
Abstract
Bacterial caseinolytic protease P subunit (ClpP) is important and vital for cell survival and infectivity. Recent publications describe and discuss the complex structure–function relationship of ClpP and its processive activity mediated by 14 catalytic sites. Even so, there are several aspects yet to be further elucidated, such as the paradoxical allosteric modulation of ClpP by peptidomimetic boronates. These compounds bind to all catalytic sites, and in specific conditions, they stimulate a dysregulated degradation of peptides and globular proteins, instead of inhibiting the enzymatic activity, as expected for serine proteases in general. Aiming to explore and explain this paradoxical effect, we solved and refined the crystal structure of native ClpP from Staphylococcus epidermidis (Se), an opportunistic pathogen involved in nosocomial infections, as well as ClpP in complex with ixazomib at 1.90 Å and 2.33 Å resolution, respectively. The interpretation of the crystal structures, in combination with complementary biochemical and biophysical data, shed light on how ixazomib affects the ClpP conformational state and activity. Moreover, SEC-SAXS and DLS measurements show, for the first time, that a peptidomimetic boronate compound also induces the assembly of the tetradecameric structure from isolated homomeric heptameric rings of a gram-positive organism.
Similar content being viewed by others
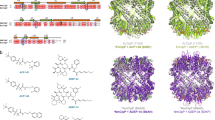


Introduction
The caseinolytic protease P (ClpP) subunit is a compartmentalized structure with two stacked heptameric rings that form a central chamber containing 14 identical catalytic sites sequestered by two protective axial pores1. In nature, ATPases, chaperones from the AAA+ superfamily2, modulate the function of ClpP3, controlling the pore access to hinder promiscuous proteolysis in the intracellular medium, stabilizing ClpP in its active state, and selecting the appropriate substrate to be degraded4.
The regulated activity of the ClpP-ATPase complex plays a key and vital role in protein turnover and homeostasis in bacteria, as this proteolytic machine degrades unfolded, misfolded, and distinct regulatory proteins5. In this context, the discovery and characterization of non-enzymatic activators make ClpP relevant for drug discovery investigations, aiming at the identification of novel and effective antimicrobial compounds and corresponding new therapies. The chaperone-free ClpP activation results in cellular self-digestion6, and this molecular mechanism has been already explored for the treatment of infections caused by multi resistant organisms, and even by dormant cells7. For example, the use of the natural antibiotic, acyldepsipeptide (ADEP) and its derivatives, in combination with rifampicin, a traditional ansamycin antibiotic, promoted the complete depletion of biofilms in vitro and in a mouse model7. ADEP, along with other ClpP modulators such as ureadepsipeptides8 and ZG1809, replace the modulation by ATPase via binding to the allosteric sites of ClpP known as “hydrophobic pockets”10. Those compounds also induce ClpP to be “locked” in its active state with enlarged axial pores11. The main advantage here, in terms of an antimicrobial effect, is that there is not any substrate selection, since the access to the protein’s catalytic chamber is independent of the regulation of a chaperone12. Thus, an uncontrolled protein degradation takes place inside the microbial cell, which affects cellular division, motility, and other biochemical processes essential for survival and growth13.
However, all data published hitherto show that the mechanism and structural aspects behind the dysregulation of ClpP activity still require further investigation. In this regard, peptidomimetic boronates, traditionally known as canonical serine protease inhibitors14, are worth being considered. The empty p orbital of boron allows it to coordinate to heteroatoms like oxygen and nitrogen, forming reversible covalent bonds with nucleophilic residues15 like the active serine (Ser98) of ClpP. Besides that, the peptidomimetic portion of those boronates interacts, via hydrogen bonds, with several amino acid residues in the catalytic clefts of ClpP. Those interactions are important for the stabilization of the tetradecameric active state of ClpP, as shown by previously published crystal structures in combination with isothermal titration calorimetry (ITC) measurements16,18.
Experiments with bortezomib, a well-known proteasome inhibitor applied as a drug for cancer treatment17, unveil that the activity of ClpP from Thermus termophilus (Tt) is affected by a peptidomimetic boronate in a paradoxical way18. The degradation of small oligopeptides increases, at low protein-to-ligand ratios, until a maximum point, and afterwards, the activity starts being reduced and inhibited as more ligand is added; proteolysis is otherwise directly proportional to the concentration of bortezomib. This kind of ambivalent activity modulation caused by that compound is described as organism dependent: ClpP from Escherichia coli, for example, is only inhibited by bortezomib18.
In the present work, we analyzed, by selecting the ClpP from Staphylococcus epidermidis (SeClpP), the influence of another peptidomimetic boronate, ixazomib, on protein structure and function. Our data and results obtained from X-ray crystallography, enzymatic assays, ITC, and nanoDSF rationalize how the ligand affects the substrate degradation performed by ClpP, from small oligopeptides to globular proteins. Moreover, our SEC-SAXS and DLS data reveal the impact of ixazomib in the oligomerization of SeClpP.
Results and discussion
The crystal structures of native SeClpP (Fig. 1a) and SeClpP-ixazomib (Fig. 1b) complex were refined to 1.90 Å and 2.33 Å, respectively. Further information related to X-ray data collection and refinement statistics can be found in Supplementary Table S1.

Crystal structures of ClpP from Staphylococcus epidermidis: apo SeClpP and SeClpP-ixazomib complex. The structures consist of two stacked heptameric rings that contain 14 identical catalytic sites in a central chamber. (a) Front and top views of apo SeClpP; (b) front and top views of SeClpP-ixazomib complex. In both cases, the protein dimensions are displayed. (c) The superposition of both tetradecamers demonstrates that there is not any significant three-dimensional difference between them. (d) Superimposition of monomers from apo SeClpP (blue) and SeClpP (orange) and their main parts.
The superimposition of both tetradecameric structures (Fig. 1c) displays that the complex is slightly more extended than the apo SeClpP, and the Cα RMSD is 1.63 Å for 1451 atoms. Moreover, as highlighted before, SeClpP has a topology that is conserved among different source organisms: 14 monomers placed in two sevenfold symmetric rings forming, a 300kDa barrel-like structure with a central catalytic chamber protected by axial pores surrounded by allosteric sites, or “hydrophobic pockets”. These sites are depicted in red (apo SeClpP) and green (SeClpP-ixazomib complex) (Figs. 1a, 1b).
When SeClpP monomers from both structures are compared (Fig. 1d), the Cα RMSD is 0.12 Å for 164 atoms. In both cases, the monomers are composed of a common structure consisting of the following main parts (Fig. 1d)19: E-helix with a Gly-rich region, catalytic site, head domain, oligomerization sensor, and N-terminal loop.
Despite the similarity between the overall structures and the high homology between amino acid sequences of ClpPs from different species (Fig. 2a), a closer look into the monomers exhibits a difference in the Gly-rich region. The structure in this region seems to be more ordered when ixazomib is present. Figure 2c shows that the apo SeClpP has a more disordered Gly-rich region with an unstructured loop and two small antiparallel beta-strands formed by two subunits from opposing heptameric rings. On the other hand, the boronate compound seems to induce the formation of longer antiparallel beta-strands (Fig. 2d) without a disordered loop, which results in a higher stabilization of the two stacked heptameric rings.

(a) ClustalW sequence alignment with SeClpP and S. aureus ClpP (SaClpP) amino acid sequences in FASTA format; (b) catalytic site of a monomer from the SeClpP-ixazomib complex and interactions between ixazomib and residues in the catalytic cleft. The amino acids that form hydrogen bonds with the ligand are labeled and colored in orange. Ixazomib is shown with the 2Fo-Fc electron density at 1.5σ. All the ClpP monomers bound to the ligand are shown in Supplementary Fig. S1. Comparison between the Gly-rich regions of different crystal structures of ClpP: (c) apo SeClpP with its partially disordered Gly-rich region; (d) SeClpP-ixazomib complex with its ordered Gly-rich region (two antiparallel beta-strands); (e) SaClpP-AV145 complex with its disordered, or unstructured, Gly-rich region.
Other examples confirm how relevant is the flexibility of the Gly-rich region to the ClpP structure. In the compressed20 and compact20 states, both inactive states of ClpP, this region is completely unfolded, resulting in less polar interactions between the heptamers. The lack of activity in those conformations also points that the degree of order of the Gly-rich region is related to the protein's function.
Moreover, till now, some compounds are known to disrupt the tetradecamer, when covalently or non covalently bound to the catalytic sites21, and others that elongate or kink (Fig. 2e) the Gly-rich region but keep the heptameric rings stacked. Ixazomib, for example, is a compound that does not affect the integrity of the barrel-like structure, as also observed in ClpPs bound to peptides or other peptidomimetic compounds16,22.
When not bound to an ATPase or a non-enzymatic modulator, the ClpP subunit acts as a peptidase, since the closed axial pores block the access of globular proteins23. In any case, even the degradation of small oligopeptides depends on some structural factors, and the most important one is the orientation and positioning of the catalytic triads formed by Ser98, His123, and Asp17224. The protein’s mode of action corresponds to that of a chymotrypsin-like protease, and it is described in two main steps: acylation and deacylation25. Firstly, the nucleophilic Ser98 attacks the substrate, resulting in a tetrahedral intermediate and then a subsequent covalent acyl-enzyme, after the C-terminal fragment is released. In the last stage, a second tetrahedral intermediate is formed, and the release of the N-terminal portion proceeds, after the hydrolysis of the acyl-enzyme. For this reaction to occur, it is generally accepted that the His123 acts as a base, obtaining a proton from the Ser98 to activate it. The proper alignment of the catalytic triad is intrinsically associated with the conformation and orientation of the E-helix and the Gly-rich region. Because of the flexibility of this region26, a transition between the extended, compact, and compressed states is possible. The shift to one of these three states promotes a particular change in the orientation of His123. In compact and compressed forms, this amino acid residue is flipped, becoming more distant from the Ser98 (4 to 6 Å apart).
As already stated, the boron atom of ixazomib is covalently bound to Ser98, while the peptide-like portion of the ligand is stabilized by forming hydrogen bonds with amino acids in the active site of ClpP (Fig. 2b), which cause to some extent cooperative structural arrangements. This interpretation is supported by enzymatic assays we performed with the substrate Suc-LY-AMC and ixazomib at low concentrations (Fig. 3b). In a range of protein-to-ligand ratios between 1:1 to 1:3, the peptidolysis increases, and from 1:5, it starts decreasing, as the inhibitory effect is predominant.

(a) Chromatograms after size-exclusion chromatography (SEC) with SeClpP sample free from glycerol (black curve) and supplemented with 20% w/v glycerol (red curve). As seen here, the use of the triol avoids the formation of heptamers (P2). When glycerol is not added to the protein sample, both tetradecameric (P1) and heptameric (P2) species are present. Suc-LY-AMC degradation rate vs. ixazomib concentration using reaction mixtures without glycerol (black bars) ( b) and with 20% w/v glycerol (red bars) (c). All assays were performed with experimental triplicates. In Supplementary Fig. S2, curves of peptidase activity (fluorescence unit) vs. time, at different ixazomib concentrations, are presented. * Only a low amount of small oligomers of ClpP can be seen in the protein sample containing 20% w/v glycerol.
Besides the importance of a structural alignment of the amino acids Ser98-His123-Asp172 for ClpP activity, the influence of ixazomib towards the Gly-rich regions is crucial here. From the literature, it is known that an extended beta-strand conformation of these regions supports the formation of well-defined substrate binding channels20. It indicates that structural changes in these channels are caused by low amounts of the ligand, which results in higher peptidolytic activity. A similar effect is seen in the study reporting data about ClpP from Thermus thermophilus and bortezomib18.
However, when the same assays are performed with a SeClpP sample supplemented with 20% w/v glycerol, an increase in peptidolytic activity is not observed (Fig. 3c). This simple triol is known as chemical chaperone and protein stabilizer26, and if used in cell lysis and purification steps, the ClpP sample becomes homogenized with a higher concentration of extended tetradecamers, as noticeable during size-exclusion chromatography (Fig. 3a, red graph), where only one peak is observed. Conversely, when SeClpP is purified in the absence of glycerol, different oligomeric states, mainly tetradecamers and heptamers, are distributed into two different peaks, P1 and P2, respectively (Fig. 3a, black graph). A lower activity is observed when the sample from peak 2 (P2) is used in enzymatic assays, once Suc-LY-AMC is poorly degraded by heptameric rings27.
Samples from P1 peak are constituted by tetradecameric structures capable of degrading the substrate without any additional ligand. Nevertheless, in the absence of glycerol, probably there is a mixture of active and inactive tetradecamers, and when ixazomib is used, it homogenizes the glycerol-free sample by extending all the ClpP species, causing the enhancement of peptidolysis, as explained previously28,29.
For further enzymatic assays, we used the tetradecameric fraction of ClpP and ixazomib, aiming to understand how the function of protein–ligand complex is modulated.
The relationship between the ClpP structure and function is complex. Besides the modification of the protein’s global conformation and the consequent alignment of the catalytic triads, the control of specific amino acid residues also influences proteolysis by regulating the diameter of the axial pores. In general, this structural control is facilitated by ClpP activators that bind to the “hydrophobic pockets”. Nevertheless, we confirmed that peptidomimetic boronate inhibitors also promote the dysregulation of ClpP proteolytic activity: it is directly proportional to the ixazomib concentration (Fig. 4), but it is not affected by low ligand concentrations as peptidolysis (Fig.4a). Experiments with SDS-PAGE and β-casein show that when the ligand concentration is higher than 500 µM (1:50 protein-to-ligand ratio) (Fig.4b), casein is degraded after passing through the open axial pores. In comparison to the substrate degradation in the presence of ONC206, an imipridone that modulates ClpP activation by binding to the “hydrophobic pockets”, the product profiles obtained in both cases are distinguishable (Fig. 4c). Proteolysis initiated by ONC206 results in products with lower molecular weight, as all the catalytic sites are free for the processive degradation of substrates throughout. Even being reversibly bound to ClpP, ixazomib still competes with β-casein for the catalytic triads.

15% SDS-PAGE after reactions with β-casein monitored for 60 minutes. (a) ixazomib and SeClpP concentrations: 200μM and 10μM, respectively; (b) ixazomib and SeClpP concentrations: 500μM and 10μM, respectively; (c) Comparative experiment with ixazomib (1mM) and ONC206 (10μM), where it is noticeable that the molecular weight (MW) of product fragments vary, according to the ligand used. A possible explanation for this difference is that ixazomib occupies the catalytic sites, affecting the processive degradation of the substrate. ONC206 only binds to the allosteric regions. Degradation products of low molecular weight can be seen in (b) and (c). *t= 0 min corresponds to the time point with no incubation, but until the complete denaturation of ClpP before the SDS-PAGE, β-casein degradation happened in the quick reaction with ONC206. The entire image of each gel is found in Supplementary Fig. S3.
The degradation of globular proteins intermediated by a serine protease inhibitor is still intriguing. So far, key amino acid residues in the hydrophobic pockets are defined as “switches” within the gating mechanism of the axial pores. To elucidate how a peptidomimetic boronate compound can support the access of bulk substrates into the active site regions, further investigations are needed. In a mutated version of ClpP from Staphylococcus aureus (Sa)28, the amino acid residue Tyr63, also present in SeClpP, was replaced by alanine. The absence of this tyrosine results in a domino effect that shifts the orientation of Asn42 side chain, from “up” to a “down” position28. As a result, the electrostatic interaction between Asn42 and Tyr21 from the neighboring monomer is affected, while the N-terminal loops assume an “open” state in the pores. The same effect on Asn42 in a wild-type structure is promoted by activators that interact with Tyr63, such as ADEP. A change in the orientation of this amino acid residue also causes the same structural arrangement of Asn4228.
A comparative analysis with the native SeClpP, the SeClpP-ixazomib complex, and SaClpP Y63A29 structures revealed that the Asn42 side chain in the complex (Fig. 5a), as well as in the mutant (Fig. 5b), is shifted to the “down” position (the hydroxyl group is approx. 5 Å distant from Tyr21). In the native protein, the residue is found in the “up” position, as expected (Fig. 5c). It indicates that the boronate compound can control the conformation of the axial pores, even if not bound to the hydrophobic pockets. Most probably, the non-covalent interactions of ixazomib play a major role in this process.

Illustration of the conformation of the Asn42 sidechain in different crystal structures of ClpP: (a) SeClpP-ixazomib complex, (b) active mutant (SaClpP Y63A) (PDB ID: 5C90)29 of ClpP from Staphylococcus aureus (Sa), and (c) native SeClpP. In (a) and (b), Asn42 is in a “down” position, with open pores (active for proteolysis). In contrast, in (c), the same amino acid residue is found in the “up” position, characteristic of closed ClpP.
Based on the previous discussion, here we show an allosteric activation caused by the inhibitor ixazomib. This ligand, at high concentrations, controls allosterically the N-terminal loops in the pores, in a way that globular proteins can be degraded inside the inner chamber of ClpP. At the same time, at low protein-to-ligand ratios, the boronate compound induces variations in the orientation of the catalytic triad, not affecting the N-terminal loops.
Additionally, our ITC analysis complements the peptidolytic assays. In the absence of glycerol (Fig. 6a), the data present a different aspect at low ixazomib concentrations, which can be a reason for the enhancement of the peptidolytic activity, as seen in Fig. 3b. This unconventional behavior was already reported in studies with ADEP2 where the reaction was more exothermic in the initial steps, resulted from a lower binding affinity. However, the conformational changes induced by the ligand at this stage promote positive cooperativity, increasing the binding affinity. It is important to consider how ixazomib affects the protein’s Gly-rich region and consequently, the substrate/ligand binding channels, as discussed before. Conversely, glycerol revokes the positive cooperativity, since it stabilizes the tetradecameric SeClpP, also organizing the binding channels. Therefore, in this latter case, only the inhibitory effect of the peptidomimetic boronate compound is pronounced and the curve obtained from the ITC titration has a sigmoidal shape (Fig. 6b).

ITC measurements with SeClpP and ixazomib, at different molar ratios, (a) in a glycerol-free environment and (b) with 20% w/v glycerol, both at 36°C (the same temperature set for the enzymatic assays). In the presence of glycerol, the ITC data could be fitted, and the following parameters were calculated: N = 9.53 ±0.0993 sites, K = 1.86E5 ±1.99E4 M−1 , \(\Delta {\text{H}}\)= -1.390E4 ±198.0 cal/mol, and \(\Delta {\text{S}}\)= -20.8 cal/mol/deg.
nanoDSF measurements also complemented our observations. Initially, the relationship between the thermal stability of SeClpP and low concentrations of ixazomib was analyzed. As shown in Fig. 7, when the protein-to-ligand ratio is 1:3.9 (gray curve), it is clear that there are two different ClpP species in solution, with melting temperatures approx. 10 units apart. In this context, the first peak, on the left, is coincident to the one of the apo SeClpP (free from ixazomib). On the other hand, the second peak, on the right, is comprised by SeClpP bound to the peptidomimetic boronate, and therefore more thermostable. Furthermore, we observed in this set-up that ixazomib starts to promote a conformational change that only allows the hydrolysis of relatively small oligopeptides, not globular proteins. Proteolysis is activated from higher ligand concentrations, according to the experiments in Fig. 4, which corresponds to the protein-to-ligand ratios 1:500 (brown curve) and 1:1000 (red curve). This outcome is also supported by recently published ITC measurements utilizing bortezomib as a modulator18, which show that the structural extension of ClpP starts at low concentrations of the ligand, but reaches a maximum point in higher amounts of the peptidomimetic boronate compound. The stabilization of the extended and more active state of SeClpP results in the highest melting temperatures (approx. 80 °C and 90 °C), according to the nanoDSF measurements.

Thermal stability analysis of SeClpP based on the first derivatives calculated from nanoDSF measurements. In Supplementary Fig. S4, a graph with more curves is presented, including a scan with the fluorescence intensity of samples that contain only ixazomib (at 1 mM) and buffer to discard any influence of this compound on the measurements.
In Fig. 8, we present data from SEC-SAXS to explore the effects of ixazomib on the overall oligomerization of SeClpP. Figure 8a confirms, once again, that, when glycerol is not added to the protein buffer, SeClpP is found as heptamers and tetradecamers. Chromatogram represented by red color shows only one peak which contains the fractions with 300 kDa variant. It indicates and confirms that ixazomib induces the assembly of a two stacked heptameric ring structure. Previous studies with heteromeric ClpP from Mycobacterium tuberculosis (ClpP1P2) and peptide analogs30 also report a similar situation, but this protein needs a modulator to form tetradecamers anyway. In the case of SeClpP, we have a homomeric protein capable of self-assembly into the active structure, even in equilibrium with another oligomeric state. Therefore, in our study, we could demonstrate this mechanism for a ClpP with identical monomers from a gram-positive bacterium.

SEC-SAXS data with SeClpP samples composed of heptamers and tetradecamers, as expected for protein samples without glycerol. (a) Chromatogram with apo SeClpP (black) and SeClpP sample incubated with 1 mM ixazomib (1:60 protein-to-ligand ratio; red). (b) Inter-atomic pair distribution functions of native SeClpP P1 (blue), native SeClpP P2 (orange), and SeClpP-ixazomib complex (green). (c) Ab initio low-resolution models calculated from SAXS scattering curves of native SeClpP P1 (blue), native SeClpP P2 (orange), and SeClpP-ixazomib complex (green). In Supplementary Table 2, there are experimental data of SAXS measurements. Guinier plots with scattering intensity graphs (I(q) vs q) are available in Supplementary Fig. S5.
The inter-atomic pair distribution functions in Fig. 8b demonstrate the oligomeric change induced by ixazomib as well. The heptamer species represented by the orange curve (RG: 3.75 ± 0.003 nm; 149.57 kDa) turn into tetradecamers (green curve; RG: 4.59 ± 0.002 nm, 318 kDa). In comparison to the tetradecamer isolated from the ixazomib-free solution (blue curve; RG: 4.47 ± 0.006 nm; 288.56 kDa), the 14-mer structure whose assembly was induced by the ligand (1:60 protein-to-ligand ratio) displays a slightly higher radius of gyration. Due to the poor solubility of ixazomib in water, the ligand amount could not be increased, as for higher concentrations of this compound, a higher DMSO concentration (a final concentration higher than 1% v/v) would be necessary, which would affect the quality of the SAXS measurements. Perhaps with increased protein-to-ligand ratios, higher RG values would be observed for the SeClpP-ixazomib complex, because of a more extended structure associated with a dysregulated proteolysis2,18.
In Fig. 8c, there are ab initio models calculated by the program GASBOR from the ATSAS suite. The models of the tetradecamer (blue) and the heptamer (orange) represent the species in the fractions P1 and P2 isolated from the sample containing native protein (black peaks) (Fig. 8a). Moreover, the green model is an illustration of the ClpP structure obtained after the P2 fraction is incubated with 2 mM ixazomib. In all cases, the χ2 values of approximately 1 confirm that the model fits well to the experimental data.
DLS is most useful to analyze the oligomeric shift caused by adding ixazomib, based on differences in the hydrodynamic radius (RH). If tetradecamers are formed from smaller oligomers, RH increases.
DLS measurements were performed, in replicates, using apo protein samples, as well as protein incubated with 1% v/v DMSO, 1 mM ixazomib, and 0.5 mM ONC206. Since this latter compound is a conventional ClpP activator, it was used in this experiment, so we could compare the outcome caused by both ligands, since ONC206 is expected to play a role in protein oligomerization. In Figs. 9a and 9b, the results corresponding to apo SeClpP (P1) and apo SeClpP (P2) are presented, respectively. The average RH of apo SeClpP P1 is approx. 5.6 nm, which is typical for tetradecamers, and the RH of apo SeClpP P2 is approximately 4.3 nm, confirming the presence of smaller oligomers in the sample.

DLS measurements with apo SeClpP from samples P1 (a) and P2 (b). Apo P1/P2 and P1/P2 supplemented with 1% v/v DMSO were selected as controls. 1 mM ixazomib and 0.5 mM ONC206 were incubated with the protein for 1 h, at 36 °C prior to the measurements. In (c,d), an unpaired t-test was applied to analyze how significant is the difference that ixazomib causes to RH values. In both cases, the change was significant (p < 0.05): p = 0.0363 (c) and p = 0.0004 (d). The last p-value shows that a higher alteration occurs when P2 is incubated with the peptidomimetic boronate.
The use of DMSO discarded any interference of this organic solvent in the hydrodynamic radius of SeClpP in both cases. Nevertheless, ixazomib affected the protein structure in P1 and P2. In the measurements with tetradecamers (P1), the ligand promoted the extension of SeClpP to approx. RH = 6.0 nm (Fig. 9c). For the P2 sample, the structural change was more drastic, as RH varied from 4.3 to 5.7 nm (Fig. 9d), a value slightly lower than the one observed for P1. This is additional evidence that the assembly of tetradecamers is induced by ixazomib. ONC206 was also capable of assembling larger oligomers of SeClpP (Fig. 9b), but the final structures do not have a RH as high as the one observed for SeClpP-ixazomib complex. As that activator binds to the “hydrophobic pockets”, maybe it is more efficient to establish interactions between adjacent subunits in the same ring, resulting in independent heptameric rings.
Conclusion
The data of the present work provide new insights about the modulation of ClpP function, dynamics, and oligomerization under the influence of boron-based peptidomimetic compounds. The high-resolution crystal structures of native ClpP and the ClpP-ixazomib complex demonstrate how the inhibitor is bound to the catalytic sites, allowing us to hypothesize the way the ligand dysregulates the proteolytic activity. Biochemical and biophysical experiments also revealed how ixazomib modulates ClpP activity and initiates global conformational alterations, particularly by affecting the Gly-rich region and the orientation of Asn42 between two adjacent ClpP monomers. An important consideration is that these alterations not only depend on the source organism of ClpP. Experimental aspects like the conformational state that is isolated after the purification steps play a major role in how ligand binding affects peptidolysis.
SEC-SAXS and DLS results showed and confirmed that the boronate inhibitor ixazomib induces the assembly of the tetradecameric structure as well. These novel results and corresponding conclusions highlight the complexity of the modulation of ClpP, showing that conformational changes in the catalytic sites also cause cooperative structural changes in more distant regions, such as the axial pores. Structural studies of this area are to some extent compromised, since the relative low electron density in this region did not allow us to resolve the flexible N-terminal loops completely. However, the opening of the gating mechanism can be inferred from the assays with β-casein, since it is the only way for a globular protein to have access to the ClpP’s catalytic chamber.
Another relevant point is how ixazomib differently affects peptidolysis and proteolysis. The explanation for the enhancement of Suc-LY-AMC degradation seems to be more straightforward, because of the structural modifications in the Gly-rich region and the cooperative effect indicated by ITC measurements at low concentrations of the ligand.
Methods
Heterologous expression of SeClpP
ClpP gene from Staphylococcus epidermidis was obtained from UniProt data bank (UniProtKB: Q8CTE0) and the corresponding open reading frame (ORF) was inserted into the vector pET28a (+) (Invitrogen, USA), after codon optimization, for posterior heterologous expression with a C-terminal His-Tag in Escherichia coli. The resulting plasmid (Biocat GmbH, Germany) was transformed into competent Escherichia coli BL21(DE3) Star cells (Invitrogen, USA), and for protein expression, bacteria were grown in LB medium supplemented with 50 µg/mL kanamycin and 0.5 mM IPTG, at 20 °C for 16 h, in an orbital shaker for constant stirring at 180 rpm.
Cell lysis and protein purification
Pellets were homogenized with buffer (A): 50 mM Tris base (pH 8.0), 100 mM NaCl, 20% w/v glycerol, or buffer (B): 50 mM Tris base (pH 8.0), 100 mM NaCl, 2 mM DTT. After sonication on ice and centrifugation, the supernatant was mixed with Ni–NTA resin (MACHEREY–NAGEL GmbH & Co. KG, Germany), previously equilibrated with (A) or (B). Protein sample eluted at 300 mM imidazole was dialyzed overnight in (A) or (B). Subsequently, SeClpP sample was submitted to size-exclusion chromatography in a HiLoad 26/600 Superdex 200 preparation grade (pg) column connected to an ÄKTA Purifier 10 system (General Electric, Sweden). The inactive mutant of SeClpP (S98A) was also expressed and purified as described in “Heterologous expression of SeClpP” and “Cell lysis and protein purification” sections. SeClpP S98A was applied to an enzymatic assay to confirm the absence of endogenous ClpP from Escherichia coli, as shown in Supplementary Fig. S6.
Protein crystallization, data collection, and refinement
Needle-like crystals were obtained by applying the hanging-drop crystallization technique, using MPD as precipitant agent. The mother liquor contained 0.1 mM sodium acetate (pH 4.5), 40% v/v MPD and 40 mM calcium chloride. Crystals with dimensions of 60 µm were obtained after 20 min, and 250 µm-crystals were observed after 48 h. To analyse the structure of the SeClpP-ixazomib complex, crystals were soaked with 10 mM ixazomib for 1 h before subsequent flash-freezing in liquid nitrogen. Diffraction data were collected at the beamline P11/PETRA III (DESY, Hamburg, Germany), at cryo conditions, up to 1.9 Å for apo SeClpP and to 2.33 Å for SeClpP in complex with ixazomib. Data merging and indexing were performed applying the XDS program package31 Data collection, processing and refinement parameters are summarized in Table S1 (supplementary material). The phase problem was solved by molecular replacement, using as search model the crystal structure of ClpP from Staphylococcus aureus (PDB: 3V5E; homology: 98.45%), and by applying the PHASER package from the Phenix suite version 1.20.1-448732. Refinement of the apo protein structure and the ClpP-ixazomib complex was performed by using phenix.refine33. Manual model building and evaluation of refined structures were done applying the program Coot34. Stereochemical parameters were analysed via the MolProbity server35.
Fluorescence assays and peptidolytic activity of SeClpP
The effect of ixazomib at different concentrations on the peptidolytic activity of SeClpP was analyzed via peptidolytic assays by applying the TECAN Infinite Pro 200 plate reader (TECAN, Austria) using 1 µM of protein and 100 µM of the substrate Suc-LY-AMC (Cayman Chemical, USA) in a reaction buffer with 50 mM Tris base (pH 7.5), 100 mM KCl and 2 mM DTT (C) or 50 mM Tris base (pH 7.5), 100 mM NaCl and 20% w/v glycerol (D), respectively. The effect of ixazomib on the peptidolytic activity, at different ligand concentrations, was analyzed. The ixazomib stock solution (100 mM in 100% v/v DMSO) was diluted in a way that the final DMSO concentration was kept constant to be 1% v/v.
SDS-PAGE experiments and proteolytic Activity of SeClpP
The globular protein β-casein from cow’s milk (Sigma-Aldrich, USA) was used as substrate to analyze the proteolytic activity dysregulated by ixazomib. 10 µM of ClpP were incubated with 200, 500, and 1000 µM of ixazomib, at 36 °C, under stirring at 900 rpm, in a ThermoMixer device (Thermo Fisher Scientific, USA). For each ixazomib concentration, five different tubes were prepared based on the reaction time scheme: 0 min, 15 min, 30 min, 45 min, 60 min. 7.5µL from the 0 min. sample were immediately mixed with 10 µL SDS-PAGE sample buffer and 2.5 µL DTT, being subsequently heated up at 95 °C for 10 min. The same procedure was performed with the remaining five tubes at the end of each corresponding reaction time interval. Following the step of protein denaturation, 15 µL of each sample were loaded into a polyacrylamide gel for subsequent SDS-PAGE analysis.
NanoDSF experiments
The influence of ixazomib on the thermostability of SeClpP was evaluated by applying nanoDSF. Samples with 10 µM SeClpP were prepared, in replicates, with different concentrations of ixazomib in buffer C, and at least 12 µL of each one was transferred to clear capillaries (NanoTemper, Germany). The measurements were done in a temperature range between 25°C and 90°C. The raw data were automatically processed using the Prometheus NT.48 software (NanoTemper, Germany) and the resulting data were further analyzed using the MoltenProt server36.
Isothermal titration calorimetry (ITC)
The interaction between SeClpP and ixazomib was also assessed by applying ITC using an Auto-iTC200 microcalorimeter (MicroCal-Malvern Panalytical, Malvern, UK). Calorimetric titrations were performed using a 800 µM ixazomib solution and a 10 µM SeClpP solution (buffer containing 50 mM Tris (pH 7.5), 150 mM KCl and 2 mM DTT). The heat evolved after injection of ixazomib was calculated from the integral of the measured calorimetric signal. Experiments were performed in replicates, and data were analyzed using Origin 7.0 (OriginLab, USA).
Size-exclusion chromatography and small-angle X-ray scattering (SEC-SAXS)
SeClpP incubated with ixazomib was utilized for SEC-SAXS measurements at the beamline P12 (EMBL/PETRAIII/DESY, Hamburg, Germany). Samples were prepared in a buffer containing 50 mM Tris base (pH 7.5), 150 mM KCl and 2mM DTT. For the size-exclusion chromatography, a Superdex 200 Increase 10/300 GL (GE Healthcare) column was used, and for all experiments, the concentration of the injected protein was 10 mg/mL. Eluted protein was automatically subjected to X-ray scattering data collection by utilizing a Dectris 2D photon-counting detector (PILATUS-6M) with 3.1 m sample-to-detector distance. Furthermore, SAXS data were collected at a constant temperature of 10 °C. The obtained scattering frames were visualized and processed using the ATSAS software suite37 and BioXTAS RAW38. Ab initio model of SeClpP as tetradecamers (P72) and heptamers (P7) were built by using the software GASBOR39.
DLS measurements
Protein samples (2.5 mg/mL) in 50 mM Tris base (pH 7.5), 150 mM KCl and 2 mM DTT were loaded into disposable plastic cuvettes with 1 mM ixazomib, 0.5 mM ONC206 or 1% v/v DMSO (as control). DLS experiments were performed applying a Wyatt Möbius laser photometer (Wyatt Technologies, USA) (532 nm and detector angle at 163.5°) at 25 °C. For each sample, 20 measurements in three independent replicates were performed, which consisted of 20 acquisitions each with an acquisition time of 20 s. Data were analysed using the software Dynamics version 7 (Wyatt Technology, USA). To evaluate how significantly ixazomib affects RH, p-values from unpaired t-tests were calculated by applying the program GraphPad Prism version 9 (GraphPad Software, USA), using an independent two-sample t-test (N = 20).
Data availability
Crystal structures of native SeClpP and SeClpP-ixazomib complex can be found on Protein Data Bank (PDB) under the following IDs: 8CJ4 and 8QYF, respectively. The authors confirm that further data supporting the findings of this study can be found within the article and its supplementary materials. In any case, other information that support our work are available from the main author, B. A. F., upon reasonable request. Please address eventual questions to bruno.franca@chemie.uni-hamburg.de.
Abbreviations
- RMSD:
-
Root-mean-square deviation
- DTT:
-
Dithiothreitol
- MPD:
-
2-Methyl-2,4-pentanediol
- Suc-LY-AMC:
-
Succinylated-leucine-tyrosine-7-amino-4-methylcoumarin
- SEC-SAXS:
-
Size-exclusion chromatography with small-angle X-ray scattering
- DLS:
-
Dynamic light scattering
References
Alexopoulos, J. A., Guarné, A. & Ortega, J. ClpP: A structurally dynamic protease regulated by AAA+ proteins. J. Struct. Biol. 179, 202–210. https://doi.org/10.1016/j.jsb.2012.05.003 (2012).
Gersch, M. et al. AAA+ chaperones and acyldepsipeptides activate the ClpP protease via conformational control. Nat. Commun. 6, 122. https://doi.org/10.1038/ncomms7320 (2015).
Moreno-Cinos, C. et al. ClpP protease, a promising antimicrobial target. Int. J. Mol. Sci., 20, 9. https://doi.org/10.3390/ijms20092232 (2019).
Dougan, D. A. Chemical activators of ClpP: Turning jekyll into hyde. Chem. Biol. 18, 1072–1074. https://doi.org/10.1016/j.chembiol.2011.09.003 (2011).
Zheng, D., Xu, Y., Yuan, G., Wu, X. & Li, Q. Bacterial ClpP protease is a potential target for methyl gallate. Front. Microbiol. 11, 31. https://doi.org/10.3389/fmicb.2020.598692 (2021).
Silber, N. et al. Cell division protein FtsZ is unfolded for n-terminal degradation by antibiotic-activated ClpP. mBio. 11, 3. https://doi.org/10.1128/mbio.01006-20 (2020).
Conlon, B. P. et al. Activated ClpP kills persisters and eradicates a chronic biofilm infection. Nature 503, 365–370. https://doi.org/10.1038/nature12790 (2013).
Griffith, E. C. et al. Ureadepsipeptides as ClpP activators. ACS Infect. Dis. 5, 1915–1925. https://doi.org/10.1021/acsinfecdis.9b00245 (2019).
Wei, B. et al. Anti-infective therapy using species-specific activators of Staphylococcus aureus ClpP. Nat. Commun. 13, 71. https://doi.org/10.1038/s41467-022-34753-0 (2022).
Ye, F., Li, J. & Yang, C. G. The development of small-molecule modulators for ClpP protease activity. Mol. Biosyst. 5, 23–31. https://doi.org/10.1039/C6MB00644B (2017).
Lee, B. G., Kim, M. K. & Song, H. K. Structural insights into the conformational diversity of ClpP from Bacillus subtilis. Mol. Cells 32, 589–595. https://doi.org/10.1007/s10059-011-0197-1 (2011).
Wong, K. S. et al. Acyldepsipeptide analogs dysregulate human mitochondrial ClpP protease activity and cause apoptotic cell death. Cell Chem. Biol. 25, 7. https://doi.org/10.1016/j.chembiol.2018.05.014 (2018).
Fedhila, S., Msadek, T., Nel, P. & Lereclus, D. Distinct clpP genes control specific adaptive responses in Bacillus thuringiensis. J. Bacteriol. 184, 5554–5562. https://doi.org/10.1128/JB.184.20.5554-5562.2002 (2002).
Tan, J. et al. De novo design of boron-based peptidomimetics as potent inhibitors of human ClpP in the presence of human ClpX. J. Med. Chem. 62, 6377–6390. https://doi.org/10.1021/acs.jmedchem.9b00878 (2019).
Ban, H. S. & Nakamura, H. Boron-based drug design. Chem. Rec. 15, 616–635. https://doi.org/10.1002/tcr.201402100 (2015).
Ju, Y. et al. Discovery of novel peptidomimetic boronate ClpP inhibitors with noncanonical enzyme mechanism as potent virulence blockers in vitro and in vivo. J. Med. Chem. 63, 3104–3119. https://doi.org/10.1021/acs.jmedchem.9b01746 (2020).
Papandreou, C. N. & Logothetis, C. J. Bortezomib as a potential treatment for prostate cancer. Cancer Res. 64, 5036–5043. https://doi.org/10.1158/0008-5472.CAN-03-2707 (2004).
Felix, J. et al. Mechanism of the Allosteric Activation of the ClpP Protease Machinery by Substrates and Active-Site Inhibitors. Sci. Adv. 5, 9. https://doi.org/10.1126/sciadv.aaw3818 (2019).
Wang, J., Hartling, J. A. & Flanagan, J. M. The structure of ClpP at 2.3 A resolution suggests a model for ATP-dependent proteolysis. Cell 91, 3. https://doi.org/10.1016/s0092-8674(00)80431-6 (1997).
Ye, F. et al. Helix unfolding/refolding characterizes the functional dynamics of Staphylococcus aureus Clp protease. J. Biol. Chem. 288, 17643–17653. https://doi.org/10.1074/jbc.M113.452714 (2013).
Gersch, M., Kolb, R., Alte, F., Groll, M. & Sieber, S. A. Disruption of oligomerization and dehydroalanine formation as mechanisms for ClpP protease inhibition. J. Am. Chem. Soc. 136, 1360–1366. https://doi.org/10.1021/ja4082793 (2014).
Kim, D. Y. & Kim, K. K. The structural basis for the activation and peptide recognition of bacterial ClpP. J. Mol. Biol. 379, 760–771. https://doi.org/10.1016/j.jmb.2008.04.036 (2008).
Brötz-Oesterhelt, H. & Vorbach, A. Reprogramming of the caseinolytic protease by ADEP antibiotics: Molecular mechanism, cellular consequences, therapeutic potential. Front. Mol. Biosci. 13, 8. https://doi.org/10.3389/fmolb.2021.690902 (2021).
Zeiler, E. et al. Structural and functional insights into caseinolytic proteases reveal an unprecedented regulation principle of their catalytic triad. Proc. Natl. Acad. Sci. U S A. 110, 11302–11307. https://doi.org/10.1073/pnas.1219125110 (2013).
Radisky, E. S., Lee, J. M., Karen Lu, C.-J. & Koshland, D. E. Insights into the Serine Protease Mechanism from Atomic Resolution Structures of Trypsin Reaction Intermediates. Proc Natl Acad Sci U S A. 103, 6835–6840. https://doi.org/10.1073/pnas.0601910103 (2006).
Perlmutter, D. H. Chemical chaperones: a pharmacological strategy for disorders of protein folding and trafficking. Pediatr. Res. 52, 832–836. https://doi.org/10.1203/00006450-200212000-00004 (2002).
Yu, A. Y. H. & Houry, W. A. ClpP: A distinctive family of cylindrical energy-dependent serine proteases. FEBS Lett. 581, 3749–3757. https://doi.org/10.1016/j.febslet.2007.04.076 (2007).
Ni, T. et al. Characterization of gain-of-function mutant provides new insights into ClpP structure. ACS Chem. Biol. 11, 1964–1972. https://doi.org/10.1021/acschembio.6b00390 (2016).
Mabanglo, M. F. et al. ClpP protease activation results from the reorganization of the electrostatic interaction networks at the entrance pores. Commun. Biol. 2, 410. https://doi.org/10.1038/s42003-019-0656-3 (2019).
Schmitza, K. R., Carneyb, D. W., Sellob, J. K. & Sauera, R. T. Crystal structure of mycobacterium tuberculosis ClpP1P2 suggests a model for peptidase activation by AAA+ partner binding and substrate delivery. Proc. Natl. Acad. Sci. U S A 111, E4587–E4595. https://doi.org/10.1073/pnas.1417120111 (2014).
Kabsch, W. XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132. https://doi.org/10.1107/S0907444909047337 (2010).
Adams, P. D. et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221. https://doi.org/10.1107/S0907444909052925 (2010).
Afonine, P. V. et al. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D Biol. Crystallogr. 68, 352–367. https://doi.org/10.1107/S0907444912001308 (2012).
Emsley, P. & Cowtan, K. Coot: Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132. https://doi.org/10.1107/S0907444904019158 (2004).
Chen, V. B. et al. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21. https://doi.org/10.1107/S0907444909042073 (2010).
Kotov, V. et al. In-depth interrogation of protein thermal unfolding data with MoltenProt. Protein Sci. 30, 201–217. https://doi.org/10.1002/pro.3986 (2021).
Panjkovich, A. & Svergun, D. I. CHROMIXS: Automatic and interactive analysis of chromatography-coupled small-angle X-ray scattering data. Bioinformatics 34, 1944–1946. https://doi.org/10.1093/bioinformatics/btx846 (2018).
Nielsen, S. S. et al. BioXTAS RAW, a software program for high-throughput automated small-angle X-ray scattering data reduction and preliminary analysis. J. Appl. Crystallogr. 42, 959–964. https://doi.org/10.1107/S0021889809023863 (2009).
Svergun, D. I., Petoukhov, M. V & Koch, M. H. J. Determination of Domain Structure of Proteins from X-Ray Solution Scattering. Biophys. J. 80, 2946–2953. https://doi.org/10.1016/S0006-3495(01)76260-1 (2001).
Acknowledgements
The authors acknowledge financial support obtained from the Cluster of Excellence ‘Advanced Imaging of Matter’ of the Deutsche Forschungsgemeinschaft (DFG)—EXC 2056—project ID 3907159, from the BMBF (Federal Ministry of Education and Research, Germany), via projects 05K19GU4 and 05K20GUB, and from the Open Access Publication Fund of Universität Hamburg. We also acknowledge the technical support by the SPC facility at EMBL Hamburg.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
B.A.F. designed the present study, coordinated manuscript writing, data processing, evaluation of experiments. S.F. assisted with data processing of SEC-SAXS data and provided some resources for experiments. C.B. and H.R. supervised the work and contributed to text correction. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Alves França, B., Falke, S., Rohde, H. et al. Molecular insights into the dynamic modulation of bacterial ClpP function and oligomerization by peptidomimetic boronate compounds. Sci Rep 14, 2572 (2024). https://doi.org/10.1038/s41598-024-51787-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-51787-0
- Springer Nature Limited