi, M., Sato, Y., Kawashima, M. & Ishiguro-Watanabe, M. KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res. 51(D1), D587–D592 (2023).
Kuleshov, M. V. et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 44(W1), W90-97 (2016).
Kustrimovic, N. et al. Parkinson’s disease patients have a complex phenotypic and functional Th1 bias: Cross-sectional studies of CD4+ Th1/Th2/T17 and Treg in drug-naive and drug-treated patients. J. Neuroinflammation 15(1), 205 (2018).
Chen, Z. L. et al. Screening and identification of lncRNAs as potential biomarkers for pulmonary tuberculosis. Sci. Rep. 7(1), 16751 (2017).
Zhang, X. et al. Comprehensive analysis of long non-coding RNAs expression pattern in the pathogenesis of pulmonary tuberculosis. Genomics 112(2), 1970–1977 (2020).
Khoruddin, N. A., Noorizhab, M. N., Teh, L. K., Mohd Yusof, F. Z. & Salleh, M. Z. Pathogenic nsSNPs that increase the risks of cancers among the Orang Asli and Malays. Sci. Rep. 11(1), 16158 (2021).
Hobbs, B. D. et al. Overlap of genetic risk between interstitial lung abnormalities and idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 200(11), 1402–1413 (2019).
Kajihara, D. et al. Analysis of splice variants of the human protein disulfide isomerase (P4HB) gene. BMC Genom. 21(1), 766 (2020).
Xia, W. et al. P4HB promotes HCC tumorigenesis through downregulation of GRP78 and subsequent upregulation of epithelial-to-mesenchymal transition. Oncotarget 8(5), 8512–8521 (2017).
Zhang, J. et al. P4HB, a novel hypoxia target gene related to gastric cancer invasion and metastasis. Biomed Res. Int. 2019, 9749751 (2019).
Wang, S. M., Lin, L. Z., Zhou, D. H., Zhou, J. X. & Xiong, S. Q. Expression of prolyl 4-hydroxylase beta-polypeptide in non-small cell lung cancer treated with Chinese medicines. Chin. J. Integr. Med. 21(9), 689–696 (2015).
Usuba, W. et al. Circulating miRNA panels for specific and early detection in bladder cancer. Cancer Sci. 110(1), 408–419 (2019).
Xuan, Z. et al. Corrigendum: TKI-resistant renal cancer secretes low-level exosomal miR-549a to induce vascular permeability and angiogenesis to promote tumor metastasis. Front. Cell Dev. Biol. 9, 726535 (2021).
Shamriz, O. et al. Respiratory manifestations in LPS-responsive beige-like anchor (LRBA) protein-deficient patients. Eur. J. Pediatr. 177(8), 1163–1172 (2018).
Azizi, G. et al. Clinical, immunologic, molecular analyses and outcomes of iranian patients with LRBA deficiency: A longitudinal study. Pediatr. Allergy Immunol. 28(5), 478–484 (2017).
Gao, Y. et al. miR-335-5p suppresses gastric cancer progression by targeting MAPK10. Cancer Cell Int. 21(1), 71 (2021).
Chen, J. et al. Comprehensive analyses of potential key genes in active tuberculosis: A systematic review. Medicine (Baltimore) 100(30), e26582 (2021).
Wu, Q. et al. Study of the association of interferon-gamma gene polymorphisms and Th1/Th2 balance in tuberculosis susceptibility. Am. J. Transl. Res. 13(5), 5533–5539 (2021).
Amelio, P. et al. Mixed Th1 and Th2 Mycobacterium tuberculosis-specific CD4 T cell responses in patients with active pulmonary tuberculosis from Tanzania. PLoS Negl. Trop. Dis. 11(7), e0005817 (2017).
Scriba, T. J. et al. Sequential inflammatory processes define human progression from M. tuberculosis infection to tuberculosis disease. PLoS Pathog. 13(11), e1006687 (2017).
Chen, Y. C. et al. Prognostic values of serum IP-10 and IL-17 in patients with pulmonary tuberculosis. Dis. Markers 31(2), 101–110 (2011).
Jurado, J. O. et al. IL-17 and IFN-gamma expression in lymphocytes from patients with active tuberculosis correlates with the severity of the disease. J. Leukoc. Biol. 91(6), 991–1002 (2012).
Acknowledgements
Acknowledgement is given to Xinjiang Tianshan Youth Project (No. 2019Q008). Researchers within the Children’s Hospital of Xinjiang Uygur Autonomous Region and The Eighth Affiliated Hospital of Xinjiang Medical University must also be acknowledged for assisting with data collection.
Funding
This work was supported by the Tianshan Talent Training Program of Xinjiang Uygur Autonomous Region (2022TSYCCX0105), the State Key Laboratory of Pathogenesis, Prevention and Treatment of Central Asian High Incidence Diseases Fund (SKL-HIDCA-2022-JH1), the Natural Science Foundation of Xinjiang Uygur Autonomous Region (2021D01E33), and Xinjiang Tianshan Youth Project (No. 2019Q008).
Author information
Authors and Affiliations
Contributions
L. designed the study. T. X., Z. L. and L. M. collected data, blood samples and lung tissue. Q. W. and L. M. extracted PBMCs from blood samples and flow cytometry. T. X. did qRT-PCR. Q. L. drafted the manuscript, performed experiments and data analysis. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, Q., Xin, T., Liu, Z. et al. Construction of ceRNA regulatory networks for active pulmonary tuberculosis. Sci Rep 14, 10595 (2024). https://doi.org/10.1038/s41598-024-61451-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-61451-2
- Springer Nature Limited