
Abstract
Plants require copper for normal growth and development and have evolved an efficient system for copper management based on transport proteins such as P1B-ATPases, also known as heavy metal ATPases (HMAs). Here, we report HMAs in eleven different Poaceae species, including wheat. Furthermore, the possible role of wheat HMAs in copper stress was investigated. BlastP searches identified 27 HMAs in wheat, and phylogenetic analysis based on the Maximum Likelihood method demonstrated a separation into four distinct clades. Conserved motif analysis, domain identification, gene structure, and transmembrane helices number were also identified for wheat HMAs using computational tools. Wheat seedlings grown hydroponically were subjected to elevated copper and demonstrated toxicity symptoms with effects on fresh weight and changes in expression of selected HMAs TaHMA7, TaHMA8, and TaHMA9 were upregulated in response to elevated copper, suggesting a role in wheat copper homeostasis. Further investigations on these heavy metal pumps can provide insight into strategies for enhancing crop heavy metal tolerance in the face of heavy metal pollution.
Similar content being viewed by others


Introduction
Industrialization has accelerated in tandem with global economic growth. Heavy metal pollution is one of the harmful consequences of rapid industrialization. Its contamination can damage water and soil resources and impact human health via food chains. These metals not only reduce the action of enzymes in cells but can also interfere with the function of crucial ion transporters, impede plant development, and disturb essential cellular processes such as the photosynthetic electron transport1. For instance, many enzyme systems are inhibited by cadmium, which affects the proper operation of organs like the liver and kidneys2.
Unlike other heavy metals such as cadmium (Cd2+), copper (Cu2+), mercury (Hg2+) and lead (Pb2+), copper does not freely bioaccumulate. Hence its toxicity to humans and other mammals is modest. However, chronic exposure to copper leads to liver toxicity, anemia, and severe neurological defects in humans3. Plants are also extremely susceptible to copper toxicity due to increased anthropogenic activities. For instance, the release of industrial wastewater with high concentrations of copper, mining, and application of copper-based fungicides on crops results in the accumulation of copper in soil and irrigation water. Plants are directly in contact with contaminated soil; hence, their toxicity in plants is higher than humans and other mammals. Copper concentrations slightly above optimal physiological concentrations lead to metabolic abnormalities and growth inhibition in plants4. Excess copper also inhibits many enzymes and disrupts vital elements involved in pigment production and membrane integrity. Its most significant effect is the inhibition of photosynthetic electron transport, which leads to lipid peroxidation5,6. Additionally, excess copper in plants can interfere with fatty acid and protein metabolism, inhibition of nitrogen fixation and respiration processes. It is also essential to consider that copper is an effective inhibitor of vegetative development and generates widespread senescence symptoms such as stunted growth of roots and shoots, chlorosis, necrosis, and leaf discoloration4.
Heavy metal tolerance has evolved in plants, as have regulatory mechanisms for ensuring an adequate supply of critical nutrients. Membrane transporters play a vital role in the uptake and transport of heavy metal cations and essential micronutrients during the complex mechanisms of heavy metal toxicity resistance. Several gene families involved in heavy metal transport have been identified and investigated. One such family is the P1B-ATPases, also known as Heavy Metal ATPases (HMAs), which belong to the P-type family of transporters. This family can selectively transport essential metal micronutrients such as Zn2+, Cu2+, and Co2+ for plant development and distribute non-essential heavy metal ions like Pb2+ and Cd2+7,8,9. Characteristic features of P1B-type ATPases include the presence of 6–8 transmembrane helices (TMs) for cation translocation, an ATP-binding domain (ATP-BD includes soluble nucleotide-binding domain and phosphorylation domains) and a soluble actuator domain (AD). The interconnections of these three domains are critical in regulating the metal ion transport10. In addition, they also possess distinct soluble metal binding domains (MBDs). These domains can bind and interact with specific metal ions, such as Pb2+ and Cd2+, at the N-terminal and C-terminus11,12,13.
HMAs have been split into two phylogenetic subgroups based on substrate specificity: a Cu/Ag subgroup and a Zn/Co/Cd/Pb subgroup13. HMAs have been investigated at the genome level in several species, including Arabidopsis and rice. The Arabidopsis HMA (AtHMA) family contains eight members, whereas the rice HMA (OsHMAs) family has nine members13,14. AtHMA1–4 and OsHMA1–3 are members of the Cu/Ag subgroup, while AtHMA5–8 and OsHMA4–9 are members of the Zn/Co/Cd/Pb subgroup13. AtHMA6/PAA1 was the first Arabidopsis cloned HMA and is involved in the copper transport framework in the chloroplasts15,16,17. More recently, 31 HMAs have been identified in Brassica napus18, 17 in Populus trichocarpa7, 11 in Zea mays and Sorghum bicolor14, 36 in Solanum tuberosum19, 20 in Glycine max20, 14 in turnip landraces21 and 32 in Triticum aestivum22. This indicates that the number of HMAs may vary from species to species. The expression of wheat TaHMA2 was studied during the cadmium stress22. The results proposed that microRNAs play a major role in regulating targeted genes such as TaHMA2 under cadmium stress. However, the role of wheat HMAs under copper stress has not been studied previously. In the present study, we aimed to elucidate the evolutionary relationship among HMAs of several Poaceae species based on phylogenetic analysis of HMA genes. We also explored segmental duplication events exhibited by TaHMA and chromosomal distributions of these genes. Furthermore, the expression of wheat HMAs was explored under copper toxicity conditions, investigating their potential roles in copper homeostasis and stress tolerance.
Results
Identification of P1B-ATPases in different members of the Poaceae family
Oryza sativa belongs to the family Poaceae, and a high degree of phenotypic synteny has been observed across the entire family23. A search was conducted in the MSU Rice Genome Annotation Project Database using the keyword “HMA,” which resulted in 68 different sequences. However, only nine contained conserved motifs and thus were considered further. LOC_Os06g48720 (OsHMA2) contained only 156 amino acids, whereas other HMAs contained more than 900 amino acids. Therefore, LOC_Os06g48720 (OsHMA2) was further analyzed on the Pfam protein database to find conserved characteristic HMA domains. No significant similarity was found for conserved domains in LOC_Os06g48720 (OsHMA2). Based on this, we considered this as a partial sequence. Thus, this protein sequence was subjected to a BlastP search on Ensembl Plants, and the full-length protein sequence of accession ID Os06t0700700-02 (1067 amino acids in length) was retrieved.
Brachypodium distachyon is more closely related to T. aestivum than O. sativa24 and has been proposed as a model species for genetic and molecular genomics studies in cereal crops and grasses25. In the case of HMAs, no previous study related to B. distachyon has been reported. Therefore, the present study used rice HMAs to identify homologs in B. distachyon (BdHMA1-9). Then HMAs homologs for other Poaceae species were identified via BlastP searches in Ensembl Plants using BdHMA1-9. This resulted in identifying 27 HMAs with conserved domains (E1–E2_ATPase domain Pfam id: 00122 and hydrolase domain Pfam id: 00702) in T.aestivum compared to 32 HMAs reported earlier22. It is important to mention that attempts were made to retrieve the 32 HMAs reported by Zhou et al. (2019) using Ensembl Plants, but no significant match was found. Moreover, the sequence IDs were also obsolete on Uniprot and inactivated on Uniparc databases. Thus, here we report the identification of 9 HMAs, hence 27 homologs of these genes in the wheat genome (Additional file S1). Furthermore, we also identified 9 HMAs in Zea mays, Sorghum bicolor, Steria italica, 8 in Oryza barthii, Oryza brachyantha, and Triticum urartu, and 7 in Saccharum spontaneum and Hordeum vulgare.
Multiple sequence alignment
Alignment of all 110 HMA protein sequences from different species revealed that they possess DKTGT, HP and CPx/SPC HMAs conserved motifs. The TGE motif was present in all HMAs except TRIUR3_31446-T1 and TraesCS7B02G337700.1. Similarly, the GDxxNDxP motif was present in all HMAs except TraesCS6B02G184800.1, HORVU4Hr1G076330.4 OBART07G08010.1, TRIUR3_18572-T, and in all HMA7 sequences, the GDxxNDxP motif was replaced by a GDxxNDxA motif.
Domains, motifs and gene structure analysis of T. aestivum HMAs
Conserved Domain Database (CDD) analysis of TaHMA1-27 revealed the presence of characteristic domains of HMAs such as hydrolase and E1-E2 ATPase. However, the presence of domains was inconsistent, and some domains were missing in wheat HMAs (Fig. 1). Further analysis of motif diversity in TaHMA1-27 showed the presence of 15 motifs (motif 1–15) within the proteins’ sequences. All TaHMA1-27 contain motifs 1, 10, 11, 13, 14 and 15, whereas a specific motif 9 was present in only clade II (Zn/Cd/Pd/Co-ATPase) and Clade III (Cu-ATPase) (Additional file S2). TaHMA1-27 length ranged from 3.4 Kbps (TaHMA3.1) to 17.78 Kbps (TaHMA2.1), their CDS ranged from 2562 to 2958 bps, and protein lengths were 916 aa to 958 aa. Gene structure analysis on GSDS tools showed that TaHMA1-27 possessed a different number of exons/introns, i.e., 6 exons (TaHMA3 and TaHMA5) to 18 (TaHMA7.2, TaHMA7.3 and TaHMA8). However, no regular changes in the number of exons/introns were found in the gene structure of all three clades (Additional file S3).

Distribution of conserved protein domains of TaHMA1-27 analyzed using NCBI CDD.
Phylogenetic analysis, predicted cellular localization, physiochemical properties and transmembrane helices of poaceae HMAs
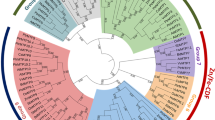
The phylogenetic relationship among the HMAs was explored using the maximum likelihood method and 1000 bootstrap replicates among eleven Poaceae species (B. distachyon, H. vulgare, O. brachyantha, O. barthii, O. sativa, S. bicolor, S. italica, S. spontaneum, T. aestivum, T. urartu, and Z. mays). All HMAs were characterized into three groups; Group A consisted of Clade I (Cu/Ca/Zn/Cd/Co-ATPase) and contained all HMA1 members; group II consisted of Clade II (Zn/Cd/Pd/Co-ATPase) and contained all members of HMA2 and HMA3; group III was comprised of clade III and IV (Cu-ATPase), containing all HMA4-9 members. Notably, within clade IV, HMA6 and HMA9 of all species formed a single cluster showing a close evolutionary relationship (Fig. 2). Predicted physiochemical properties of the TaHMA1-27 showed that the molecular weight of these proteins ranged from 42.548 kDa (TaHMA1.2) to 107.491 kDa (TaHMA5.2), pI ranged from 5.23 (TaHMA6.1) to 7.23 (TaHMA7.2), and GRAVY ranged from -0.162 (TaHMA2.1) to 0.325 (TaHMA6.1). Most TaHMA contained 6–8 transmembrane helices (TMH). However, the number of TMH in eleven out of 27 identified TaHMAs (TaHMA1.1, TaHMA1.2, TaHMA1.3, TaHMA3.2, TaHMA3.3, TaHMA4.1, TaHMA4.2, TaHMA4.3, TaHMA8.1, TaHMA8.2, and TaHMA8.3) were less than six (Additional file S3). The WoLF PSORT prediction algorithm predicted varied cell localization for TaHMAs1-27. These were predicted to localize to the plasma membrane, except TaHMA1.2, an extracellular matrix protein. All TaHMA1-27 members were also predicted to localize to the endoplasmic reticulum except TaHMA3.3, TaHMA4.1, TaHMA5.1, TaHMA5.2, and TaHMA5.3. Furthermore, TaHMA3.3 and TaHMA4.1 were also predicted to show some expression in the vacuole. Only two proteins, TaHMA2.2 and TaHMA2.3, were predicted to localize to the cytoplasm (Fig. 3).

Phylogenetic analysis of P1B-type ATPases genes in eleven different hosts of the family Poaceae. Four distinct clades are formed. Clade I comprises Cu/Ca/Zn/Cd/Co-ATPase and contains all HMA1 homologs. Clade II comprises Zn/Cd/Pd/Co-ATPase and contains all HMA2 and HMA3 homologs. Clade III and IV (represented as a blue color arc) comprise Cu-ATPase and contain all HMA4-9 homologs. All HMAs classes are represented with different colors, whereas to distinguish OsHMAs, reference sequences are presented with red, bold, and large fonts. Bootstrap values of the phylogenetic tree are represented in different color gradients on the left.

Heat map of predicted localization of each TaHMA1-27 homolog in the cell by WoLF PROST. Map was generated using TB tools software. Here color gradients represent the signals intensity of proteins within the cell. Red means the least expression, whereas green means the greatest expression. 1. Nucleus 2. Cytoplasm 3. Mitochondria 4. Vacuole 5. Cytoskeleton 6. Chloroplast 7. Endoplasmic Reticulum 8. Plasma membrane 9. Golgi bodies 10. Peroxisomes 11. Extracellular protein A: Cu/Ca/Zn/Cd/Co-ATPase B: Zn/Cd/Pd/Co-ATPase C: Cu-ATPase.
Chromosomal localization and gene duplication analysis of TaHMA1-27
A PhenoGram plot of the chromosomal localization revealed that TaHMA1-27 were positioned on 15 chromosomes and distributed on ABD sub-genomes (Fig. 4). Tandem and segmental duplication events are considered important factors of gene family expansion. Analysis revealed that the segmental duplication events related to 15 gene pairs occurred in all studied sub-genomes, whereas no tandem duplication events were found in any of the TaHMA1-27 homologs (Fig. 5). Furthermore, the ka/ks ratios of the segmentally duplicated TaHMA1-27 pair ranged from 0.466 to 1.533, with an average value of 0.2895. The ka/ks ratio of TaHMA5.3/5.2 and TaHMA5.3/5.1 were > 1, indicating positive selection. However, the ka/ks ratio of the remaining TaHMA pairs was less than 1, which suggested that these genes underwent extensive purifying selection26,27.

Genomic distribution of TaHMAs across sets of ABD genomes. The blue circles represent group A, which is involved in the translocation of Cu/Ca/Zn/Cd/Co ions; green circles represent group B, which involves the translocation of Zn/Cd/Pb/Co ions and red represents group C which is involved in translocation of Cu ions.

Segmental duplication events exhibited by TaHMAs. The 27 segmental gene duplication pairs are connected by different color lines and labeled on 14 wheat chromosomes. Black arc presents wheat chromosomes with chromosome numbers labeled black, whereas names are given in different colors corresponding to the clades of HMAs in Fig. 1.
Phenotypic analysis of T. aestivum seedlings grown under copper stress
Wheat seedlings were grown hydroponically on Lombnaes media to investigate the effects of copper toxicity28. Two different concentrations of copper were tested (200 µM and 400 µM). Prominent symptoms of copper toxicity chlorosis were observed with stunted growth and reduced weight in copper-treated plants compared to controls. Copper toxicity symptoms such as chlorosis were more pronounced in plants that were exposed to 400 µM as compared to 200 µM copper treatment. Similarly, plants grown under 400 µM copper treatment were more stunted and showed reduced total fresh weight compared to 200 µM copper-treated plants.
The data for root length and shoot obtained on day 7, day 14 and day 21 of the set I (200 µM) and set II (400 µM) was analyzed by two-way ANOVA. The statistical analysis shows copper treatment (set II) significantly affected root length on day 21. In contrast, the shoot length (set I) and (set II) was significantly reduced by the treatment on day 14 and day 21 when compared with the control group. Shoot fresh weight data analysis revealed that the treatment in the set I and set II groups for 21 days significantly decreased the shoot length compared to the control group. The fresh root weight data demonstrated a significant interaction between treatment × days. However, post-hoc analysis showed that the mean difference between the groups for fresh root weight did not reach the statistically significant value (Fig. 6a,b). Based on these observations, plants grown under 400 µM copper were further selected for gene expression analysis using qRT-PCR.

(a) Length data of root (A) and shoot (B) was analyzed by two-way ANOVA with repeated measure design with Bonferroni post-hoc test. *is showing P-values < 0.05, ** is showing P-values < 0.01 as compared to the control group. Whereas three biological replicates were harvested from each treatment group on day 7, day 14, and day 21 for phenotypic study. (b) Fresh weight data of root (A) and shoot (B) was analyzed by two-way ANOVA with repeated measure design with Bonferroni posthoc test. * shows P-values < 0.05 as compared to the control group. At the same time, three biological replicates were harvested from each treatment group on day 7, day 14, and day 21 for phenotypic study.
Gene expression analysis in plants grown under copper stress (400 µM)
Statistical analysis of qRT-PCR data shows a significant decrease in the expression of HMA1 by the treatment in the shoot on days 14 and 21. The HMA3 expression was significantly downregulated in shoots on day 7 while upregulated in root on day 14. A marked upregulation of HMA5 was observed in both shoot and root following the treatment on the 7th, 14th, and 21st day. Compared to the controls, the expression of HMA6 in the shoot on day 21 and the root on day 14 was significantly upregulated in test groups. In the shoot, the expression of HMA7 was significantly increased on day 7. However, in the root, the HMA7 expression gradually increased on days 7 and 14 and drastically increased on days 21. Upregulation of HMA8 expression in shoot and roots on days 7, 14 and 21 days in test samples was observed. A strong upregulation in the expression of HMA9 was observed in both shoot and root on days 7, 14, and 21 in the test group compared to the control samples. The analysis did not reveal significant expression differences for HMA4 in shoot and root. A heat map of the expression pattern of HMAs generated from quantitative real-time PCR data under copper stress is represented in Fig. 7.

The expression of T. aestivum HMAs under elevated copper stress where A = expression of TaHMAs in shoots and B = expression of TaHMAs in roots. Here color gradients represent signal intensity. Blue means the least expression, whereas red means the greatest expression. Bonferroni test revealed an increase in the expression of TaHMA5 and TaHMA9 in both shoot and root on days 7, 14, and 21 (P-values < 0.01) in the test group compared to the control samples. Expression of TaHMA8 was upregulated on days 7, 14 and 21 (P-values < 0.01) in roots only. However, in shoots, the expression of TaHMA8 increased on day 7 (P-values < 0.05), 14 (P-values < 0.05) and 21 (P-values < 0.01). Statistical analysis further showed that expression of TaHMA7 was increased on day 7 and day 21 (P-values < 0.05) in shoots whereas, in roots, TaHMA7 was upregulated significantly on day 7 (P-values < 0.05), 14 (P-values < 0.05) and 21 (P-values < 0.01) as compared to control.
Discussion
A range of heavy metals, i.e., copper and zinc, are pivotal to plants due to their roles in various physiological processes. However, when these heavy metals exceed optimal physiological concentrations in plants, these can have detrimental effects. Plants grown on soils contaminated with such heavy metals tend to cope with these stresses by adopting various mechanisms. HMAs play a significant role in the trafficking and sequestration of heavy metal field29. Still, more information must be given regarding these heavy metal transporters' expression patterns and functioning in wheat under copper stress. In the present study, we identified HMAs in different Poaceae species, conducted phylogenetic analysis and analyzed HMA's specific domains and motifs. Furthermore, chromosomal localization was predicted, and synteny analysis was done to discover possible duplication events in T. aestivum HMAs. The expression of T. aestivum HMAs under copper toxicity was also determined in T. aestivum roots and shoots.
Phylogenetic analysis of Poaceae HMAs revealed four clades based on the specification of the ions they translocate. TaHMA1 was in clade I whereas, TaHMA2 and TaHMA3 were in clade II.
The clades III and IV comprised TaHMA4, TaHMA5, TaHMA6, TaHMA7, TaHMA8, and TaHMA9. Previous studies have identified eight HMAs in A. thaliana and nine HMAs in O. sativa13. Similar heavy metal genes were identified in our study in different Poaceae species, except in S.spontaneum and H. vulgare, where seven have been identified. In S.spontaneum and H. vulgare, we initially found nine HMAs for both plants. However, four were redundant and hence removed from phylogenetic analysis.
There are three primary mechanisms of evolution in gene duplication events: segmental duplication, tandem duplication, and transposition. These elements are mainly responsible for the expansion of the plant gene family30. Moreover, plants have maintained various duplicated chromosomal blocks, which results in a high frequency of segment duplication27. In this analysis, all identified 27 HMAs were located on five out of seven chromosomes across the three sub-genomes (ABD), revealing that the segmental duplication event had participated in the expansion of the TaHMA1-27 homologs. Bread wheat is an allohexaploid (BBAADD genomes, 2n = 6x = 42) that has undergone hybridization events involving three different progenitors of the genera Triticum and Aegilops. The donor of sub-genomes A and B are Triticum urartu and Aegilops speltoides, derived ~ 7 million years ago from a common ancestor. The first hybridization between sub-genomes A and B resulted in homoploid hybrid speciation of sub-genome D approximately 5.5 million years ago. The second occurred less than 0.8 million years ago, and sub-genomes A and B underwent the event of polyploidization, leading to the formation of the AABB genome. Then the third event, allopolyploidization, gave rise to modern-day wheat less than 0.4 million years ago31,32,33. In the present research, the gene pairs of TaHMA1-27 homologs were analyzed for an approximate time of segmental duplication using the ka/ks rate. It can be concluded from the calculated Ka/Ks rate that the paralogous pair of TaHMA1-27 homologs emerged from a recent event of segmental duplication during the formation of the three sub-genomes (ABD) through purification and positive selection27,34. Notably, a different number of exons and introns were observed in TaHMA1-27. Variations in these exons/introns regions are plausibly due to the consequence of gene fragment integration and realignment. Indeed, this contributes significantly to the evolution of gene families35. Previous investigations have revealed that P-type ATPase contains conserved regions DKTGT, PxxK, GDGxNDxP, and S/TGE. In addition, 6–8 transmembrane segments, a CPx/SPC, and an HP locus motif are specific to P1B-ATPase and are essential for the metal transport13,36. In the present study, all identified TaHMA1-27 contain conserved hydrolase domain, E1-E2 ATPase domain, DKTGT, HP, CPx/SPC, GDxxNDxP, and TGE motifs. However, the number of TMH in TaHMA1.1, TaHMA1.2, TaHMA1.3, TaHMA3.2, TaHMA3.3, TaHMA4.1, TaHMA4.2, TaHMA4.3, TaHMA8.1, TaHMA8.2 and TaHMA8.3 are found to be less than 6. Therefore, TaHMAs without 6–8 transmembrane domains cannot be assigned as functional HMAs.
In dicots, HMAs are assumed to play a vital part in metal detoxification using efflux mechanisms from the cytosol13. Despite their significance, no information is available about the possible role of HMAs in T. aestivum in response to copper. Regulation of copper is required for normal growth, development, and normal cellular functionality of plants. However, excessive copper may hamper the availability of other nutrients for plants37. In the current research, qPCR was used to investigate the expression of wheat HMAs under copper stress. The real-time results analysis showed that the copper stress could increase or decrease the TaHMA expression. This revealed that T. aestivum exhibits diverse mechanisms of tolerance under copper stress. In the present study, an upregulation of TaHMA5 and TaHMA9 was found after seven days of copper treatment. The expression of two other HMAs, namely, TaHMA7 and TaHMA8, also significantly increased after 21 days of copper treatment in wheat roots. This indicates that TaHMA5 and TaHMA9 may provide a more immediate homeostatic response in wheat following copper exposure.
Most organisms contain P1B-type- ATPases, which are key for metal homeostasis. The copper-transporting P-type ATPases are likely to be key players considering their ability for contra electrochemical gradient transport and unique features conserved in plants11. In this study, the upregulation of TaHMA5, TaHMA7, TaHMA8 and TaHMA9 in wheat in response to copper stress suggests their role in regulating copper ions. Previous studies have reported that HMA transporter functions are related to their subcellular localization. For example, studies conducted on Arabidopsis thaliana AtHMA1 are localized at the chloroplast envelope. AtHMA1 is found to be involved in the copper translocation into the chloroplast and detoxification of heavy metal zinc38,39.
Similarly, Oryza sativa OsHMA3 and OsHMA4 are localized at the tonoplast and limit the accumulation of copper and cadmium in seeds and roots vacuole40,41. OsHAM9 is a copper efflux protein localized to the plasma membrane. It is expressed in roots and helps in lead, zinc and copper transport42,43.OsHMA2 and OsHMA5 are located at the plasma membrane and perform the function of heavy metal transport from root to shoot44,45. The earlier investigation also suggested that AtHMA7 is a post-golgi protein that delivers copper to an endoplasmic reticulum-associated protein (ethylene receptors ETR1). These two proteins interact between the sub-compartments of the inner membrane of both organelles46. Another study on two ecotypes of Silene vulgaris showed upregulation of SvHMA7 under copper stress, highlighting the role of HMA7 in the cellular copper detoxification47. Our analysis of predicted subcellular localization using WoLF PSORT revealed that TaHMA5, TaHMA7, TaHMA8 and TaHMA9 are localized to the plasma membrane, which are the orthologues of OsHMA5, OsHMA7, OsHMA8 and OsHMA9.
Moreover, WoLF PSORT also predicted the subcellular localization of TaHMA7 and TaHMA8 to the endoplasmic reticulum. Therefore, it is predicted that the four TaHMA (TaHMA5, TaHMA7, TaHMA8 and TaHMA9) have similar functions to other reported HMAs. However, further experimental studies based on genome engineering are necessary to reveal the molecular mechanism and localization of these TaHMA in response to copper stress.
Conclusion
This study demonstrated the identification of HMAs in 11 different plant species from the Poaceae family and their phylogenetic evolutionary relationship. Identified members were divided into 4 main clades based on the type of metal ions translocation. Three genes, TaHMA7, TaHMA8, and TaHMA9, were significantly upregulated by elevated levels of copper. Overall, this study indicates that these HMAs play an important role in regulating and translocating copper in wheat. Our results will provide a foundation for a better understanding of TaHMA members in wheat for functional characterization.
Methods
Cultivation of wheat plants for phenotypic study under copper stress
As reported previously, Triticum aestivum seedlings (Var. Sehar-06) were grown hydroponically using the Lombnaes media Field28. The use of plants/plant parts/seeds in the present study complies with international, national and/or institutional guidelines. The seedlings were grown under controlled conditions with temperatures at 21 °C/16 °C (day/night) and humidity at 55–65%. The photoperiod was maintained at 16 h at a quantum flux density (PAR) of 220 μmol m−2 s−1. After 14 days on the control media, three plants (roots and shoots) were harvested, and their fresh weight and length were noted. Plants were then snap-frozen using liquid nitrogen and stored at − 80 °C. The remaining twenty-seven plants were divided into three treatment groups (nine plants in each): control (2 µM CuCl2), Set I (200 µM CuCl2), and Set II (400 µM CuCl2). Plants were grown for 21 days under their respective treatments. Three plants were harvested from each group on day 7, day 14 and day 21 after the start of the treatment, and fresh weight and length of shoots and roots were determined. A two-way ANOVA with repeated measure design followed by Bonferroni post-hoc analysis was used to determine whether there were significant changes between copper treatments. P-values < 0.05 were considered significant.
RNA extraction and cDNA synthesis
RNA was extracted as previously described by48. Dnase I (Thermo Fisher Scientific, USA) was used to treat RNA samples to ensure any potential genomic contamination was removed. The first complementary DNA (cDNA) strand was created using ThermoFisher Scientific (USA) cDNA synthesis kit per the manufacturer’s instructions.
Quantitative real-time PCR (qRT-PCR)
The expression of wheat HMAs under copper stress was investigated using qRT-PCR. Three replicates were used per treatment per timepoint for qPCR. The primers for wheat HMAs were created using Primer3Plus (version 2.4.2)49 and validated using Primer-BLAST of NCBI. Actin was used as the reference gene50. All the primers utilized in this study are listed in Table 1. The ThermoFisher scientific SYBR Green Kit (Thermofisher scientific, USA) was used to execute the real-time PCR reaction in a 96 well-plate. To prepare a reaction mixture, 2.5 ng cDNA template, 0.3 M of reverse and forward primers, 1X SYBR-green master mix, and sterile 18 MΩ H2O (up to 20 µL) were employed. The reaction was run on an Opticon DNA Engine Continuous Fluorescence Detector (Applied Biosystems 7000 Real-time PCR system) under the following conditions: initial denaturation at 95 °C for 2 min, followed by 40 cycles of amplification comprising of denaturation for 50 s at 95 °C, annealing for 5 min at 70 °C and extension for 4 min at 68 °C. The final extension period was carried out for 10 min at 71 °C. The Pfaffl method calculated relative gene expression values51. The results were analyzed using two-way ANOVA with repeated measure design followed by Bonferroni post-hoc analysis. Significance was taken at P-values < 0.05. Finally, a gene expression heat map was generated using TB tools software52.
Database screening for sequence retrieval
Nine Oryza sativa HMA (OsHMA1-9) previously reported by Zhiguo et al. (2018) were retrieved from MSU Rice Genome Annotation Project Database (http://rice.uga.edu/)14,26 and crossed checked with the ARAMEMNON plant membrane database (http://aramemnon.uni-koeln.de/)53. These amino acid sequences were then used to retrieve putative HMA amino acid sequences of Brachypodium distachyon (BdHAM1-9) using Ensembl Plants (https://plants.ensembl.org/index.html)54. Information like Ensembl accession numbers, amino acid length, genomic length, and percentage similarity was also noted. HMAs of the remaining species (Hordeum vulgare, Oryza brachyantha, Oryza barthii, Sorghum bicolor, Steria italica, Saccharum spontaneum, Triticum aestivum, Triticum urartu, and Zea mays) were retrieved from Ensembl Plants through BLAST (Additional file S5). The Multiple Expectation Maximization for Motif Elucidation (MEME) webserver (https://meme-suite.org/meme/tools/meme)55 was used to determine HMA conserved motifs. These motifs were DKTGT, HP CPx/SPC, TGE, and GDxxNDxP, as previously reported by Fang et al. (2016)20. The distribution type of motifs was set as zero (0) or ‘one occurrence per sequence’ (zoops). The number of motifs and the minimum and maximum widths were set as 6, 15, and 50, respectively.
Multiple sequence alignment (MSA) and phylogenetic analysis
MSA of all amino acids was performed using the web server NGPhylogen.fr (https://ngphylogeny.fr/)56. A Maximum Likelihood (MLH) tree was constructed using advanced workflow. MAFFT for MSA57, BMGE for MSA file trimming58, and Fast-Tree for phylogenetic MLH tree with 1000 bootstrap value were selected59,60. The resulting tree was visualized and refined using the iTOL online tool (http://itol.embl.de/index.shtml)61.
Physiochemical properties, gene structure and domain identification in TaHMA1-27
Sequence information was obtained for TaHMA1-27 from the Uniprot database, while chromosomal location and genomic length were from Ensembl Plants (https://www.uniprot.org/uniprot/62, https://plants.ensembl.org/index.html63). Other physiochemical properties like protein mass in Daltons (Da), hypothetical isoelectric point, and grand average of hydropathicity (GRAVY) were calculated using the ProtParam tool of ExPaSy (https://web.expasy.org/protparam/)64. The NCBI conserved domain database (CDD) (https://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi)65 with search option Pfam V33.1-18271 PSSMs (http://pfam.xfam.org/)66 was used for distribution analysis of HMA conserved protein domains. The resultant domains diagram was drawn using the TB tools software52. The composition and position of exon/introns in TaHMA1-27 homologs were analyzed by comparing the CDS of TaHMA1-27 homologs to its corresponding genomic sequences using the online tool Gene Structure Display Server (GSDS) (http://gsds.gao-lab.org/)67.
Subcellular localization and transmembrane helixes prediction
The subcellular localization of HMAs was predicted using the WoLF PSORT protein localization predictor (https://wolfpsort.hgc.jp/)68. Resulting cellular signal values were used to construct a heatmap using the Tbtools software52. The TMHMM Server V. 2.0 (http://www.cbs.dtu.dk/services/TMHMM/)69 was used to predict the putative transmembrane helices of all TaHMA1-27 members.
TaHMA chromosomal location, synonymous (Ka) and non-synonymous rates and synthetic relationship
Chromosomal location and synthetic relationship were illustrated using a phenoGram plot (http://visualization.ritchielab.psu.edu/phenograms/plot)70 and Circos v0.6771, respectively. The substitution rate of synonymous (Ks) and nonsynonymous (Ka) was calculated using the Ka/Ks calculator of TB tools software for the selective pressure determination52. The approximate divergence time of the duplication events for each paralogous gene pair a million years ago (MYA = 10−6) was determined using the formula T = Ks/2x*MYA where x is the mean synonymous substitution rate which is 6.56 × 10−972.
Ethics approval
Wheat seeds (var. Sehar-06) were collected from the seed bank of the National Agricultural Research Council (NARC), Islamabad. This variety is widely cultivated in Pakistan.
Data availability
The databases used in this research are open and publicly available. No administrative permissions are needed for accessing and using the data. The links to the databases are given below. Ensembl Plants (https://plants.ensembl.org/index.html). MEME webserver (https://meme-suite.org/meme/tools/meme). NGPhylogen.fr (https://ngphylogeny.fr/). iTOL online tool (http://itol.embl.de/index.shtml). Uniport database (https://www.uniprot.org/uniprot/). ProtParam tool of ExPaSy (https://web.expasy.org/protparam/). NCBI conserved domain database (CDD) (https://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi). Pfam V33.1-18271 PSSMs (http://pfam.xfam.org/). Gene Structure Display Server (GSDS) (http://gsds.gao-lab.org/). WoLF PSORT (https://wolfpsort.hgc.jp/). TMHMM Server V. 2.0 (http://www.cbs.dtu.dk/services/TMHMM/). Phenogram plot (http://visualization.ritchielab.psu.edu/phenograms/plot).
References
Yruela, I. Copper in plants. Braz. J. Plant. Physiol. 17, 145–156. https://doi.org/10.1590/S1677-04202005000100012 (2005).
Choppala, G. et al. Cellular mechanisms in higher plants governing tolerance to cadmium toxicity. Crit. Rev. Plant Sci. 33, 374–391. https://doi.org/10.1080/07352689.2014.903747 (2014).
Karim, N. Copper and human health—a review. Eur. J. Soil Sci. 8, 117–122. https://doi.org/10.1111/j.1365-2389.1997.tb00558.x (2018).
Fernandes, J. & Henriques, F. J. T. b. r. Biochemical, physiological, and structural effects of excess copper in plants. Bot. Rev. 57, 246–273. https://doi.org/10.1007/BF02858564 (1991).
Hippler, F. W. R. et al. Oxidative stress induced by Cu nutritional disorders in Citrus depends on nitrogen and calcium availability. Sci. Rep. 8, 1–13. https://doi.org/10.1038/s41598-018-19735-x (2018).
Li, Q. et al. Excess copper effects on growth, uptake of water and nutrients, carbohydrates, and PSII photochemistry revealed by OJIP transients in Citrus seedlings. Environ. Sci. Pollut. Res. 26, 30188–30205. https://doi.org/10.1007/s11356-019-06170-2 (2019).
Li, D. et al. Genome-wide analysis and heavy metal-induced expression profiling of the HMA gene family in Populus trichocarpa. Front. Plant Sci. 6, 1149. https://doi.org/10.1007/s11104-018-3637-2 (2015).
Østerberg, J. T. & Palmgren, M. Heavy metal pumps in plants: Structure, function and origin. Adv. Bot. Res. 87, 57–89. https://doi.org/10.1016/bs.abr.2018.09.004 (2018).
Zorrig, W., Abdelly, C. & Berthomieu, P. The phylogenetic tree gathering the plant Zn/Cd/Pb/Co P1B-ATPases appears to be structured according to the botanical families. Plant Biol. Pathol. 334, 863–871. https://doi.org/10.1016/j.crvi.2011.09.004s (2011).
Smith, A. T., Smith, K. P. & Rosenzweig, A. C. Diversity of the metal-transporting P1B-type ATPases. J. Biol. Inorg. Chem. 19, 947–960. https://doi.org/10.1007/s00775-014-1129-2 (2014).
Argüello, J. M., Eren, E. & González-Guerrero, M. The structure and function of heavy metal transport P1B-ATPases. Biometals 20, 233–248. https://doi.org/10.1007/s10534-006-9055-6 (2007).
Bækgaard, L. et al. A combined zinc/cadmium sensor and zinc/cadmium export regulator in a heavy metal pump. J. Biol. Chem. 285, 31243–31252. https://doi.org/10.1074/jbc.M110.111260 (2010).
Williams, L. E. & Mills, R. F. P1B-ATPases—an ancient family of transition metal pumps with diverse functions in plants. Trends Plant Sci. 10, 491–502. https://doi.org/10.1016/j.tplants.2005.08.008 (2005).
Zhiguo, E., Tingting, L., Chen, C. & Lei, W. Genome-wide survey and expression analysis of P1B-ATPases in rice, maize and sorghum. Rice Sci. 25, 208–217. https://doi.org/10.1016/j.rsci.2018.06.004 (2018).
Shikanai, T., Müller-Moulé, P., Munekage, Y., Niyogi, K. K. & Pilon, M. PAA1, a P-type ATPase of Arabidopsis, functions in copper transport in chloroplasts. Plant Cell 15, 1333–1346. https://doi.org/10.1105/tpc.011817 (2003).
Catty, P. et al. Biochemical characterization of AtHMA6/PAA1, a chloroplast envelope Cu (I)-ATPase. J. Biol. Chem. 286, 36188–36197. https://doi.org/10.1074/jbc.M111.241034 (2011).
Boutigny, S. et al. HMA1 and PAA1, two chloroplast-envelope PIB-ATPases, play distinct roles in chloroplast copper homeostasis. J. Exp. Bot. 65, 1529–1540. https://doi.org/10.1093/jxb/eru020 (2014).
Li, N. et al. Genome-wide analysis and expression profiling of the HMA gene family in Brassica napus under cd stress. Plant Soil 426, 365–381. https://doi.org/10.1007/s11104-018-3637-2 (2018).
He, G. et al. Heavy metal transporters-associated proteins in Solanum tuberosum: Genome-wide identification, comprehensive gene feature, evolution and expression analysis. Genes 11, 1269. https://doi.org/10.3390/genes11111269 (2020).
Fang, X. et al. Genome-wide characterization of soybean P 1B-ATPases gene family provides functional implications in cadmium responses. BMC Genomics 17, 1–15. https://doi.org/10.1186/s12864-016-2730-2 (2016).
Wu, Y. et al. Comparative expression analysis of heavy metal ATPase subfamily genes between Cd-tolerant and Cd-sensitive turnip landraces. Plant Diversity 41, 275–283. https://doi.org/10.1016/j.pld.2019.02.001 (2019).
Zhou, M. et al. The genome-wide impact of cadmium on microRNA and mRNA expression in contrasting Cd responsive wheat genotypes. BMC Genomics 20, 1–19. https://doi.org/10.1186/s12864-019-5939-z (2019).
Devos, K. M. & Gale, M. D. Genome relationships: The grass model in current research. Plant Cell 12, 637–646. https://doi.org/10.1105/tpc.12.5.637 (2000).
Faris, J. D., Zhang, Z., Fellers, J. P. & Gill, B. S. Micro-colinearity between rice, Brachypodium, and Triticum monococcum at the wheat domestication locus Q. Funct. Integr. Genomics 8, 149–164. https://doi.org/10.1007/s10142-008-0073-z (2008).
Girin, T. et al. Brachypodium: A promising hub between model species and cereals. J. Exp. Bot. 65, 5683–5696. https://doi.org/10.1093/jxb/eru376 (2014).
Kawahara, Y. et al. Improvement of the Oryza sativa Nipponbare reference genome using next generation sequence and optical map data. Rice 6, 1–10. https://doi.org/10.1186/1939-8433-6-4 (2013).
Qiao, X. et al. Gene duplication and evolution in recurring polyploidization–diploidization cycles in plants. Genome Biol. 20, 1–23 (2019).
Lombn s, P., Singh, B. R. & Science, S. Varietal tolerance to zinc deficiency in wheat and barley grown in chelatorbuffered nutrient solution and its effect on uptake of Cu, Fe, and Mn. J. Plant Nutr. 166, 76–83. https://doi.org/10.1002/jpln.200390015 (2003).
Sinha, D., Tandon, P. K., Srivastava, G. P., Srivastava, S. K. & Mukherjee, S. in Plant Metal and Metalloid Transporters 109–131 (Springer, 2022).
Hurles, M. Gene duplication: the genomic trade in spare parts. PLoS Biol. 2, e206. https://doi.org/10.1371/journal.pbio.0020206 (2004).
Feldman, M. & Levy, A. A. Genome evolution due to allopolyploidization in wheat. Genetics 192, 763–774. https://doi.org/10.1534/genetics.112.146316 (2012).
Huo, N. et al. Gene duplication and evolution dynamics in the homeologous regions harboring multiple prolamin and resistance gene families in hexaploid wheat. Front. Plant Sci. 9, 673. https://doi.org/10.3389/fpls.2018.00673 (2018).
Consortium, I. W. G. S. et al. Shifting the limits in wheat research and breeding using a fully annotated reference genome. Science 361, eaar7191. https://doi.org/10.1126/science.aar7191 (2018).
Yu, J. et al. Genome evolutionary dynamics followed by diversifying selection explains the complexity of the Sesamum indicum genome. BMC Genomics 18, 1–14. https://doi.org/10.1186/s12864-017-3599-4 (2017).
Xu, G., Guo, C., Shan, H. & Kong, H. Divergence of duplicate genes in exon–intron structure. Proc. Natl. Acad. Sci. 109, 1187–1192. https://doi.org/10.1073/pnas.1109047109 (2012).
Williams, L. E., Pittman, J. K. & Hall, J. Emerging mechanisms for heavy metal transport in plants. Biochim. Biophys. Acta Biomembranes 1465, 104–126. https://doi.org/10.1016/S0005-2736(00)00133-4 (2000).
Seeda, A., Abou El-Nour, E., Mervat, G. & Zaghloul, S. Interaction of copper, zinc, and their importance in plant physiology: Review, acquisition and transport. Middle East J. Appl. Sci. 10, 407–434 (2020).
Seigneurin-Berny, D. et al. HMA1, a new Cu-atpase of the chloro plast envelope, is essential for growth under adverse light conditions. J. Biol. Chem. 281, 2882–2892 (2006).
Kim, Y. Y. et al. AtHMA1 contributes to the detoxification of excess Zn (II) in Arabidopsis. Plant J. 58, 737–753. https://doi.org/10.1111/j.1365-313X.2009.03818.x (2009).
Miyadate, H. et al. OsHMA3, a P1B-type of ATPase affects root-to-shoot cadmium translocation in rice by mediating efflux into vacuoles. New Phytol. 189, 190–199. https://doi.org/10.1111/j.1469-8137.2010.03459.x (2011).
Huang, X.-Y. et al. A heavy metal P-type ATPase OsHMA4 prevents copper accumulation in rice grain. Nat. Commun. 7, 1–13. https://doi.org/10.1038/ncomms12138 (2016).
Lee, S., Kim, Y.-Y., Lee, Y. & An, G. Rice P1B-type heavy-metal ATPase, OsHMA9, is a metal efflux protein. Plant Physiol. 145, 831–842. https://doi.org/10.1104/pp.107.102236 (2007).
Wenli, Z. et al. Rice heavy metal P-type ATPase OsHMA6 is likely a copper efflux protein. Rice Sci. 27, 143–151 (2020).
Takahashi, R. et al. The OsHMA2 transporter is involved in root-to-shoot translocation of Zn and Cd in rice. Plant Cell Environ. 35, 1948–1957. https://doi.org/10.1111/j.1365-3040.2012.02527.x (2012).
Deng, F., Yamaji, N., Xia, J. & Ma, J. F. A member of the heavy metal P-type ATPase OsHMA5 is involved in xylem loading of copper in rice. Plant Physiol. 163, 1353–1362. https://doi.org/10.1104/pp.113.226225 (2013).
Zhou, X., Liu, Q., Xie, F. & Wen, C.-K. RTE1 is a Golgi-associated and ETR1-dependent negative regulator of ethylene responses. Plant Physiol. 145, 75–86. https://doi.org/10.1104/pp.107.104299 (2007).
Baloun, J. et al. Characterization of the HMA7 gene and transcriptomic analysis of candidate genes for copper tolerance in two Silene vulgaris ecotypes. J. Plant Physiol. 171, 1188–1196. https://doi.org/10.1016/j.jplph.2014.04.014 (2014).
Aslam, R., Williams, L. E., Bhatti, M. F. & Virk, N. J. B. p. b. Genome-wide analysis of wheat calcium ATPases and potential role of selected ACA s and ECA s in calcium stress. BMC Plant Biol. 17, 1–14. https://doi.org/10.1186/s12870-017-1112-5 (2017).
Untergasser, A. et al. Primer3—new capabilities and interfaces. Nucleic Acids Res. 40, e115–e115. https://doi.org/10.1093/nar/gks596 (2012).
Giménez, M. J., Pistón, F. & Atienza, S. G. Identification of suitable reference genes for normalization of qPCR data in comparative transcriptomics analyses in the Triticeae. Planta 233, 163–173. https://doi.org/10.1007/s00425-010-1290-y (2011).
Pfaffl, M. W. A new mathematical model for relative quantification in real-time RT–PCR. Nucleic Acids Res. 29, e45–e45. https://doi.org/10.1093/nar/29.9.e45 (2001).
Chen, C. et al. TBtools: an integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 13, 1194–1202. https://doi.org/10.1016/j.molp.2020.06.009 (2020).
Schwacke, R. et al. ARAMEMNON, a novel database for Arabidopsis integral membrane proteins. Plant Physiol. 131, 16–26. https://doi.org/10.1104/pp.011577 (2003).
Bolser, D., Staines, D. M., Pritchard, E., & Kersey, P. Ensembl plants: Integrating tools for visualizing, mining, and analyzing plant genomics data. Plant Bioinf. Methods. 115–140 (2016).
Bailey, T. L., Williams, N., Misleh, C. & Li, W. W. MEME: Discovering and analyzing DNA and protein sequence motifs. Nucleic Acids Res. 34, W369–W373. https://doi.org/10.1093/nar/gkl198 (2006).
Lemoine, F. et al. NGPhylogeny. fr: New generation phylogenetic services for non-specialists. Nucleic Acids Res. 47, W260–W265. https://doi.org/10.1093/nar/gkz303 (2019).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. https://doi.org/10.1093/molbev/mst010 (2013).
Criscuolo, A. & Gribaldo, S. J. B. E. B. BMGE (Block Mapping and Gathering with Entropy): A new software for selection of phylogenetic informative regions from multiple sequence alignments. BMC Evol. Biol. 10, 1–21. https://doi.org/10.1186/1471-2148-10-210 (2010).
Price, M. N., Dehal, P. S. & Arkin, A. P. FastTree 2–approximately maximum-likelihood trees for large alignments. PloS one 5, e9490. https://doi.org/10.1371/journal.pone.0009490 (2010).
Lemoine, F. et al. Renewing Felsenstein’s phylogenetic bootstrap in the era of big data. Nature 556, 452–456. https://doi.org/10.1038/s41586-018-0043-0 (2018).
Letunic, I. & Bork, P. Interactive Tree Of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 49, W293–W296. https://doi.org/10.1093/nar/gkab301 (2021).
Apweiler, R. et al. UniProt: The universal protein knowledgebase. Nucleic Acids Res. 32, D115–D119. https://doi.org/10.1093/nar/gkh131 (2004).
Zerbino, D. R. et al. Ensembl 2018. Nucleic Acids Res. 46, D754–D761. https://doi.org/10.1093/nar/gkx1098 (2018).
Artimo, P. et al. ExPASy: SIB bioinformatics resource portal. Nucleic Acids Res. 40, W597–W603. https://doi.org/10.1093/nar/gks400 (2012).
Lu, S. et al. CDD/SPARCLE: The conserved domain database in 2020. Nucleic Acids Res. 48, D265–D268. https://doi.org/10.1093/nar/gkz991 (2020).
Mistry, J. et al. Pfam: The protein families database in 2021. Nucleic Acids Res. 49, D412–D419. https://doi.org/10.1093/nar/gkaa913 (2021).
Hu, B. et al. GSDS 2.0: An upgraded gene feature visualization server. Bioinformatics 31, 1296–1297. https://doi.org/10.1093/bioinformatics/btu817 (2015).
Horton, P. et al. WoLF PSORT: Protein localization predictor. Nucleic Acids Res. 35, W585–W587. https://doi.org/10.1093/nar/gkm259 (2007).
Krogh, A., Larsson, B., Von Heijne, G. & Sonnhammer, E. L. Predicting transmembrane protein topology with a hidden Markov model: Application to complete genomes. J. Mol. Biol. 305, 567–580 (2001).
Wolfe, D., Dudek, S., Ritchie, M. D. & Pendergrass, S. A. Visualizing genomic information across chromosomes with PhenoGram. BioData Min. 6, 1–12. https://doi.org/10.1186/1756-0381-6-18 (2013).
Krzywinski, M. et al. Circos: An information aesthetic for comparative genomics. Genome Res. 19, 1639–1645. https://doi.org/10.1101/gr.092759.109 (2009).
He, Y. et al. Genome-wide identification and expression analysis of two-component system genes in tomato. Int. J. Mol. Sci. 17, 1204. https://doi.org/10.1007/s11104-018-3637-2 (2016).
Acknowledgements
The authors acknowledge Dr. Zehra Batool (PCMD, ICCBS, Uok, Karachi, Pakistan) for her constructive efforts in statistical analysis.
Funding
This work was financially supported by Higher Education Commission, Pakistan.
Author information
Authors and Affiliations
Contributions
T.S.B. and M.I.: performed experiments, data collection, analysis, and manuscript writing. A.G. and R.A.: designed and supervised all research. F.M., R.A., and R.Z.P.: participated in data analysis and results writing. L.E.W., and K.D.A.: revised manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Batool, T.S., Aslam, R., Gul, A. et al. Genome-wide analysis of heavy metal ATPases (HMAs) in Poaceae species and their potential role against copper stress in Triticum aestivum. Sci Rep 13, 7551 (2023). https://doi.org/10.1038/s41598-023-32023-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-32023-7
- Springer Nature Limited