
Abstract
Members of the family Trichomeriaceae, belonging to the Chaetothyriales order and the Ascomycota phylum, are known for their capability to inhabit hostile environments characterized by extreme temperatures, oligotrophic conditions, drought, or presence of toxic compounds. The genus Knufia encompasses many polyextremophilic species. In this report, the genomic and morphological features of the strain FJI-L2-BK-P2 presented, which was isolated from the Mars 2020 mission spacecraft assembly facility located at the Jet Propulsion Laboratory in Pasadena, California. The identification is based on sequence alignment for marker genes, multi-locus sequence analysis, and whole genome sequence phylogeny. The morphological features were studied using a diverse range of microscopic techniques (bright field, phase contrast, differential interference contrast and scanning electron microscopy). The phylogenetic marker genes of the strain FJI-L2-BK-P2 exhibited highest similarities with type strain of Knufia obscura (CBS 148926T) that was isolated from the gas tank of a car in Italy. To validate the species identity, whole genomes of both strains (FJI-L2-BK-P2 and CBS 148926T) were sequenced, annotated, and strain FJI-L2-BK-P2 was confirmed as K. obscura. The morphological analysis and description of the genomic characteristics of K. obscura FJI-L2-BK-P2 may contribute to refining the taxonomy of Knufia species. Key morphological features are reported in this K. obscura strain, resembling microsclerotia and chlamydospore-like propagules. These features known to be characteristic features in black fungi which could potentially facilitate their adaptation to harsh environments.
Similar content being viewed by others


Introduction
The ability of microorganisms to survive and proliferate in extreme environments is facilitated by numerous characteristics; chief among them is their capacity to withstand the competition and stress presented by their natural habitats1,2. These microorganisms are capable of adapting to both physical and chemical stressors via phenotypic plasticity3,4. Some fungi, for example, can survive under harsh conditions by developing specialized morphological and reproductive structures, such as cysts, conidia, or spores5,6. The detection of resilient black fungi persisting under oligotrophic conditions in cleanrooms of spacecraft assembly facilities (SAFs) may pose a risk of forward contamination to planets targeted for space exploration. If these fungi inadvertently hitch a ride on the components of planned space missions designed for life-detection efforts, they could potentially compromise the integrity of such explorations.
Certain fungi, categorized as polyextremotolerant, inhabit environments that are inhospitable to most other organisms, due to factors such as extreme temperatures, desiccation, radiation, chemical exposure, and nutrient scarcity7. Other polyextremophilic fungi have been isolated from a diverse range of environments, including areas with high concentrations of monoaromatic and volatile hydrocarbons, gasoline, and diesel fuel8,9,10. Moreover, certain species can thrive in the presence of polyphenols, shikimic acids, monoaromatic compounds, depsides, depsidones, diphenyl ethers, and dibenzofurans11.
The members of the genus Knufia are black fungi within the family Trichomeriaceae, order Chaetothyriales, and phylum Ascomycota12. Chaetothyriales also encompasses other families of black fungi, namely Herpotrichiellaceae, Cyphellophoraceae, and Coccodiniaceae, which are likewise regarded as polyextremotolerant13,14. Species within the family Trichomeriaceae rock-inhabiting fungi colonize rock surfaces15,16. They have also been detected on stone sculptures17,18. These fungi demonstrate physiological versatility that allows them to survive on human-made structures and in extreme habitats, such as cold and hot deserts, salterns, caves, dishwashers, roofs, solar panels, and tombs. The black fungi of the order Chaetothyriales are distinctively known for producing highly specialized polymers e.g., polyphenols (including melanins), chitin, and glucans. They also produce enzymes, such as cytochrome P450, glutathione S-transferases, catalases, and superoxide dismutases. Furthermore, they possess an efficient DNA repair system that shields them from a wide variety of toxic compounds and radiation19,20,21,22.
Knufia species are melanized fungi that grow slowly and display limited morphological differentiation. Given their highly convergent morphology, molecular analyses are often employed for their identification and phylogeny23. Recently, Knufia was established as a separate genus within the family, a decision supported by both molecular and morphological data24,25. Elucidating the genomic and phenotypic characteristics of a Knufia species enhances our understanding of the adaptive strategies they employ in extreme environments. It is important to note that as of now, only seven genomes of the family Trichomeriaceae are available in databases, such as the National Center for Biotechnology Information (NCBI), FungiDB, or Mycocosm, with the majority of them lacking genome annotation.
The objective of this study was to characterize the strain FJI-L2-BK-P2, which was isolated from the SAF used for the Mars 2020 mission at the National Aeronautics and Space Administration's (NASA) Jet Propulsion Laboratory (JPL). The morphological features of strain FJI-L2-BK-P2 were compared to those of the type strain of Knufia obscura CBS 148926T that was isolated from a the gasoline tank of a car in Italy26. The whole genome sequences (WGS) for both strains of K. obscura were generated, assembled, annotated, and subsequently used for multi-locus sequence analysis (MLSA) and WGS phylogeny. Additionally, we performed a thorough analysis of those morphological features in K. obscura FJI-L2-BK-P2, which might be potentially significant for the survival and adaptation of Knufia species in harsh environments. The genomic and morphological characteristics of the K. obscura strains discussed in this study further refine both molecular and traditional taxonomical identification of other Knufia species.
The results of this study showed that FJI-L2-BK-P2, characterized by septate hyphae, unique conidiophores, and chlamydospores, genetically aligns closely with K. obscura CBS 148926T based on molecular phylogeny analyses. In addition, FJI-L2-BK-P2 withstands extreme UV-C, indicating its importance for NASA planetary protection and provides insight into the resilience of this black fungus.
Results
In previous work27, we isolated 65 strains from two NASA SAFs and by sequencing the internal transcribed spacer (ITS) region 28 unique fungal species were identified, including a strain belonging to the genus Knufia. The ITS sequence of this Knufia isolate, FJI-L2-BK-P2, did not align with any known fungal species until 2022 when K. obscura was described by molecular phylogeny and published as a new species26. Subsequent molecular analyses revealed that the strain FJI-L2-BK-P2 was found to be closely related to the type strain of K. obscura26.
Morphology
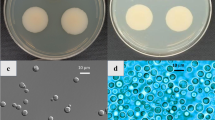
Morphological features of K. obscura FJI-L2-BK-P2 are shown in Fig. 1. On day 14, colonies on potato dextrose agar (PDA) were irregular, ~ 20 mm in diameter, dark black with yellow pigmentation near the periphery. The center of colony appeared yellowish brown and cottony (Fig. 1A). The reverse side of colonies on PDA appeared dark black with yellow pigmentation that encircled the entire colony (Fig. 1B). On day 21, colonies on oatmeal agar (OMA) were more regular (circular) in shape, ~ 25 mm in diameter, dark brown with cottony appearance in the center, and light brown on the periphery (Fig. 1C). The back of colonies on OMA appeared dark brown in the center and light brown on the periphery (Fig. 1D).

Colony morphology of Knufia obscura FJI-L2-BK-P2. Colonies after incubation at 25 °C on PDA medium (14 days after inoculation; A,B) and on OMA medium (21 days after inoculation; C,D). (A) and (C) are front side of the plates whereas (B) and (D) are back side of the plates.
Differential interference contrast (DIC) microscopic images of K. obscura FJI-L2-BK-P2 are shown in a number of figures. Hyphae and conidiophores are septate. Mature hyphae exhibit swollen structure and often appear like arthroconidia prior to separation, as recently seen in genus Pasadenomyces (Fig. 2A,B, swollen forms, Figs. 3 and 4F,G)28. Conidiophores are smooth walled (Fig. 2C) unlike the regular hyphae and seem to originate from mature hyphae (Fig. 2A). FJI-L2-BK-P2 has distinguishing features from other Knufia species by having swollen chlamydospore propagules intercalary and pear-shaped, ~ 30 µm in length, at apex of the mature hyphae (Figs. 2A,B and 5F). Conidia (oval; ~ 20 µm in diameter) are noticed on the conidiophores (Fig. 2C,D). Conidiogenous cells are monoblastic. Conidiophores are light colored unlike the dark colored chlamydogenous hyphae. Conidiophores are directly differentiated to form conidiogenous cells, which produce single conidia (Fig. 2D). In contrast, conidiophores also differentiate into branches of variable length and apical cells give rise to bulbous conidia. In short, conidia are either attached directly onto the conidiophore or on the new branch (Fig. 2D). So far, such conidiophores and conidia formation have not been reported in strains of Knufia species (or these features remained undetected or not reported). In addition, the pear-shaped chlamydospore observed at terminal ends and intercalary positions is a distinguishing morphological feature of FJI-L2-BK-P2 compared to the type strain CBS 148926T. These structures are also analogous to the microsclerotia reported in members of Phialocephala29. Two types of microsclerotia formation were observed in this strain (Figs. 3 and 4). Vegetative hyphae containing arthroconidia and chlamydogenous hyphae intermingle separately or together with each other to form dense/hard microsclerotia (Fig. 5).

Microscopic features shown by DIC microscopy for Knufia obscura FJI-L2-BK-P2. (A,B). Mature hyphae with multicellular globose apex containing large pear shaped chlamydospores. (C,D) Conidia and conidiophores (arrows). Image (A) and (C) are at ×400 total magnification and image (B) and (D) are taken at ×1000 total magnification.

Bright field (A,B) and phase contrast microscopy (C) provides evidences for presence of microsclerotia in Knufia obscura FJI-L2-BK-P2: (A,B) Extensive hyphal branching and hyphae intermingle to form dense microsclerotia type network. (C) Closely attached multi-celled chlamydospores intermingled form microsclerotia indicated by using phase contrast microscopy. Images (A) and (B) are at ×200 and image (C) is at ×1000 total magnification.

Bright field microscopy revealed chlamydospore featured mature microsclerotia. (A) Microsclerotium forms a dense clump of biomass (encircled). (B) Microsclerotia containing intermingled arthroconidia and chlamydospores. (C,D) Arthroconidia released (arrow). (E) Germinating conidia (arrow). (F,G) Arthroconidia. Images (A) and (C) are taken at ×200, (B), (D) and (E) are taken at ×400 and image (F) is taken at ×1000 total magnification.

SEM images. (A–C) Arthroconidia (encircled). (D,E) Microsclerotia. (F) Chlamydospore/chlamydospore like propagules. (G) Microsclerotia network creates dense surface layer over the fungal colony. (H) Microsclerotia.
UV-C Irradiation Resistance of Knufia obscura FJI-L2-BK-P2
Survival of the fungus was assessed as a binary outcome (survival: yes/no) across a range of UV-C exposure doses. Growth was observed for K. obscura FJI-L2-BK-P2 after exposure to UV-C doses ranging from 1,000-9,999 J/m2, confirming the strain’s resistant to UV-C (up to doses of 9,999 J/m2). This assay was based on a qualitative response across all treatments.
Variations of several molecular markers among closely related Knufia species
Sequences of two marker genes (ITS and large subunit [LSU] of rRNA) of FJI-L2-BK-P2 with the type strain of K. obscura were compared. The LSU sequence comparison showed that strain FJI-L2-BK-P2 has maximum sequence identity of 99.64% with K. obscura CBS 148926T, followed by K. hypolithi (98.48%). This data suggests that strain FJI-L2-BK-P2 is genetically related to K. obscura CBS 148926T. Similarly, ITS sequence comparison shows that strain FJI-L2-BK-P2 has maximum sequence identity (99.46%) with K. obscura CBS 148926T, followed by K. hypolithi (98.48%). In the NCBI database, only the ITS and LSU sequences were available for the K. obscura CBS 148926T type strain. To verify whether FJI-L2-BK-P2 is similar to or different from K. obscura CBS 148926T, we extracted other marker gene sequences from the WGS data for comparison. The sequence identities for tef1 (99.68%), actin (100%), rpb1 (100%), and ß-tubulin (100%) were notably high between the CBS 148926T and NASA SAF strain and confirming that FJI-L2-BK-P2 strain is K. obscura.
MLSA and whole genome phylogeny
Next, we validated the results obtained by BLASTN using MLSA analysis of rDNA and nuclear ITS, LSU, tef1, rpb1, and ß-tubulin markers that are commonly used to diagnose species within Chaetothyriales. As per MLSA, K. hypolithi, K. mediterranea, and K. aspidiotus were the closest species to K. obscura including the strain FJI-L2-BK-P2. According to our reconciled phylogenetic tree (or species tree), the Knufia clade is monophyletic and harbors at least 19 species (Fig. 6). The strain FJI-L2-BK-P2 is phylogenetically related to K. obscura CBS 148926T, thus, we diagnosed FJI-L2-BK-P2 as K. obscura. It is worth noting that K. obscura shares a common ancestor with K. hypolithi and forms a triad along with K. mediterranea. In addition to MLSA, the evolutionary relationships between K. obscura CBS 148926T and FJI-L2-BK-P2 was characterized via WGS-based phylogeny (Fig. 7). We noticed that the strains are almost identical and map next to the K. petricola clade. The Knufia clade based on WGS is monophyletic and in agreement with MLSA phylogeny. The two K. obscura strains are partitioned with strong bootstrap/aLRT support. However, the two K. obscura isolates and K. petricola are similar in terms of branch length compared to the branch that separates the two species. The branches are proportional to the number of mutations and 1000 ultrafast bootstraps. Shimodaira–Hasegawa [SH]-like approximate likelihood ratio test (SH-aLRT) confirmed the branch support and values were added to each corresponding branch of the tree. The MLSA tree was rooted with Arthrocladium fulminans CBS 136243 and we included Exophiala dermatitidis UT8656 for the WGS phylogenetic tree. At the time of analyzing data for this paper, only 4 Knufia WGSs were available in public databases. Additional genomes are needed to better comprehend the evolutionary relationships among members of the Knufia genus, as well as within the Trichomeriaceae family.

The MLSA of Knufia obscura. The genes ITS, LSU, tef1, rpb1 and ß-tubulin were used to investigate phylogenetic placement of the K. obscura via ML tree on the IQTREE2 software.

The WGS based phylogenomic analysis for K. obscura. Phylogenomic tree was constructed for Knufia strains whose genomes were available. Maximum-likelihood tree was constructed using the RAxML and ASTRAL software packages. Exophiala dermatitidis was set as the outgroup and the branches are proportional to the number of mutations. Branch fidelity used posterior probabilities, which were added next to the corresponding branches.
Genomic features of Knufia obscura
Assembled genomes of K. obscura FJI-L2-BK-P2 and CBS 148926T were assessed and found to be ~ 91% complete with similar characteristics. A summary of genomic features and annotations of the two draft genomes is presented in Table 1. The genomes of FJI-L2-BK-P2 and CBS 148926T were assembled into 140 and 54 contigs, respectively, and yielded 30.5 Mb and 30.3 Mb in size (Table 1). K. obscura appears to have 10 chromosomes according to the number of eukaryotic repeats found in the terminal end of the contigs (Table 1). The total number of genes, introns, and exons predicted (including mRNA and tRNA), the average of gene, protein, and exon lengths, as well as the annotation features, such as gene ontology (GO) terms, Interpro domains, EggNOG, MEROPS, CAZYmes, BUSCO, and secreted proteins were similar (Table 1).
Extraction of gene information from GenBank files
Apart from the above insights the number of gene identified in both the genomes were 2640 (Table S1) after eliminating all hypothetical proteins from the dataset.
Genomic clustering and shared features
Genomic clustering analysis demonstrated a substantial overlap in features between strains FJI-L2-BK-P2 and CBS 148926T. A shared 10,737 number of genes were identified in both the genomes, with 9 unique genes in FJI-L2-BK-P2 and 14 in strain CBS 148926T (Fig. 8).

Shared and Unique Genomic Clusters between Knufia obscura strains. Venn diagram depicting the shared and unique genomic clusters between Knufia obscura FJI-L2-BK-P2 and CBS 148926T strains. The figure presents a Venn diagram that illustrates the comparative genomic analysis of Knufia obscura strains, highlighting the shared and unique clusters of each.
Functional annotation and gene ontology analysis
The GO analysis highlighted predicted differences in functional annotations between the strains (Table S1). Strain FJI-L2-BK-P2 has lost functions associated with RNA 3'-end processing, transferase activity, fatty acid beta-oxidation, and sphingolipid metabolism. In contrast, strain CBS 148926T has gained functions in the urea catabolic process, metal ion transport, response to copper ion, ribosome biogenesis, and DNA replication processes (Table S2).
Gene ontology enrichment analysis
For strain CBS 148926T, gene ontology (GO) enrichment analysis revealed significant functional enrichments in various biological processes and molecular functions. Notably, CBS 148926T showed enrichment in ubiquitin-dependent endocytosis (GO:0070886), urea metabolic process (GO:0019627), protein serine/threonine kinase activity (GO:0004674), and the Wnt signaling pathway (GO:0016055). In contrast, no significant GO enrichments were observed for strain FJI-L2-BK-P2.
KEGG pathway annotations highlighting adaptability and survival
The KEGG pathway analysis for the two K. obscura strains reveals an extensive repertoire of metabolic and regulatory pathways, which are instrumental for adaptability and survival in diverse environments (Table S3).
Metabolic flexibility and energy conservation
Central to survival is metabolic flexibility, which in CBS 148926T and FJI-L2-BK-P2, is evidenced by the annotation of carbohydrate metabolic processes (GO:0005975) and organic acid metabolic processes (GO:0006082; Table S2). These pathways suggest that both strains can utilize a range of substrates for energy and biomolecule synthesis. The presence of genes involved in the metabolism of alcohols and cellular aldehydes indicates a capacity for utilizing various organic compounds, which is particularly beneficial in nutrient-scarce environments30.
Stress response and detoxification
The xenobiotic metabolic process (GO:0006805) annotations indicate that both strains have developed mechanisms to cope with potentially harmful substances, which could include natural toxins or anthropogenic pollutants (Table S3). This capability is crucial for survival in extreme conditions, where exposure to damaging agents can be frequent31.
Genetic information processing
Nucleobase-containing compound metabolic processes (GO:0006139) and DNA replication (GO:0006260) are foundational for the growth and reproduction of these organisms (Table S2). The ability to replicate and repair DNA is essential for maintaining genomic integrity, especially in environments that can cause DNA damage, such as those with high radiation or desiccation32.
Cellular processes and organization
Annotations related to cellular component organization (GO:0016043) and biogenesis, such as ribosome biogenesis (GO:0042254), indicate that these strains invest in the maintenance and efficient assembly of cellular machinery (Table S3). This investment is crucial for rapid response and adaptation to environmental changes.
Transport mechanisms
The extensive annotations related to transport (GO:0006810), including ion transport (GO:0006811) and amino acid transport (GO:0006865) (Table S3), point to sophisticated mechanisms for the uptake of nutrients and disposal of waste products. Efficient transport systems are vital for survival in environments where nutrients may be patchy or in limited supply33.
Signal transduction and inter-organismal interactions
Signal transducer activity (GO:0004871) annotations highlight the ability of these strains to perceive and respond to their environment. Furthermore, the presence of genes associated with interspecies interactions (GO:0044419) suggests these strains may engage in complex ecological relationships, potentially including symbiosis or competition, which could be key to their survival in diverse microbial communities.
Discussion
Black fungi, known for their resilience, are found in some of the planet's most extreme environments, including the desolate cold of Antarctica, arid deserts, and the crushing depths of deep-sea hydrothermal vents34. These fungi have evolved a suite of adaptive mechanisms that enable them to cope with severe environmental stressors. Among these adaptations are pigmentation and specialized dormant structures like conidia and chlamydospores, which allow the fungi to withstand prolonged periods of desiccation35. In addition to these morphological adaptations, black fungi have developed unique metabolic pathways that support their survival in habitats where most life forms would struggle21,36.
K. obscura, a species within the resilient Trichomeriaceae family, is particularly adept at enduring extreme conditions, ranging from intense temperatures, drought, and nutrient scarcity to the presence of toxic compounds. Strains of K. obscura have been isolated from a variety of environments, which include human-made habitats (e.g., gasoline tank of the car)26, and natural environments (e.g., soils of Antarctica)26. The broad ecological range and adaptability suggest that K. obscura has the potential to be inadvertently present in sensitive environments, such as an SAF. The extremophilic capacity of K. obscura raises the question about its introduction, whether through environmental exposure or human-mediated transfer, and its survivability in the oligotrophic conditions of cleanrooms. To fully understand the risks posed by such extremophiles and their adaptability, further research into the genomic diversity of Knufia species is vital. Such investigations will provide insights into their potential to hitchhike spacecraft surfaces and endure the rigors of space.
To date, MycoBank lists 22 recognized species within the genus Knufia. Fungorum catalogues 21 species, omitting K. aloicola as reported in MycoBank. Genomic data for these species are scarce, with only the genomes of two K. petricola strains are currently available. The lack of WGS for the remaining 21 validly described Knufia species underscores the pressing need for extensive genetic research to clarify the evolutionary relationships within this genus and the wider Trichomeriaceae family.
In this study, we have not only described the morphological characteristics of the K. obscura strain FJI-L2-BK-P2 but also generated WGS for this strain along with the type strain CBS 148926T. Through rigorous phylogenetic analysis, employing MLSA and WGS methodologies, we have verified the phylogenetic affiliation of FJI-L2-BK-P2. The use of both Illumina and Oxford Nanopore Technology (ONT) sequencing methods was not to compare the DNA sequence composition accuracy between these methods, as their consistency is well-established37. The short-read sequencing (Illumina) of the ITS region of FJI-L2-BK-P2 showed a 99.46% identity to the corresponding region in the type strain CBS 148926T that was sequenced using long read sequencing by the ONT. The MLSA and WGS-based analyses showed that both strains were phylogenetically identical.
The functional analysis predict substantially shared genomic features reflects the core genome conservation, while the presence of unique genes may be representative of strain-specific adaptations. The significant enrichment of ubiquitin-dependent endocytosis in strain CBS 148926T predicts the advanced intracellular trafficking capability meant for contributing regulation of its cellular processes38,39,40. The identification of the predicted urea metabolic process can equip the species for an efficient nitrogen utilization pathway in nutrient-limited environments41,42. The presence of predicted protein serine/threonine kinase activity enrichment is indicative of it’s abilities to facilitate several cellular activities for growth and response to environmental stimuli43. The lack of significant GO enrichment and specific gene functions in FJI-L2-BK-P2 could also be due to the evolutionary trajectory of FJI-L2-BK-P2, or the sensitivity of the sequencing performed for it.
The KEGG pathway annotations underscore the genetic potential of strain CBS 148926T and FJI-L2-BK-P2 for metabolic versatility, environmental resilience, and interactive complexity. The core genome's conservation highlights essential survival traits, while genomic differences underscore the species' adaptive flexibility and their potential utility in biotechnological applications. Further research into the ecological implications of the unique genomic traits may provide deeper understanding of the evolutionary strategies of these fungi.
A systematic review of available literature for Knufia species suggests that the morphological features of conidiophores and conidia observed in FJI-L2-BK-P2 have not been noticed or reported in other members of Knufia, whereas similar conidiophores and conidia formations were prominently described in several members of Phialocephala, a genus of the order Helotiales29. Chlamydospore-like propagules were noticed in K. hypolithi in terminal and intercalary positions. However, these propagules found in K. hypolithi were subcylindrical to ovoid-ellipsoid44. These structures in K. walvisbayicola had subcylindrical to elongated ellipsoid-ovoid shape44. Arthroconidia were reported in K. vaticanii CBS 139722 and K. marmoricola CBS 139726. Enlarged, thick walled and brown bodies, containing endoconidia were reported in K. marmoricola CBS 139726 and K. mediterranea CBS 139721, which seem similar to chlamydospore-like propagules of strain FJI-L2-BK-P215. Thick-walled rugose cells and terminal and intercalary multicellular bodies containing endoconidia were also observed in K. karalitana CBS 13972045, which seem similar to chlamydospore-like propagules described above by various authors. In K. separata CGMCC 3.17337, swollen cells like arthroconidia were reported globose to sub-globose. Multicellular bodies globose, ellipsoidal, or irregular, dark brown were also reported in K. separata, which seem similar to the endoconidia containing cells or chlamydospore-like propagules45. Microsclerotium-like structures, round to ovoid multicellular bodies, and chlamydospore-like propagules were also observed in K. obscura type strain CBS 148926T26. Despite minimal phylogenetic differences between the strains FJI-L2-BK-P2 and K. obscura CBS 148926T, the former displays a more varied morphology not found in the latter26. In conclusion, there are swollen cells in FJI-L2-BK-P2 (as seen in Fig. 4A–C), which bear similarity to those reported in Knufia species and are often referred to as arthroconidia46. More enlarged thick bodies containing endoconidia were also documented15 which are very similar to the chlamydospores seen in Fig. 2A,B (and might contain endoconidia inside). In phase contrast images we observed structures that were similar to chlamydospores and endoconidial forms (Fig. 3C; encircled). The terminal chlamydospore propagules in FJI-L2-BK-P2 were swollen, pear-shaped, and multicellular. Table 2 contains comparative information about all morphological features of Knufia strains discussed above.
In some fungi, melanized multicellular globose structures similar to the chlamydospore-like propagules are part of firm, frequently rounded mass of hyphae. They are abundant in aerial mycelia and later form crust on colony surfaces with or without the addition of host tissue or soil34. They are termed microsclerotia, which survive longer periods in adverse habitats (up to 15 years)29,47,48. Formation of microsclerotia was observed in members of Phialocephala29, which appears to be a convergent morphological characteristic of adaptation. These structures were described differently by various taxonomists and a uniform protocol would be desirable to avoid confusion. It is noteworthy that microsclerotium-like structures were also observed in some recently defined novel fungal species from SAF28.
The environments from which Knufia species have been reported are extremely harsh (e.g., petroleum hydrocarbons in a gas tank), characterized by low nutrient availability (e.g., cleanrooms), high salinity, and severe temperature fluctuations—conditions that present significant challenges for most organisms49. These fungi have developed a number of mechanisms to cope with such conditions50. For instance, their conidia and other specialized structures, such as chlamydospores, allow these fungi to enter a dormant state for survival in desiccated conditions over long periods51,52,53. Moreover, the presence of chlamydospore-like propagules and microsclerotia observed in K. obscura FJI-L2-BK-P2, and in some members of Phialocephala have been reported to be adaptive traits that aid in tolerating adverse environments34.
The resilience of K. obscura to UV-C radiation is in line with the capabilities of extremophiles, which are adept at surviving in conditions considered extreme by human standards. The biochemical and physiological changes that extremophiles undergo, such as producing extremolytes and extremozymes, are essential for understanding the boundaries of life on Earth and are of significant interest in astrobiology. The survival mechanisms inferred from the genomes of K. obscura and its isolation highlight the necessity for rigorous decontamination procedures in SAFs to avoid potential biological contamination during space missions. Furthermore, understanding their survival mechanisms could potentially provide insights into how lifeforms persist in harsh and seemingly inhospitable conditions in extraterrestrial environments. Therefore, further research in black fungi and their survival strategies is crucial for the effective implementation of planetary protection measures.
Materials and methods
Sample collection and isolation of fungi
The strain FJI-L2-BK-P2 was isolated from samples collected at JPL-SAF in April 2018 as previously described28. Samples were spread onto potato dextrose agar (PDA, Difco, #213400) containing chloramphenicol (25 µg/mL) and incubated at 25 °C for 7 days. Subsequent culturing was also carried out on PDA medium (unless noted otherwise).
Morphological analysis
To characterize colony morphology, the fungal strain FJI-L2-BK-P2 was streaked onto PDA and oatmeal agar (OMA, Difco, #255210) and incubated at 25 °C. Colony diameter (in mm), structure, pigmentation, and other morphological characteristics were recorded after 14 days and 21 days. Photographs of the colonies were taken using a smartphone camera (Samsung Galaxy A32, Model No. SM-A325M/DS). The slide culture technique (~ 30 days growth on PDA) was used to observe cell morphology by employing bright-field light microscopy with DIC54. The DIC images were generated on a Nikon Ti2-E inverted microscope with Nikon NIS Elements software. In addition, to gain more morphological insight phase contrast microscopy was used on a loopful of scraped mycelium mixed with a drop of water or lactophenol cotton blue (Z68, Hardy Diagnostics). Phase contrast images were generated on an Olympus BX53 microscope equipped with an Olympus DP25 camera and Olympus cellSens software. Measurements of fungal morphological structures were taken with either the Olympus cellSens or Nikon NIS Elements software.
Scanning electron microscopy
Sample preparation and scanning electron microscopy (SEM) were performed as described previously28. In brief, fungal samples were obtained from cultures cultivated on PDA plates. These samples were then immersed in chilled 2.5% glutaraldehyde (Ted Pella Inc.) solution in 0.1 M sodium cacodylate buffer (Sigma–Aldrich) and incubated at 4 °C for 1 h. Subsequently, the samples were subjected to three washes with 0.1 M sodium cacodylate buffer to remove any excess fixative. Dehydration of the samples was carried out using isopropyl alcohol (IPA) with incremental concentrations from 50 to 100% (50%, 70%, 80%, 90%, 95%, and three times 100%). Following dehydration, the samples were stored at 4 °C in 100% IPA. As previously described55, critical point drying was performed to prepare the samples, which were then subjected to SEM using an FEI Quanta 200F scanning electron microscope (Thermo Fisher Scientific).
Internal transcribed spacer (ITS)-based fungal identification
Initial identification of the fungus was performed by sequencing the ITS amplicon. Fungal biomass grown on PDA plates was collected and genomic DNA (gDNA) extracted with a Maxwell-16 MDx automated system, following the manufacturer’s instructions (Promega). The ITS region was amplified by polymerase chain reaction (PCR) with primers ITS 1F (5ʹ-CTT GGT CAT TTA GAG GAA GTA A-3′)56, and Tw13 (5′-GGT CCG TGT TTC AAG ACG-3′)57. The PCR conditions and sample preparation steps for sequencing were described elsewhere58. The ITS sequences were also included in the MLSA-based analysis.
Whole genome sequencing
To sequence the genome of FJI-L2-BK-P2, gDNA was extracted from 1 g of wet biomass scraped from a PDA plate, using the ZymoBIOMICS MagBead DNA kit (Zymo Research) and a Precellys homogenizer (Bertin Technologies). Quality of genomic DNA was verified by gel electrophoresis and quantified by spectrophotometric measurement (Nanophotometer NP80 Mobile, Implen). Library preparation followed the Illumina Nextera Flex Protocol (Illumina document # 1000000025416 v07). The DNA was used for library preparation, and 5 to 12 cycles of PCR amplification were carried out to normalize the output depending on the input DNA concentration. The amplified genomic DNA fragments were indexed and pooled in a 384-plex configuration with dual-index adapters. WGS was performed on a NovaSeq 6000 S4 flow cell PE 2 × 150 platform with a paired-end module.
The genome was assembled after filtering with NGS QC Toolkit v2.3 for high-quality (HQ) vector and adaptor-free reads (cutoff read length for HQ 80%; cutoff quality score 20)59. The filtered reads were used with the Automatic Assembly for the Fungi (AAFTF v 0.3.3) pipeline60, as follows: (i) raw Illumina reads were trimmed using BBDuk module of BBMap (v 38.95)61; (ii) contaminant reads were removed using BBMap; (iii) the polished reads were assembled, using SPAdes v 3.15.4; (iv) contaminant contigs were identified and purged with vectrim and sourpurge steps, using the nucleotide Basic Local Alignment Search Tool (BLASTN)62; (v) duplicated contigs were identified and removed, using minimap263; (vi) assembled contigs were polished with the Pilon v1.24 software64; (vii) the final scaffolds were sorted by length and the fasta headers renamed for deposit at the National Center for Biotechnology Information (NCBI). The assembly quality was verified with QUAST 5.1.065.
For Knufia obscura CBS 148926T, gDNA was extracted from PDA-grown mycelium, using the DNeasy PowerSoil Kit (Qiagen) and following manufacturer’s protocol. Oxford Nanopore Technologies sequencing was performed on a GridION MK1 sequencer using a R10.4flow cell (FLO-MIN114) and a library synthesized from Q20 + EA (early access) ligation reagents (SQK-LSK114). The raw reads were base-called using MinKNOW v29.10.8 with a mean quality score of 16.3 and a mode of 18.03. All reads went through base calling with Guppy v5.1.1366. The genome was assembled with Flye assembler (version 2.9.2) by setting up the parameters g-31.5m and i-2. Assembly quality and genome features were verified with QUAST 5.1. 065.
Multi locus sequence analysis (MLSA)
We used the MLSA approach for the phylogenetic characterization of the two fungal strains. Initially, ITS amplicon sequences were used to determine preliminary genus assignment using NCBI BLAST62. After WGS was available, we extracted the desired nuclear markers according to the Chaetothyriales MLSA scheme from each genome, using a python wrapper script with the BLASTN tool62. Once the target genes were located and extracted from each genome, we performed a BLASTN analysis against the NCBI nucleotide database to confirm the homology with other Chaetothyriales species62. The gene sequences of ITS, LSU, translational elongation factor (tef1), RNA polymerase I subunit (rpb1) and ß-tubulin were retrieved from various Trichomeriaceae species (Table S4), and Arthrocladium fulminans CBS 136243 was used as outgroup to generate the phylogenetic tree67. Individual alignments for each genetic marker were generated by Practical Alignments with the Sate and TrAnsitivity (PASTA) software68. Next, we used ClipKIT to trim uninformative sites from the DNA alignments according to the smart-gap mode69. Species tree inference was reconciliated from these alignments using the concatenation approach via IQ-TREE v 2.1.170. The best DNA substitution model was inferred with the ModelFinder approach71, and branch support was calculated using ultrafast bootstraps and an SH-aLRT72,73. The tree topologies were then visualized with FigTree v 1.4.474.
Genome annotation
The genomes of the two K. obscura strains were annotated with the funannotate v1.8.1 pipeline75. We masked the repetitive DNA sequences using TANTAN via the funannotate mask command76. We predicted gene structure and content using different evidence-based and ab initio gene prediction algorithms with the funannotate predict command. Briefly, the steps funannotate performs start with using BUSCO (Chaetothyriales_odb10 database) to find conserved gene models for training the ab initio predictors Augustus77, glimmerhmm78, and SNAP79. We also used GeneMark-ES gene finder in iterative unsupervised mode, using the option for fungal genomes. Next, funannotate computed consensus gene models with EVidenceModeler to obtain a weighted consensus gene structure79; all predictors had weight = 1, but Augustus HiQ models were set to weight = 2. Gene models with less than 50 amino acids (aa) in length or identified as transposable elements were removed. Funannotate used tRNAscan-SE to predict tRNAs80. The two K. obscura strains’ annotation also applied previous functional analysis from Interproscan81, Eggnog82, Pfam83, CAZYme84, MEROPS85, BUSCO77, and Secreted proteins86. Telomers were identified based on repeat monomer pattern ([TAA[C]+]) occurring in the end of scaffolds with minimum number of 2. Gene Ontology (GO) terms were appended to the final annotation file in ASN.1 format for deposit into NCBI GenBank. The assembled genomes were submitted to fungiSMASH, the fungal-specific pipeline for antiSMASH87, for prediction of the presence of biosynthetic gene clusters. The predicted proteomes of the two Knufia obscura strains were compared using the OrthoVenn3 tool88. The shared and unique ortholog protein clusters were submitted to KEGG analysis revealing common and unique biological features of those two close related taxa89,90,91. Gene gain and loss were also evaluated using the CAFE5 pipeline92. GO enrichment analysis were calculated using the log2 Ratio of Classes option as a metric and selecting the significantly enriched terms.
Extraction of gene information from GenBank (.gbk) files
A comprehensive methodology to extract and analyze gene information from GenBank files was developed by using Python (version 3.x), Biopython (version 1.78)93, and Pandas (version 1.2.x) for automated data processing94. GenBank files were parsed using the SeqIO module from Biopython to extract gene and coding sequence (CDS) features, including gene names, product descriptions, and genomic locations. We structured this information into tabular datasets, focusing on removing rows lacking gene names and entries corresponding to hypothetical proteins to ensure the analysis focused on genes with known or predicted functions. Further data cleanup steps were implemented to refine the datasets, including renaming columns with unique identifiers, merging datasets based on gene names using an outer join to preserve all unique gene entries, and creating a consolidated 'Gene Name' column. Additional filters were used to eliminate rows with NaN values in specific columns, remove duplicates in specific gene identifiers, and exclude entries without product/proteins names. The final, cleaned dataset was exported to an Excel spreadsheet with Pandas' to_excel method94.
Whole genome-based phylogenetic tree
To gain a comprehensive understanding of the phylogenetic positioning of the two strains of K. obscura, we conducted a phylogenomic analysis utilizing conserved genes within the fungal kingdom. The PHYling pipeline was employed for this purpose95. Protein models representing the Trichomeriaceae family were obtained from the Mycocosm portal96. For genomes lacking annotations, contigs from each species were retrieved (Table S5), and gene models were predicted using the same approach as described earlier for both K. obscura strains. Subsequently, the hmmsearch tool, available in the HMMER v3.3.2 software, was employed to identify homologous sequences present in the fungi_odb10 database. These sequences serve as a universal benchmark for evolutionary investigations in fungi. Individual protein alignments were constructed using the hmmbuild function of HMMER v3.3.2, and any erroneous positions in the alignments were removed using the ClipKIT v1.3.0 tool with the smartgap function. A phylogenetic tree was then generated using the IQTREE2 software70. In summary, a species tree was created based on 686 protein markers. Additionally, individual gene trees were generated to calculate concordance factors for each branch. We also determined ultrafast bootstraps and conducted an SH-aLRT to assess branch support72. Finally, the resulting tree was visualized using FigTree v1.4.4.
UV-C resistance evaluation
The radiation resistance in strain FJI-L2-BK-P2 was evaluated using methods previously employed for the strains Pasadenomyces melaninifex and Floridaphiala radiotolerans except for the different cfu counts per coupon and wide range of UV-C doses28. Round aluminum coupons (Diameter = 13 mm, thickness 2 mm) loaded with a suspension (150 μL) of arthroconidia and chlamydospores (153/150 μL of sterile water) were exposed to UV-C doses ranging from 1000 to 9999 J/m2 (1000, 2000, 3000, 5000, 7000, 8000, and 9999 J/m2) using a UV Crosslinker CL-1000 (UVP Inc). For each radiation dose point, the experiment was conducted with three technical replicates. After radiation exposures, the test coupon was placed into potato dextrose broth and incubated at 25 °C for 7 days. The survival of fungi was recorded as a binary outcome (yes/no).
Informed consent statement
This manuscript was prepared as an account of work sponsored by NASA, an agency of the US Government. The US Government, NASA, California Institute of Technology, Jet Propulsion Laboratory, and their employees make no warranty, expressed or implied, or assume any liability or responsibility for the accuracy, completeness, or usefulness of information, apparatus, product, or process disclosed in this manuscript, or represents that its use would not infringe upon privately held rights. The use of, and references to any commercial product, process, or service does not necessarily constitute or imply endorsement, recommendation, or favoring by the US Government, NASA, California Institute of Technology, or Jet Propulsion Laboratory. Views and opinions presented herein by the authors of this manuscript do not necessarily reflect those of the US Government, NASA, California Institute of Technology, or Jet Propulsion Laboratory, and shall not be used for advertisements or product endorsements.
Data availability
This Whole Genome Shotgun project for strain CBS 148926T and FJI-L2-BK-P2 has been deposited at DDBJ/ENA/GenBank under the accessions JAVHJV000000000 and JAKLMC000000000. The versions described in this paper are version JAVHJV010000000 and version JAKLMC010000000.
References
Muggia, L., Ametrano, C. G., Sterflinger, K. & Tesei, D. An overview of genomics, phylogenomics and proteomics approaches in Ascomycota. Life https://doi.org/10.3390/life10120356 (2020).
Chander, A. M. et al. Genome insight and comparative pathogenomic analysis of Nesterenkonia jeotgali strain CD08_7 isolated from duodenal mucosa of celiac disease patient. Front. Microbiol. 8, 129. https://doi.org/10.3389/fmicb.2017.00129 (2017).
Min, K., Neiman, A. M. & Konopka, J. B. Fungal pathogens: Shape-shifting invaders. Trends Microbiol. 28, 922–933. https://doi.org/10.1016/j.tim.2020.05.001 (2020).
Rong, M. et al. Phenotypic plasticity of Staphylococcus aureus in liquid medium containing vancomycin. Front. Microbiol. 10, 809. https://doi.org/10.3389/fmicb.2019.00809 (2019).
Corona Ramirez, A. et al. Assessment of fungal spores and spore-like diversity in environmental samples by targeted lysis. BMC Microbiol. 23, 68. https://doi.org/10.1186/s12866-023-02809-w (2023).
Amiri Fahliyani, S., Rastegari, A. A., Yadav, N. & Yadav, A. N. Human fungal pathogens: Diversity, genomics, and preventions. Recent Trends Mycol. Res. Vol. 1 Agric. Med. Perspect. 371–394 (2021).
Isola, D., Scano, A., Orru, G., Prenafeta-Boldu, F. X. & Zucconi, L. Hydrocarbon-contaminated sites: Is there something more than Exophiala xenobiotica? New insights into black fungal diversity using the long cold incubation method. J. Fungi https://doi.org/10.3390/jof7100817 (2021).
Medaura, M. C., Guivernau, M., Moreno-Ventas, X., Prenafeta-Boldu, F. X. & Vinas, M. Bioaugmentation of native fungi, an efficient strategy for the bioremediation of an aged industrially polluted soil with heavy hydrocarbons. Front. Microbiol. 12, 626436. https://doi.org/10.3389/fmicb.2021.626436 (2021).
Grube, M., Muggia, L. & Gostinčar, C. Niches and adaptations of polyextremotolerant black fungi. In Polyextremophiles: Life Under Multiple Forms of Stress, 551–566 (2013).
Rodriguez-Rodriguez, C. E., Rodriguez, E., Blanco, R., Cordero, I. & Segura, D. Fungal contamination of stored automobile-fuels in a tropical environment. J. Environ. Sci. 22, 1595–1601. https://doi.org/10.1016/s1001-0742(09)60294-1 (2010).
Yousuf, S. & Choudhary, M. I. Lichens: Chemistry and biological activities. Stud. Nat. Prod. Chem. 43, 223–259 (2014).
Tesei, D. et al. Effects of simulated microgravity on the proteome and secretome of the polyextremotolerant black fungus Knufia chersonesos. Front. Genet. 12, 638708 (2021).
Schoch, C. L. et al. A multigene phylogeny of the Dothideomycetes using four nuclear loci. Mycologia 98, 1041–1052 (2006).
Quan, Y. et al. The origin of human pathogenicity and biological interactions in Chaetothyriales. Fungal Divers. 1–22 (2023).
Isola, D. et al. Extremotolerant rock inhabiting black fungi from Italian monumental sites. Fungal Divers. 76, 75–96 (2016).
Selbmann, L. et al. Shed light in the daRk lineagES of the fungal tree of life—STRES. Life 10, 362 (2020).
Wang, Y., Zhang, H., Liu, X., Liu, X. & Song, W. Fungal communities in the biofilms colonizing the basalt sculptures of the Leizhou Stone Dogs and assessment of a conservation measure. Herit. Sci. 9, 1–12 (2021).
De Leo, F., Marchetta, A. & Urzì, C. Black fungi on stone-built heritage: Current knowledge and future outlook. Appl. Sci. 12, 3969 (2022).
Karentz, D. Beyond xeroderma pigmentosum: DNA damage and repair in an ecological context. A tribute to James E. Cleaver. Photochem. Photobiol. 91, 460–474 (2015).
Cordero, R. J. & Casadevall, A. Functions of fungal melanin beyond virulence. Fungal Biol. Rev. 31, 99–112 (2017).
Teixeira, M. D. M. et al. Exploring the genomic diversity of black yeasts and relatives (Chaetothyriales, Ascomycota). Stud. Mycol. 86, 1–28 (2017).
Oren, A. & Gunde-Cimerman, N. Mycosporines and mycosporine-like amino acids: UV protectants or multipurpose secondary metabolites?. FEMS Microbiol. Lett. 269, 1–10 (2007).
de Hoog, G. S., Vicente, V. A. & Gorbushina, A. A. The bright future of darkness–the rising power of black fungi: Black yeasts, microcolonial fungi, and their relatives. Mycopathologia 175, 365–368. https://doi.org/10.1007/s11046-013-9666-8 (2013).
Zakharova, K. et al. Microcolonial fungi on rocks: A life in constant drought?. Mycopathologia 175, 537–547. https://doi.org/10.1007/s11046-012-9592-1 (2013).
Nai, C. et al. Nutritional physiology of a rock-inhabiting, model microcolonial fungus from an ancestral lineage of the Chaetothyriales (Ascomycetes). Fungal Genet. Biol. 56, 54–66. https://doi.org/10.1016/j.fgb.2013.04.001 (2013).
Isola, D. et al. Knufia obscura sp. nov. and Knufia victoriae sp. nov., two new species from extreme environments. Int. J. Syst. Evol. Microbiol. https://doi.org/10.1099/ijsem.0.005530 (2022).
Blachowicz, A. et al. The isolation and characterization of rare mycobiome associated with spacecraft assembly cleanrooms. Front. Microbiol. 13, 777133. https://doi.org/10.3389/fmicb.2022.777133 (2022).
Chander, A. M. et al. Description and genome characterization of three novel fungal strains isolated from Mars 2020 mission-associated spacecraft assembly facility surfaces—recommendations for two new genera and one species. J. Fungi 9, 31 (2023).
Tanney, J. B., Douglas, B. & Seifert, K. A. Sexual and asexual states of some endophytic Phialocephala species of Picea. Mycologia 108, 255–280 (2016).
Sun, W. et al. Metabolic engineering of an acid-tolerant yeast strain Pichia kudriavzevii for itaconic acid production. Metab. Eng. Commun. 10, e00124. https://doi.org/10.1016/j.mec.2020.e00124 (2020).
Khan, M. F., Hof, C., Niemcova, P. & Murphy, C. D. Recent advances in fungal xenobiotic metabolism: Enzymes and applications. World J. Microbiol. Biotechnol. 39, 296. https://doi.org/10.1007/s11274-023-03737-7 (2023).
Raghuraman, M. K. et al. Replication dynamics of the yeast genome. Science 294, 115–121. https://doi.org/10.1126/science.294.5540.115 (2001).
Mitchison-Field, L. M. Y. et al. Unconventional cell division cycles from marine-derived yeasts. Curr. Biol. 29, 3439-3456.e3435. https://doi.org/10.1016/j.cub.2019.08.050 (2019).
Biju, C. N. et al. Significance of microsclerotia in the epidemiology of black pepper anthracnose and an approach for disease management in nurseries. J. Phytopathol. 165, 342–353. https://doi.org/10.1111/jph.12567 (2017).
Martin, I., Galvez, L., Guasch, L. & Palmero, D. Fungal pathogens and seed storage in the dry state. Plants https://doi.org/10.3390/plants11223167 (2022).
Quan, Y. et al. Black fungi and ants: A genomic comparison of species inhabiting carton nests versus domatia. IMA Fungus 13, 4. https://doi.org/10.1186/s43008-022-00091-5 (2022).
Linde, J. et al. Comparison of Illumina and Oxford Nanopore Technology for genome analysis of Francisella tularensis, Bacillus anthracis, and Brucella suis. BMC Genomics 24, 258. https://doi.org/10.1186/s12864-023-09343-z (2023).
Piper, R. C. & Katzmann, D. J. Biogenesis and function of multivesicular bodies. Annu. Rev. Cell Dev. Biol. 23, 519–547. https://doi.org/10.1146/annurev.cellbio.23.090506.123319 (2007).
Hicke, L. & Dunn, R. Regulation of membrane protein transport by ubiquitin and ubiquitin-binding proteins. Annu. Rev. Cell Dev. Biol. 19, 141–172. https://doi.org/10.1146/annurev.cellbio.19.110701.154617 (2003).
Raiborg, C. & Stenmark, H. The ESCRT machinery in endosomal sorting of ubiquitylated membrane proteins. Nature 458, 445–452. https://doi.org/10.1038/nature07961 (2009).
Marzluf, G. A. Genetic regulation of nitrogen metabolism in the fungi. Microbiol. Mol. Biol. Rev. 61, 17–32. https://doi.org/10.1128/mmbr.61.1.17-32.1997 (1997).
Scazzocchio, C. The fungal GATA factors. Curr. Opin. Microbiol. 3, 126–131. https://doi.org/10.1016/s1369-5274(00)00063-1 (2000).
Barske, T. et al. The role of serine/threonine-specific protein kinases in cyanobacteria—SpkB is involved in acclimation to fluctuating conditions in Synechocystis sp. PCC 6803. Mol. Cell Proteomics 22, 100656. https://doi.org/10.1016/j.mcpro.2023.100656 (2023).
Crous, P. W. et al. Fungal planet description sheets: 1182–1283. Persoonia Mol. Phylogeny Evol. Fungi 46, 313 (2021).
Sun, W. et al. Unveiling the hidden diversity of rock-inhabiting fungi: Chaetothyriales from China. J. Fungi 6, 187 (2020).
Song, Z. Fungal microsclerotia development: Essential prerequisites, influencing factors, and molecular mechanism. Appl. Microbiol. Biotechnol. 102, 9873–9880 (2018).
Fan, R. et al. Vayg1 is required for microsclerotium formation and melanin production in Verticillium dahliae. Fungal Genet. Biol. 98, 1–11. https://doi.org/10.1016/j.fgb.2016.11.003 (2017).
Hu, D. F. et al. Whole genome wide expression profiles on germination of Verticillium dahliae microsclerotia. PLoS ONE https://doi.org/10.1371/journal.pone.0100046 (2014).
Isola, D., Bartoli, F., Meloni, P., Caneva, G. & Zucconi, L. Black fungi and stone heritage conservation: Ecological and metabolic assays for evaluating colonization potential and responses to traditional biocides. Appl. Sci. https://doi.org/10.3390/app12042038 (2022).
Tesei, D. Black fungi research: Out-of-this-world implications. Encyclopedia 2, 212–229 (2022).
Romanazzi, G. & Feliziani, E. Postharvest decay 131–146 (Elsevier, 2014).
McConnaughey, M. Physical chemical properties of fungi. (2014).
Wang, F. et al. Transcription in fungal conidia before dormancy produces phenotypically variable conidia that maximize survival in different environments. Nat. Microbiol. 6, 1066. https://doi.org/10.1038/s41564-021-00922-y (2021).
Riddell, R. W. Permanent stained mycological preparations obtained by slide culture. Mycologia 42, 265–270 (1950).
Parker, C. W. et al. Genomic characterization of Parengyodontium torokii sp. nov., a biofilm-forming fungus isolated from Mars 2020 assembly facility. J. Fungi https://doi.org/10.3390/jof8010066 (2022).
Checinska Sielaff, A. et al. Characterization of the total and viable bacterial and fungal communities associated with the International Space Station surfaces. Microbiome 7, 1–21 (2019).
Taylor, D. L. & Bruns, T. D. Community structure of ectomycorrhizal fungi in a Pinus muricata forest: Minimal overlap between the mature forest and resistant propagule communities. Mol. Ecol. 8, 1837–1850. https://doi.org/10.1046/j.1365-294x.1999.00773.x (1999).
Blachowicz, A. et al. Human presence impacts fungal diversity of inflated lunar/Mars analog habitat. Microbiome 5, 1–16 (2017).
Patel, R. K. & Jain, M. NGS QC Toolkit: A toolkit for quality control of next generation sequencing data. PLoS ONE 7, e30619 (2012).
Stajich, J. & Palmer, J. (2019).
Bushnell, B. BBMap: A Fast, Accurate, Splice-Aware Aligner (Lawrence Berkeley National Lab. (LBNL), 2014).
Met, J. NCBI BLAST: A better web interface. Nucleic Acids Res. 36, W5–W9 (2008).
Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100. https://doi.org/10.1093/bioinformatics/bty191 (2018).
Walker, B. J. et al. Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 9, e112963 (2014).
Gurevich, A., Saveliev, V., Vyahhi, N. & Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075 (2013).
Wick, R. R., Judd, L. M. & Holt, K. E. Performance of neural network basecalling tools for Oxford Nanopore sequencing. Genome Biol. 20, 1–10 (2019).
Quan, Y. et al. A re-evaluation of the Chaetothyriales using criteria of comparative biology. Fungal Divers. 103, 47–85 (2020).
Mirarab, S., Nguyen, N. & Warnow, T. In Research in Computational Molecular Biology: 18th Annual International Conference, RECOMB 2014, Pittsburgh, PA, USA, April 2–5, 2014, Proceedings 18. 177–191 (Springer).
Steenwyk, J. L., Buida, T. J. 3rd., Li, Y., Shen, X. X. & Rokas, A. ClipKIT: A multiple sequence alignment trimming software for accurate phylogenomic inference. PLoS Biol. 18, e3001007. https://doi.org/10.1371/journal.pbio.3001007 (2020).
Minh, B. Q. et al. IQ-TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 37, 1530–1534. https://doi.org/10.1093/molbev/msaa015 (2020).
Kalyaanamoorthy, S., Minh, B. Q., Wong, T. K. F., von Haeseler, A. & Jermiin, L. S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 14, 587–589. https://doi.org/10.1038/nmeth.4285 (2017).
Anisimova, M., Gil, M., Dufayard, J. F., Dessimoz, C. & Gascuel, O. Survey of branch support methods demonstrates accuracy, power, and robustness of fast likelihood-based approximation schemes. Syst. Biol. 60, 685–699. https://doi.org/10.1093/sysbio/syr041 (2011).
Minh, B. Q., Nguyen, M. A. & von Haeseler, A. Ultrafast approximation for phylogenetic bootstrap. Mol. Biol. Evol. 30, 1188–1195. https://doi.org/10.1093/molbev/mst024 (2013).
Rambaut, A. FigTree, a graphical viewer of phylogenetic trees (Version 1.4. 4) (Institute of Evolutionary Biology, University of Edinburgh, 2018).
Palmer, J. M. & Stajich, J. Funannotate v1. 8.1: Eukaryotic genome annotation. Zenodo https://doi.org/10.5281/zenodo.4054262 (2020).
Frith, M. C. A new repeat-masking method enables specific detection of homologous sequences. Nucleic Acids Res. 39, e23. https://doi.org/10.1093/nar/gkq1212 (2011).
Seppey, M., Manni, M. & Zdobnov, E. M. BUSCO: Assessing genome assembly and annotation completeness. Methods Mol. Biol. 1962, 227–245. https://doi.org/10.1007/978-1-4939-9173-0_14 (2019).
Delcher, A. L., Harmon, D., Kasif, S., White, O. & Salzberg, S. L. Improved microbial gene identification with GLIMMER. Nucleic Acids Res. 27, 4636–4641. https://doi.org/10.1093/nar/27.23.4636 (1999).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 9, R7. https://doi.org/10.1186/gb-2008-9-1-r7 (2008).
Chan, P. P. & Lowe, T. M. tRNAscan-SE: Searching for tRNA genes in genomic sequences. Methods Mol. Biol. 1962, 1–14. https://doi.org/10.1007/978-1-4939-9173-0_1 (2019).
Blum, M. et al. The InterPro protein families and domains database: 20 years on. Nucleic Acids Res. 49, D344–D354. https://doi.org/10.1093/nar/gkaa977 (2021).
Huerta-Cepas, J. et al. eggNOG 5.0: A hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 47, D309–D314. https://doi.org/10.1093/nar/gky1085 (2019).
Mistry, J. et al. Pfam: The protein families database in 2021. Nucleic Acids Res. 49, D412–D419. https://doi.org/10.1093/nar/gkaa913 (2021).
Drula, E. et al. The carbohydrate-active enzyme database: Functions and literature. Nucleic Acids Res. 50, D571–D577. https://doi.org/10.1093/nar/gkab1045 (2022).
Rawlings, N. D. et al. The MEROPS database of proteolytic enzymes, their substrates and inhibitors in 2017 and a comparison with peptidases in the PANTHER database. Nucleic Acids Res. 46, D624–D632. https://doi.org/10.1093/nar/gkx1134 (2018).
Choi, J. et al. Fungal secretome database: Integrated platform for annotation of fungal secretomes. BMC Genomics 11, 105. https://doi.org/10.1186/1471-2164-11-105 (2010).
Blin, K. et al. antiSMASH 6.0: Improving cluster detection and comparison capabilities. Nucleic Acids Res. 49, W29–W35. https://doi.org/10.1093/nar/gkab335 (2021).
Sun, J. et al. OrthoVenn3: An integrated platform for exploring and visualizing orthologous data across genomes. Nucleic Acids Res. 51, W397–W403. https://doi.org/10.1093/nar/gkad313 (2023).
Kanehisa, M. & Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30. https://doi.org/10.1093/nar/28.1.27 (2000).
Kanehisa, M. Toward understanding the origin and evolution of cellular organisms. Protein Sci. 28, 1947–1951. https://doi.org/10.1002/pro.3715 (2019).
Kanehisa, M., Furumichi, M., Sato, Y., Kawashima, M. & Ishiguro-Watanabe, M. KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res. 51, D587–D592. https://doi.org/10.1093/nar/gkac963 (2023).
Mendes, F. K., Vanderpool, D., Fulton, B. & Hahn, M. W. CAFE 5 models variation in evolutionary rates among gene families. Bioinformatics 36, 5516–5518. https://doi.org/10.1093/bioinformatics/btaa1022 (2021).
Cock, P. J. et al. Biopython: Freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics 25, 1422–1423. https://doi.org/10.1093/bioinformatics/btp163 (2009).
McKinney, W. In Proceedings of the 9th Python in Science Conference. 51–56.
Stajich, J. stajichlab/PHYling_unified: Release for REPET2.5 and running on UCR HPCC (v1.0). Zenodo https://doi.org/10.5281/zenodo.1257002 (2018).
Grigoriev, I. V. et al. MycoCosm portal: Gearing up for 1000 fungal genomes. Nucleic Acids Res. 42, D699-704. https://doi.org/10.1093/nar/gkt1183 (2014).
Acknowledgements
The research was carried out at the Jet Propulsion Laboratory, California Institute of Technology, under a contract with the National Aeronautics and Space Administration (80NM0018D0004). © 2023 All rights reserved. We would like to thank Snehit Mhatre for isolating the strains. We thank Biotechnology and Planetary Protection Group members, especially Cynthia Ly for collecting samples. DIC imaging was performed at California State Polytechnic University, Pomona, CA with the support of Wei-Jen Lin and Craig W. LaMunyon. We also acknowledge the JPL supercomputing facility staff, notably Narendra J. Patel (Jimmy) and Edward Villanueva, for their continuous support in providing the best possible infrastructure for BIG-DATA analysis. Genome assembly and annotation were performed on the IIGB High-Performance Computing Cluster supported by NSF DBI-1429826 and NIH S10-OD016290 grants. J.E.S. is a CIFAR Fellow in the program Fungal Kingdom: Threats and Opportunities. We gratefully acknowledge the critical support and infrastructure provided for this work by The Kavli Nanoscience Institute at Caltech.
Funding
The research described in this manuscript was funded by the NNH18ZDA001N-PPR award 18-PPR18-0011 to KV.
Author information
Authors and Affiliations
Contributions
K.V. and N.K.S. conceived and designed the experiments. K.V. isolated the strain. C.E.M. and S.T. performed shotgun sequencing using the NovaSeq and ONT platforms, respectively. M.d.M.T., and N.K.S. carried out phylogenetic analysis and alignment for a positional description of organisms in the tree. A.M.C. cultivated and incubated fungal strains for imaging and phenotypic characterization. M.P.W. conducted DIC imaging experiments, added scale bars on images. A.M.C., selected images and drafted descriptions. T.T. along with A.M.C. characterized the morphological characteristics of the fungus. M.d.M.T., N.K.S., J.E.S., and A.C.S. extracted marker genes from genomes. A.M.C. generated the draft assembly of the genomes and submitted them in NCBI. A.M.C. wrote the first draft of the manuscript (Introduction, results, and discussion). A.M.C. has done bioinformatics and data engineering part of project. K.V. and A.M.C. compiled the contribution of write-ups from all authors associated with microscopy (C.W.P.), genotype and table generation (M.d.M.T.), and phylogenetic trees and figure generation (M.d.M.T.) and genomic characteristics (M.d.M.T. and J.E.S.). All authors have read and agreed to the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chander, A.M., de Melo Teixeira, M., Singh, N.K. et al. Genomic and morphological characterization of Knufia obscura isolated from the Mars 2020 spacecraft assembly facility. Sci Rep 14, 12249 (2024). https://doi.org/10.1038/s41598-024-61115-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-61115-1
- Springer Nature Limited