
Abstract
Some recent studies showed that severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infections and idiopathic pulmonary fibrosis (IPF) disease might stimulate each other through the shared genes. Therefore, in this study, an attempt was made to explore common genomic biomarkers for SARS-CoV-2 infections and IPF disease highlighting their functions, pathways, regulators and associated drug molecules. At first, we identified 32 statistically significant common differentially expressed genes (cDEGs) between disease (SARS-CoV-2 and IPF) and control samples of RNA-Seq profiles by using a statistical r-package (edgeR). Then we detected 10 cDEGs (CXCR4, TNFAIP3, VCAM1, NLRP3, TNFAIP6, SELE, MX2, IRF4, UBD and CH25H) out of 32 as the common hub genes (cHubGs) by the protein–protein interaction (PPI) network analysis. The cHubGs regulatory network analysis detected few key TFs-proteins and miRNAs as the transcriptional and post-transcriptional regulators of cHubGs. The cDEGs-set enrichment analysis identified some crucial SARS-CoV-2 and IPF causing common molecular mechanisms including biological processes, molecular functions, cellular components and signaling pathways. Then, we suggested the cHubGs-guided top-ranked 10 candidate drug molecules (Tegobuvir, Nilotinib, Digoxin, Proscillaridin, Simeprevir, Sorafenib, Torin 2, Rapamycin, Vancomycin and Hesperidin) for the treatment against SARS-CoV-2 infections with IFP diseases as comorbidity. Finally, we investigated the resistance performance of our proposed drug molecules compare to the already published molecules, against the state-of-the-art alternatives publicly available top-ranked independent receptors by molecular docking analysis. Molecular docking results suggested that our proposed drug molecules would be more effective compare to the already published drug molecules. Thus, the findings of this study might be played a vital role for diagnosis and therapies of SARS-CoV-2 infections with IPF disease as comorbidity risk.
Similar content being viewed by others

Introduction
The severe acute respiratory syndrome corona virus 2 (SARS-CoV-2) is the main cause of COVID-19 pandemic that brings a major threat for human life as well as economy around the world1,2. It was first detected from Wuhan town in China at the end of 2019 and rapidly spread all over the world with several symptoms like as cough, fever, and pneumonia diarrhea, severe respiratory diseases and become a complex and deadly health concern3,4. The World Health Organization (WHO) announced it as a momentous pandemic of twenty-first century on March 11, 2020. It is highly homologous to the SARS coronavirus (SARS-CoV) which was responsible for the respiratory pandemic during the 2002–2003 period5,6. Coronaviruses are single-stranded RNA viruses of ~ 30 kb. Based on their genomic structures, they are generally classified into four genera known as α, β, γ, and δ. The life cycle of SARS-CoV-2 with the host consists of the five steps classified as attachment, penetration, biosynthesis, maturation and release. The SARS-CoV-2 enters in to the host cells through the membrane fusion or endocytosis (penetration) after binding to the host receptor proteins (attachment). Once viral proteins (biosynthesis) including the major protease (MPro/3ClPro), the papain-like protease (PLpro) and the RNA-dependent RNA polymerase (RdRp) are released inside the host cells, viral RNA enters to the nucleus for replication. Thus, create new viral particles (maturation) and released. Coronaviruses consist of four structural proteins; Spike (S), membrane (M), envelop (E) and nucleocapsid (N)7. The spike (S) protein of SARS-CoV-2 interacts with the host ACE2 (angiotensin-converting enzyme 2) receptor protein to stimulate the infection8,9,10. ACE2 is highly expressed in lung, heart, kidney, ileum and bladder11,34. In the case of lung, ACE2 is highly expressed in lung epithelial cells which leads the interstitial lung damage including epithelial and endothelial injury with excessive fibroproliferation12. Until September 2022, there have been around 614,825,354 confirmed cases of COVID-19, including 6,536,284 million deaths13. Though different vaccination programs reduced the severity of SARS-CoV-2 infections worldwide, however, it is yet one of the most severe risk factor for some chronic diseases including idiopathic pulmonary fibrosis (IPF) disease, since they stimulate each other very much12,14. The IPF disease is a chronic, progressive lung syndrome which leads to the respiratory collapse and decline the lung function12. The primary symptoms of IPF are dry cough and breathing complexity15. The average survival time of a patients suffering from IPF is approximately 3 years after the first diagnosis and therapies. Initially, IPF has been considered as a chronic inflammatory process16, but recent studies showed that abnormally activated alveolar epithelial cells (AECs) are the main factor responsible for the fibrotic response because they release cytokines that stimulate the fibroblasts17. IPF is typified by the progressive and fatal accumulation of fibroblasts and extracellular matrix (ECM) in the lung, leading to distortion of the lung architecture and reduction in lung function. So, Individuals with inflammatory lung disease are at a higher risk of death from COVID-1918,19. Therefore, identification of SARS-CoV-2 and IPF diseases causing shared genes is required for diagnosis and therapies of COVID-19 patients with IPF disease as comorbidity.
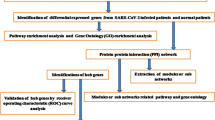
Taz et al.14 tried to identify shared genomic biomarkers for diagnosis and therapies of COVID-19 patients with IPF disease as comorbidity. They analyzed bulk RNA-Seq profiles for SARS-CoV-2 infections and microarray gene-expression profiles for IPF disease and found only 11 common differentially expressed genes (cDEGs) to separate both COVID-19 and IPF patients from control samples. They identified top-ranked 5 genes as common hub-genes (cHubGs) by protein–protein interaction (PPI) analysis, where only 2 cHubGs were detected from the cDEGs and the rest 3 cHubGs did not belongs to their cDEGs-set. Also, they did not examine their common differential expression patterns by from any other databases, which indicates that their cHubGs-set were not so representative of their cDEGs-set. Another drawback in their study was that they used microarray data instead of RNA-Seq data for identification of differentially expressed genes (DEGs) between IPF disease and control samples, though RNA-Seq data perform better than microarray data in identifying DEGs20. Therefore, in this study, an attempt was made to explore comparatively more representative and effective cHubGs-set for SARS-CoV-2 and IPF diseases from RNA-Seq profiles for diagnosis and therapies of COVID-19 patients with IPF disease as comorbidity by using the integrated bioinformatics analyses. The pipeline of this study is given in Fig. 1.

The pipeline of this study.
Materials and methods
Data sources and descriptions
We used both original data and meta-data to reach the goal of this study as described below.
Collection of RNA-Seq profiles for SARS-CoV-2 infections, IPF disease and control samples
We collected RNA-Seq profiles for SARS-CoV-2 infections, IPF disease and control samples from the National Center for Biotechnology Information (NCBI) Gene Expression Omnibus (GEO) database (http://www.ncbi.nlm.nih.gov/geo/) to explore both diseases causing common genes. The SARS-CoV-2 infected patient’s RNA-Seq profiles were downloaded from GPL18573 platform of Illumina NextGen 500 (Homo sapiens) with the GEO accession numbers GSE147507 published by Blanco-Melozet al.21. We collected 18 case and 29 control samples of RNA-Seq profiles from 3 cell lines (1) normal human bronchial epithelium, alveolar cells, transformed lung-derived Calu-3 cells of lung tissues for SARS-CoV-2 infections. On the hand, the IPF patient’s RNA-Seq profiles were downloaded from GPL11154 platform of Illumina HiSeq 2000 (Homo sapiens) with the GEO accession numbers GSE52463 published by Nance et al.22. This dataset contained 16 samples including 8 IPF and 8 control samples collected from lung tissues.
Collection of meta-drug agents for exploring candidate drugs
Beck et.al.23 suggested SARS-CoV-2 main protease (3CLpro)-guided top-ranked 90 drug molecules out of 3410 FDA approved anti-viral drugs for the treatment against COVID-19 by molecular docking analysis. In our study, we collected their suggested top-ranked 90 drug molecules as the meta-drug agents [see Table S1(I)]. We also collected host transcriptome-guided 95 meta-drug agents that are recommended for COVID-19 or IPF diseases [see Table S1(II)] in different published articles. Thus, we considered total 90 + 95 = 185 drugs to explore most probable candidate drug agents by molecular docking with our proposed receptors.
Collection of independent meta-receptors for in-silico validation of the proposed drugs
We selected the top-ranked 11 hub-genes (independent meta-receptors) as the independent meta-receptors associated with COVID-19 and IPF disease by reviewing 23 published articles [see Table S1(III)] for in-silico validation of the proposed drug molecules by molecular docking.
Identification of DEGs from RNA-Seq profiles by using edgeR
To explore DEGs between case and control samples from RNA-Seq profiles GSE147507 and GSE52463 of NCBI-GEO database, we considered a popular statistical approach known as edgeR. In order to introduce edgeR, let \({X}_{gi}\) denote the total number of read counts for gth gene (g = 1, 2, …, G) in the ith sample (i = 1, 2, …, n), which is assumed to be followed negative binomial (NB) distribution in the edgeR setting24,25. That is, \(X\sim NB\left({\mu }_{gi}, {\delta }_{g}\right),\) where the parameters are described by the mean and variance as \({\mu }_{gi}=E\left({X}_{gi}\right)={M}_{i}{\pi }_{gi}\) and \(V\left({X}_{gi}\right)={\mu }_{gi}(1+{\mu }_{gi}{\delta }_{g})\), respectively. Here, \({M}_{i}\) is the total number of short reads of RNA-Seq profiles, \({\pi }_{gi}\) denote the fraction of all cDNA fragments for gth gene in the ith sample so that \(\sum_{g=1}^{G}{\pi }_{gi}=1,\) \({\delta }_{g}\) is the squired coefficient of variation of \({\pi }_{gi}\) based on the replicates i. The NB distribution convert to Poisson distribution when \({\delta }_{g}=0\). According to the generalized linear model (GLM) approach, the mean response, \({\mu }_{gi}\), is linked to a linear predictor as \({Log(\mu }_{gi})={{{\varvec{z}}}_{i}^{T}{\varvec{\beta}}}_{g}{+Log(M}_{i}),\) where zi is the ith column of the design matrix (Z) including the covariates (e.g. batch effects, experimental conditions, etc.), \({{\varvec{\beta}}}_{g}\) is a q × 1 vector of regression parameters for differential expression patterns. The regression vector \({{\varvec{\beta}}}_{g}\) is estimated by maximum likelihood estimation (MLE) and calculated iterative way as
where \({D}_{g}\) is the diagonal matrix of working weights, \({u}_{gi}=({x}_{gi}-{\mu }_{gi})/(1+{\delta }_{g}{\mu }_{gi})\). The dispersion parameter \({\delta }_{g}\) is calculated as
where
is the adjusted profile likelihood (APL) in terms of \({\delta }_{g}\), penalized for the estimation of the regression parameters \({\beta }_{g}\), \({\mathrm{X}}_{g}\) is the vector of counts for gene g,\({\widehat{\beta }}_{g}\) is the estimated coefficient vector, L(·) is the log-likelihood function, \({I}_{g}\) is the Fisher information matrix and |·| is the determinant, \(\tau\) is the prior degree of freedom afforded to the shared likelihood and
Then the edgeR approach allows us for testing the significance of any parameter or any contrast or linear combination of the parameters in the linear model. Gene-wise hypothesis testing are conducted by computing likelihood ratio (LRT) statistic \(L({\theta }_{g,0}|X)/L({{\varvec{\theta}}}_{g}|X)\) to compare the null hypothesis (H0) that \({{\varvec{\beta}}}_{{\varvec{g}}}=0\) (insignificant gene) against the two-sided alternative (H1), \({\varvec{\beta}}\ne 0\) (indicating DEGs), where \({{\varvec{\theta}}}_{g}=\{{\varvec{\beta}},\boldsymbol{ }\delta \}\) and \({\theta }_{g,0}=\delta\). The log-LRT follows asymptotically chi-square distribution under H0. Adjusted P.values are computed to control the FDR (false discovery rate) by the Benjamini and Hochberg approach26. We implemented this algorithm to identify DEGs from our downloaded RNA-Seq count datasets GSE147507 and GSE52463 for SARS-CoV-2 and IPF diseases, respectively, by using edgeR, an R-package in Bioconductor25. To separate up and down-regulated DEGs, we for the combined data as follows
where \(\mathrm{adj}.{P}_{g}.\mathrm{value}\) is the adjusted P.value and \(({\mathrm{aFC})}_{\mathrm{g}}={\overline{X} }_{g}^{\mathrm{case}}/{\overline{X} }_{g}^{\mathrm{control}}\): fold change on average expressions, for gth gene.
Identification of SARS-CoV-2 and IPF diseases causing common DEGs (cDEGs)
Let \({\mathrm{C}}_{\mathrm{UR}}\) and \({\mathrm{C}}_{\mathrm{DR}}\) indicates the upregulated and downregulated DEGs-sets respectively for Covid-19 patients. Again let \({\mathrm{I}}_{\mathrm{UR}}\) and \({\mathrm{I}}_{\mathrm{DR}}\) be the upregulated and downregulated DEGs-sets respectively, for IPF patients. Then, we defined common upregulated gene-set as \({G}_{UR}=({C}_{UR}\cap {I}_{UR})\) and common downregulated as \({G}_{DR}=({C}_{DR}\cap {I}_{DR})\). Finally, we considered common DEGs (cDEGs) set as \(\mathrm{cDEG}=({\mathrm{G}}_{\mathrm{UR}}\cup {\mathrm{G}}_{\mathrm{DR}})\) that can differentiate both group of patients from the control samples. Therefore, in this study, we considered this cDEGs-set for further investigation.
Protein–protein interaction (PPI) network analysis of cDEGs
In order to explore both SARS-CoV-2 and IPF diseases causing common hub-genes (cHubGs), we performed protein–protein interaction (PPI) network analysis of cDEGs by using the STRING databases27. To build the PPI network, the distance "D" between the proteins (u, v) is computed as,
where \({N}_{u}\) and \({N}_{v}\) are the neighbor sets of u and v, respectively. To improve the quality of PPI networks, we used the Cytoscape web-tool28. The Cytoscape plugin cytoHubba was used to select the Hub-Genes (HubGs) or Hub-Proteins (HubPs) from PPI networks28,29. The PPI network provides a number of nodes and edges, which indicates proteins and their interactions, respectively. The HubGs were selected from the PPI network by using five topological measures including Degree (Deg)30, BottleNeck (BN)31, Betweenness (BC)32, Stress (Str)33, and Closeness (Clo)34.
Regulatory network analysis of cHubGs
To explore key transcription factors (TFs) and micro-RNAs (miRNAs) as the transcriptional and post-transcriptional regulators of cHubGs, we performed TFs-cHubGs, miRNAs-cHubGs, and TFs-miRNAs-cHubGs interaction networks based on the JASPAR35, TarBase36 and RegNetwork37 databases, respectively by using the NetworkAnalyst web-tool38. The Cytoscape software28 was used to improve the quality of networks. The key regulators were selected by using two topological measures (Degree (Deg)30 and Betweenness (BC)32) of networks.
GO terms and KEGG pathway enrichment analysis of cDEGs highlighting cHubGs
To explore the pathogenetic processes of cDEGs, we performed enrichment analysis of cHubGs with different gene ontology (GO) terms (BPs: biological processes, MFs: molecular functions and CCs: cellular components)39 and Kyoto encyclopedia of genes and genomes (KEGG) pathways40. Here, BPs are different types of molecular activities that are essential for the functioning of integrated living entities including cells, tissues, and organs. MFs are different types of fundamental molecular activity of a gene product, including catalysis. CCs are different types of components of a cell or the extracellular space. The KEGG pathway database consists of a set of pathway maps that represent the molecular interaction and relationship networks for genetic information processing, metabolism, human diseases, and drug development. Let Si denote the reference/annotated gene-set in the ith GO-term or KEGG-pathway, Mi denote the total number genes in Si (i = 1, 2,…, r); N denote the total number of reference/annotated genes that construct the whole-set \(S=\bigcup_{i=1}^{r}{S}_{i}={S}_{i}\bigcup {S}_{i}^{c}\) such that \(N\le \sum_{i=1}^{r}{M}_{i} ;\) where \({S}_{i}^{c}\) is the complement set of Si. Again, let n represent the total number of cDEGs and ki represent the number of cDEGs that are a part of the annotated gene-set Si. To examine the enrichment of ith GO-term or KEGG-pathway by the cDEGs-set, the following contingency table (Table 1) is constructed.
We considered three web tools (Enrichr41, DAVID42,43, and GeneCodis44) to explore significantly enriched GO-terms and KEGG pathways by using the chi-square (\({\chi }^{2})\) or Fisher’s exact testing procedures. The \({\chi }^{2}\)-testing procedure based on \(2\times 2\) contingency table, is used in GeneCodis to calculate the p-values, while the Fisher’s exact testing procedure is used in both Enrichr and DAVID web-tools. Fisher’s exact testing procedure is formulated based on hypergeometric distribution. This distribution is used to estimate the probability of overlapping exactly ki cDEGs with the reference genes in the ith GO-term or pathway (Si) out of n cDEGs. Then the p value for testing the significance of ki cDEGs in ith GO-term or pathway is calculated as,
The ki cDEGs in Si is considered as a significantly enriched gene-set if its adjusted p value (pi) < 0.05 at 5% level of significance. We adjusted the p values for each of three procedures by using the Benjamini and Hochberg procedure26.
Association of cHubGs with other disease risk
We performed diseases-cHubGs association analysis by using the Enrichr web tool45 with DisGeNET database46,47 to explore other diseases that can increase the severity of COVID-19 and IPF both diseases through cHubGs. The DisGeNET database is a comprehensive discovery platform to address association of a gene-set with different disease risk47. The necessary data for this database has been collected from different online sources including UniProt, Comparative Toxicogenomics Database (CTD), Mouse Genome Database (MGD), Rat Genome Database (RGD) and, peer-reviewed publications on Genome Wide Association Studies (GWAS), Genetics Association Database (GAD), literature of human gene-disease networks (LHGDN), and the BeFree datasets. To measure the gene-disease association (GDA), the DisGeNET web-tool compute a score (S) by using the following formula.
where
and L: LHGDN, GAD, or BeFree, \({n}_{gd}\) is the number of publications reported a GDA in the source and \({N}_{L}\) is the total number of publications in the source.
Obviously, the DisGeNET score (S) lies between 0 to 1.
Prognostic performance of cHubGs
To investigate the prognostic performance of cHubGs with lung cancer, we performed multivariate survival analysis of lung cancer patients based on the expressions of cHubGs by using SurvExpress web-tool48. The TCGA database (https://tcga-data.nci.nih.gov) is used in SurvExpress web-tool for survival analysis with a gene-set. In this analysis, patient samples were divided in to low-risk and high-risk groups by using the median of risk-scores (RSs) which is defined by the linear component of the Cox regression model. That is, \({\mathrm{RS}=\upbeta }_{1}{\mathrm{X}}_{1}+{\upbeta }_{2}{\mathrm{X}}_{2}+\dots +{\upbeta }_{\mathrm{n}}{\mathrm{X}}_{\mathrm{n}}\), where \({\mathrm{X}}_{\mathrm{i}}\) is the expression of ith gene, \({\upbeta }_{\mathrm{i}}\) is calculated by using the Cox regression approach49. Generally, a patient whose risk-score (RS) is greater than median of RSs was considered in the high-risk group, otherwise, the patient belongs to the low-risk group. Then the Kaplan–Meier survival plot for each risk group is constructed for each risk group. The significant difference between two risk groups was investigated by using the concordance indexes (CI), hazard-ratio (HR) and log-rank test50. The significance level was set to p value < 0.05.
Drug repurposing by molecular docking
To explore cHubGs-guided FDA approved repurposable drug molecules for the treatment against SARS-CoV-2 infections in presence of IPF risk, we employed molecular docking analysis of cHubGs and its TFs with 185 drug agents as introduced previously in the data sources and descriptions section [see Table S1(I, II)]. The molecular docking analysis requires 3-Dimensional (3D) structures of both receptor proteins and drug agents/ligands. We downloaded 3D structure of all targeted proteins from Protein Data Bank (PDB)51, AlphaFold Protein Structure Database52 and SWISS-MODEL53. The 3D structures of all drug agents were downloaded from PubChem database54. The 3D structures of the target proteins were visualized by using the Discovery Studio Visualizer 201955. Further, the receptor protein was prepared by removing ligand heteroatoms and water molecules and by addition of polar hydrogens on AutoDock tools 1.5.756. The drug agents/ligands were prepared by setting the torsion tree and rotatable, nonrotatable bonds present in the ligand through AutoDock tools 1.5.7. Then, pairwise binding affinities between the target proteins and drug agents were calculated using the AutoDock Vina57. The exhaustiveness parameter was set to 10. Discovery Studio Visualizer 201955 and PyMol58 were used to analyze the docked complexes for its surfaces, types and distances of non-covalent bonds. Let Bij is the binding affinity (BA) score corresponding to the ith receptor (i = 1, 2, … , p) and jth drug (j = 1, 2, … , q). The receptors and drug agents were ordered by the decreasing order of their average BA scores for selecting the top-ordered potential drug agents. We compared the binding performance of our suggested drugs with previously suggested candidate drugs by Taz et al.14 by molecular docking analysis against the Taz et al., suggested receptors as well as top-ranked independent receptors. Finally, we looked into the effectiveness of our suggested drugs with randomly selected independent receptor proteins that were not associated with SARS-CoV-2 infections in the presence of IPF risk.
Results
Identification of cDEGs
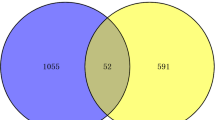
The dataset GSE147507 was analyzed by using the edgeR r-package to identify DEGs between SARS-CoV-2 infections and control samples. A total of 851 DEGs were identified by satisfying the cutoff criteria of adjusted p value < 0.05 and |log2(aFC)|> 1, where 712 DEGs are upregulated and 139 DEGs are downregulated. The GSE52463 dataset was analyzed to identify DEGs between IPF diseases and control samples. A total of 668 DEGs were found according to the same criteria, where 391 DEGs are upregulated and 277 are downregulated. Then we commonly found 27 upregulated cDEGs {TNFAIP3, SELE, MX2, PTX3, CH25H, UBASH3A, EREG, BIRC3, ZBP1, NLRP3, LIF, PRDM1, ADM, VCAM1, FOSL1, CXCR4, CCL22, IRF4, SLC5A5, SYTL3, ADAMTS4, UBD, CCL17, CPNE5, TNFAIP6, IKZF3, TNIP3} and 5 downregulated cDEGs {MVD, KIF12, RAAG1GAP, SLC27A3, TMEM160} that can separate both COVID-19 and IPF patients from the control samples (see Fig. 2).

Venn diagrams for visualizing the upregulated and downregulated cDEGs that can separate both COVID-19 and IPF patients from the control samples.
PPI network analysis of cDEGs for identification of cHubGs
The PPI network of cDEGs was constructed using STRING database (Fig. 3) which contains 27 nodes and 115 edges. We selected top ranked 10 cHubGs {CXCR4, TNFAIP3, VCAM1, NLRP3, TNFAIP6, SELE, MX2, IRF4, UBD, CH25H} by applying five topological measures (Deg, BN, BC, Str and Clo) in the PPI network (see Table S2).

Protein–protein interaction (PPI) network of cDEGs for COVID-19 and IPF disease to identify cHubGs. The red color nodes indicate the cHubGs.
The regulatory network analysis of cHubGs
The cHubGs-TFs interaction network detected top-ranked two sets of TFs proteins {JUN, SRF and NFKB1} and {JUN, SPI1 and NFKB1} based on JASPAR and RegNetwork databases respectively, as the key transcriptional regulatory factors for cHubGs, where two TFs proteins JUN and NFKB1were common in both-sets (see Fig. 4B,C). Based on two databases, we observed that JUN is a regulator of 7 cHubGs (TNFAIP3, TNFAIP6, IRF4, SELE, TNFAIP6, NLRP3, VAM1) and NFKB1 for 6 cHubGs (TNFAIP3, IRF4, CXCR4,UBD, SELE, VAM1). The cHubGs-miRNAs interaction network detected top-ranked two sets of miRNAs {hsa-mir-155-5p, hsa-mir-27a-5p, hsa-mir-107, hsa-mir-21-3p, hsa-mir-129-2-3p} and { hsa-mir-155-5p, hsa-mir-154, hsa-mir-613, hsa-mir-21-3p, hsa-mir-132} based on TarBase and RegNetwork databases respectively, as the key post-transcriptional regulatory factors for cHubGs, where two miRNAs hsa-mir-155-5p and hsa-mir-21-3p were common in both-sets (see Figs. 4A,C). Based on two databases, we observed that hsa-mir-155-5p regulates 6 cHubGs {IRF4, VAM1, TNFAIP3, TNFAIP6, SELE, CXCR4} and hsa-mir-21-3p regulates 5 cHubGs {VAM1, TNFAIP3, TNFAIP6, MX2, UBD}.

The regulatory network of cHubGs with TFs and miRNAs. The red, light blue and green color nodes represent the cHubGs, TFs and miRNAs. The names of cHubGs, key regulatory TFs and miRNAs were displayed only. (A) The cHubGs-TFs interaction network based on JASPAR database. (B) The miRNA-cHubGs interaction network based on TarBase database. (C) The cHubGs-TFs-miRNAs interaction network based on RegNetwork database.
GO functions and KEGG pathway enrichment analysis of cDEGs highlighting cHubGs
The Table 2 displayed the top five commonly enriched GO terms (BPs, MFs and CCs) and KEGG pathways by cDEGs with three web-tools and databases Enrichr41, DAVID42,43 and GeneCodis44 (p value < 0.001). Among the top five GO terms of BPs, four BPs including inflammatory response, regulation of inflammatory response, response to tumor necrosis factor and response to cytokine were enriched by the cHubGs-sets with all of three databases. The other BP-term (response to virus) was enriched by the cHubGs-sets with each of two databases (DAVID42,43 and GeneCodis44). Three MFs (sequence-specific DNA binding, CCR chemokine receptor binding and chemokine activity) out of four, were commonly enriched by the cHubGs-sets with all of three databases. The other MF-term (ubiquitin binding59) was enriched by the cHubGs-sets with each of two databases (Enrichr41 and GeneCodis44). Among the top five significantly enriched CCs terms, four terms including early endosome, extracellular space, cytosol and extracellular region were commonly enriched by the cHubGs-sets with two databases out of 3. Only one CC-term (tertiary granule lumen) was enriched by the cHubGs-sets with all of 3 databases. Interestingly, all of the top five KEGG pathways (TNF signaling pathway, IL-17 signaling pathway, NF-kappa B signaling pathway, NOD-like receptor signaling and Cytokine-cytokine receptor interaction) were commonly enriched by the cHubGs-sets with all of 3 databases.
Association of cHubGs with other disease risks
We performed cHubGs-disease association analysis by using the Enrichr web-tool with DisGeNET database to explore other diseases that may increase the severity of both COVID-19 and IPF diseases simultaneously or separately, through the influence of cHubGs. This analysis significantly detected top-ranked 20 diseases (Epstein-Barr virus infections, Myocardial Infarction, Degenerative polyarthritis, Arthritis, Diabetes, Juvenile arthritis, Inflammation, Eczema, Dermatitis, Atopic, Lymphoma, Non-Hodgkin and Arthritis, multiple sclerosis, Acute Fulminating, Inflammatory disorder, Lupus Nephritis, Hypercholesterolemia, thrombocytopenia due to platelet alloimmunization and Hereditary Autoinflammatory ) that may stimulate both SARS-CoV-2 and IPF diseases through the influence of cHubGs (see Fig. 5A and Table S3). The multivariate survival probability curves with cHubGs significantly separated the lung cancer patients (high risk group) from the control groups (low risk group) (see Fig. 5B), which indicates the cHubGs stimulate the lung cancer also.

(A) cHubGs-disease association results. Red box indicates significant association. (B) The multivariate survival probability curves of lung cancer patients based on cHubGs. Red indicates the survival curve with high risk (cancer) group and green indicates the low risk (control) group.
Drug repurposing by molecular docking
To explore cHubGs-guided candidate drug molecules by docking analysis, we downloaded 3D structure of cHubGs-mediated 10 proteins (CXCR4, IRF4, MX2, NFKB1, NLRP3, SELE, SRF, TNFAIP3, TNFAIP6, UBD) and two TFs proteins (JUN, VCAM1) as the receptor proteins from the Protein Data Bank (PDB)51 with source codes 2k04, 2dll, 1jnm, 4 × 0r, 2o61, 3qf2, 1esl, 1k6o, 2eqf, 1o7c, 6gf1, and 1vca, respectively. The 3D structure of CH25H target proteins were downloaded from SWISS-MODEL53 by using UniProt92 ID of O95992. The 3D structures of 185 drug agents (see Table S1(I, II)) were downloaded from PubChem database54 as mentioned previously. Then pairwise drug-target molecular docking analysis were carried out to calculate their binding affinity score (kcal/mol) for each pair of receptors and agents. Then, we ordered the target proteins in descending order of row average of the binding affinity matrix A = (Aij) and drug agents according to the column average to select the top-ranked drug agents as the candidate drugs (see Fig. 6A). Thus, we selected top-ranked 10 drug agents (Tegobuvir, Nilotinib, Digoxin, Proscillardin, Simeprevir, Sorafenib, Torin 2, Rapamycin, Vancomycin and Hesperidin) as candidate drug agentss with average binding affinity scores − 14.5 kcal/mol against the proposed 13 receptors. The Table 3 showed the summary results of interacting properties of top 3 candidate drugs (Tegobuvir, Nilotinib, Digoxin) with our proposed top 3 receptors (IRF4, MX2, and NFKB1) that produced the negatively highest binding affinity scores.

Molecular docking analysis results, where red colors indicated the strong binding affinities (A) Image of binding affinities based on the ordered top ranked 50 drug agents out of 185 against the ordered 13 proposed receptor proteins, (B) Cross-validation performance results of our proposed candidate drugs compare to the Taz et al. (2020) suggested drugs against their suggested receptors. (C) Cross-validation performance results of our proposed candidate drugs compare to the Taz et al. (2020) suggested drugs, against the top-ranked independent receptors published by others.
Cross-validation (CV) of the proposed drugs
We investigated the resistance performance of our selected 10 candidate drugs compare to the Taz et al.14 suggested 10 drugs (mentioned introduction section) against their utilized 11 receptors as well as stat-of-art alternatives publicly available top-ranked 11 independent receptors by molecular docking. The 3D structures of 9 Taz et al. suggested receptors14 PI3, MMP9, SOD2, SAA1, S100A8, SERPINA3, ICAM1, S100A12 and S100A9 were retrieved from PDB with codes p0dji9, 2rel, 1l6j, 1ap6, 4ip8, q9nr00, 4ggf, 1qmn, 2oz4, 2wcb, and 1irj respectively. The 3D structures of their 2 receptors SAA2 and C8orf4 were downloaded from SWISS-MODEL53 using UniProt92 ID P0DJI9 and Q9NR00, respectively. The 3D structures of top-ranked 11 independent receptor proteins ICAM1, IRF7, MX1, NFKBIA, STAT1, IL6, TNF, CCL20, CXCL8, VEGFA and CASP3 were retrieved from PDB with codes 5mza, 2o61, 3szr, 1nfi, 1bf5, 1il6, 1tnf, 2jyo, 1ikl, 1cz8 and 1gfw, respectively. The docking results were displayed in Fig. 6B,C. We observed that our proposed candidate drugs show much better binding performance compare to the Taz et al. (2020) suggested drugs against their utilized receptors (see Fig. 6B) as well as independent receptors also (see Fig. 6C). We also investigated the binding performance of our selected 10 candidate drugs with randomly chosen 7 independent receptor proteins that were not belongs to the DEGs-set (see Fig. S1). Among them, 3D structures of POSTN, TNC, COL1A1, TACSTD2, LAMC1, COL1A2 were retrieved from PDB with ID 5WT7, 1TEN, 5CTI, 2MAE, 5XAU, and 5CTD, respectively. The “AlphaFold Protein Structure Database” was used to retrieve the 3D structures TVP23C with Uniport ID K7EK95. We observed that our suggested drug molecules do not significantly bind to those randomly selected unimportant independent receptors. Therefore, binding affinity scores (docking scores) play an important role to select the potential drug molecules.
Discussion
The SARS-CoV-2 infections and IPF disease stimulate each other through the common genetic factors for which patients goes to the severe condition14. To investigate the genetic influence between SARS-CoV-2 infections and IPF disease, we identified 32 shared cDEGs. Among them, we detected 10 cDEGs (CXCR4, TNFAIP3, VCAM1, NLRP3, TNFAIP6, SELE, MX2, IRF4, UBD and CH25H) as cHubGs/cHubPs highlighting their functions, pathways, regulators, associated other diseases and candidate drug molecules. Some individual studies also supported the association of our proposed cHubGs with the SARS-CoV-2 infections and IPF disease as displayed in Fig. 7A. The literature review showed that CXCR4 is highly expressed in the lung cell of COVID-19 affected patients and also able to regulated the dense T cell infiltration93. In addition, CXCR4 signaling is considered as the key molecular regulator of different COVID-19 symptom including hypoxia and intussusceptive angiogenesis94. It is believed that fibrocytes, which differentiate into matrix produce myofibroblasts after transferring from the circulation into the lung, are considered as the major source of fibroblasts in IPF95. According to one study, the majority of circulating fibrocytes contains CXCR496, and more importantly, fibrocytes are not present in healthy lungs cell97. TNFAIP3 overexpression could also lead to severe tissue damage and inflammation98. Some studies found that a number of neutrophils is significantly higher in severe SARS-CoV-2 infected patients with TNFAIP3 overexpression than the patient with no or mild infection98. The interaction between TNFAIP3 and glycogen synthase kinase-3β increased the C/EBP\(\upbeta\) dysregulation in alveolar macrophages which considered as the major risk factor for IPF99. Serum Levels of VCAM-1 is significantly increased in endothelial cell of COVID-19 affected patients with mild symptom, dramatically increase in severe case and also displayed a massive reduction after antiviral treatment100. The gene VCAM-1 is upregulated in the IPF patients and displayed negative correlation with two major lung function such that pulmonary diffusion capacity for carbon monoxide and forced vital capacity. VCAM1 mainly found in the blood vessels and fibrotic foci of IPF affected lung with a high expression101. NLRP3 was found as an upregulated gene in SARS-COV2 affected patients and considered as a major biomarker to increase the cytokines level in the plasma of COVID-19 patients100,102. The inflammasome of NLRP3 plays a vital role in the pathology of bleomycin induced DNA damage via oxidative injury, cell death of alveolar macrophages and epithelial cells and lung injury of IPF patients103,104,105. The gene TNFAIP6(TSG-6) is a potential biomarker of mesenchymal stromal cells (MSCs) which can decrease the cytokine storm produced by SARS-CoV-2 infection. This MSCs based therapy may be potential therapeutic strategy for COVID-19 and also MSCs may be able to improve pulmonary fibrosis and lung function106,107,108. Inflammatory cytokines are considered as the major key player of SELE upregulation109. It has been widely suggested that the soluble form of E-selectin also known as SELE, which is released during inflammation, is a biomarker of endothelial dysfunction during COVID-19 disease110. The SELE gene has an important role in the pathogenesis of IPF disease111. An abnormal type I interferon (IFN) response is exhibited with the COVID-19 patients, which results in the deregulation of interferon-stimulated genes(ISGs) including MX2 and leads the COVID-19 severity112,113. The MX2 gene is significantly enriched in type I IFN signaling pathway which plays a vital role for IPF disease114. The IRF4 gene may regulate various altered expression of long noncoding RNAs (lncRNAs) in response to SARS-CoV-2 infection115 and it regulates M2 macrophages which play an important role in IPF116,117. The upregulations of UBD, a known inhibitor of the antiviral interferon host response, is also seen, as an increased permissiveness to the SARS-CoV-2-VSV pseudovirus118. Another ISGs, CH25H transforms cholesterol into 25-hydrocholesterol (25HC), and 25HC exhibits widespread anti-coronavirus behavior by preventing membrane fusion119. Thus, it was seen that both COVID-19 and IPF disease might be stimulated each other through the influence of cHubGs. We also investigated other diseases as the comorbidities risk factors that may stimulate both COVID-19 and IPF diseases through the influence of cHubGs by using the disease-cHubGs association studies and multivariate survival analysis of lung cancer patients with cHubGs. We found that different lung diseases including cancer120, Epstein-Barr virus121, arthritis122, diabetes12, Inflammation122 etc. might be the significant risk factors for severity of both COVID-19 and IPF diseases.

Verification of cHubGs and candidate drugs in favor of COVID-19 and IPF disease by the literature review (A) Verification of the proposed cHubGs: elliptical nodes with green color indicates cHubGs, and each connected network with white color node indicates the reference of COVID-19 and yellow color node indicates the reference of IPF, (B) Verification of the proposed candidate drugs: elliptical nodes with green color indicate FDA approved & wet-lab validated for both COVID-19 and IPF diseases, light purple color indicates FDA approved drugs, deep blue color indicate investigational drugs and light red color indicate unapproved drugs. Each connected network with white color node indicates the reference of COVID-19 and yellow color node indicates the reference of IPF.
The cHubGs regulatory network analysis based on two databases commonly revealed two TFs proteins (JUN and NFKB1) and two miRNAs (hsa-mir-155-5p and hsa-mir-21-3p) as the transcriptional and post-transcriptional regulatory factors of cHubGs, respectively that were introduced previously in the “Results” section. Some previous studies also supported our finding that JUN is an important molecular signature for both SARS-CoV-2 infection and IPF disease123,124. The NFKB1 protein is considered as a potential biomarker for both SARS-COV-2 infection and IPF disease125,126. To investigate the common pathogenetic processes of cHubGs, we selected top five common GO terms for each of BPs, MFs and CCs, and KEGG pathways that are significantly enriched by cHubGs in at least two of three databases (see Table 3). Among them, the association of the detected BPs (inflammatory response, response to virus, regulation of inflammatory response, response to tumor necrosis factor, response to cytokine) with COVID-19 and IPF diseases were supported by several independent studies60,61,62,63,64,65,66,67,68,69,70,71,72,73,74. The SARS-CoV-2 infection is influenced by a severe inflammatory response with the release of a huge amount of cytokine storm known as pro-inflammatory cytokines. A number of studies suggested that the cytokine storms are associated directly with multi-organ failure, lung injury and unfavorable prognosis of SARS-CoV-2 infections60,61,62,63,64. Within the respiratory tract, persistent inflammatory response underlies the pathogenesis of a number of chronic pulmonary diseases including pulmonary fibrosis, asthma and chronic obstructive pulmonary disease65. The top five commonly enriched MFs (ubiquitin binding, sequence-specific DNA binding, CCR chemokine receptor binding, chemokine activity) by cHubGs, are significantly associated with both COVID-19 and IPF disease59,75,76. Similarly, the association of top four CCs (tertiary granule lumen, early endosome, extracellular space, cytosol, extracellular region) with COVID-19 and IPF disease were supported by the literature review66,77,78,79,80,81,82,83. The top five commonly enriched KEGG pathways (TNF signaling, IL-17 signaling, NF-kappa B signaling, NOD-like receptor signaling and Cytokine-cytokine receptor interaction pathways) are also significantly associated with COVID-19 and IPF diseases69,84,85,86,87,88,89,90,91. These pathways are associated with the inflammatory response, immune response and the pathogen-related molecular modes recognition. The IL-17 signaling pathway is related with acute respiratory distress syndrome and critical for clearing extracellular pathogens85. A study reported that the expression levels of different inflammatory factors might be increased in the SARS-CoV-2 infected patients due to the activation of NF-kappa B signaling88. The degradation of SARS-CoV-2 genome and its inhibition are influenced by the elevated NOD-like receptor genes89. On the other hand, some studies also claimed that the TNF signaling, IL-17 signaling, NF-kappa B signaling, NOD-like receptor signaling and cytokine-cytokine receptor interactions pathways were significantly enriched for IPF disease69,90.
De-novo drug discovery is a time consuming, laborious and expensive procedure. In this case, drug repurposing reduces time and cost both. To explore repurposable candidate drug molecules for the treatments against SARS-CoV-2 infections with IPF disease as comorbidity risk, we considered our proposed 10 cHubGs and its regulatory 2 key TFs-proteins as the drug target receptors and performed their docking analysis with 185 meta-drug agents [see Table S1(I, II)]. Then we selected top-ranked 10 drugs (Tegobuvir127,128,129,130,131, Nilotinib132,133,134,135,136,137,138, Digoxin139,140,141,142,143,144,145, Proscillaridin146,147,148,149,150,151, Simeprevir152,153,154,155, Sorafenib113,156,157,158., Torin 2159, Rapamycin160,161, Vancomycin162 and Hesperidin163,164,165,166,167,168 as the candidate drug molecules (see Fig. 6A), where the first three drugs showed strong binding affinities with all target proteins. Then we investigated the binding performance of our proposed drug molecules compared to the Taz et al. 2020 suggested molecules by docking analysis against the Taz et al. 2020 suggested receptors14 and observed that our proposed drugs show much better binding performance than their suggested drugs (see Fig. 6B). Then we investigated the resistance performance of both the proposed and Taz et al. suggested drugs against the state-of-the-art alternatives top-ranked 11 independent receptors published by others for SARS-CoV-2 infections. In that case, we also observed that our proposed candidate drugs are computationally more effective compare to the TAZ et al. suggested drugs against the independent receptors (see Fig. 6C). Thus, by molecular docking approach, we observed that our proposed cHubGs-guided drug molecules show much better performance compare to the TAZ et al. (2020)14 suggested cDEGs-guided drug molecules. Some individual studies also recommended our proposed 10 drug molecules for the treatment against SARS-CoV-2 infections and IPF disease individually (see Fig. 7B) in which 7 molecules (Nilotinib, Digoxin, Simeprevir, Sorafenib, Rapamycin, Vancomycin and Hesperidin) are FDA approved for other diseases. Among the 7 FDA approved molecules, 4 molecules (Nilotinib, Digoxin, Sorafenib, and Hesperidin) are experimentally validated for the treatment against SARS-CoV-2 infections and IPF disease individually as discussed below.
An in vitro study reported that the drug molecule ‘Nilotinib’ (Dose: 400 mg twice daily) show a strong antiviral activity against SARS-CoV-2 and it has a safety profile that has been established for humans use and is comparatively well tolerated134. It also reduces the antifibrotic activity as well as pulmonary fibrosis in animal model138. An experimental study reported that the Digoxin drug effectively prevents over 99% of SARS-CoV-2 replication, which resulted in viral inhibitory activity at the post entry stage of the viral life cycle139,148. An in vitro and an in-vivo study found that the PI3K/Akt signaling pathway is inhabits by digoxin (100 nmol/L and 50 nmol/L in 12 mice model group) which may be able to regulate the activation of fibroblasts and prevent the pulmonary fibrosis that bleomycin-induced in mice145. The drug proscillaridin drug plays a significant role against SARS-COV-2 viral activities with 50% inhibitory concentrations values 2.04 μM148. An in vitro study showed that the Sorafenib drug showed antiviral activities against SAR-COV-2 by inhibiting growth factor receptor signaling pathways in non-toxic conditions158. The approved drugs Sorafenib one per day at a dose of 5 mg/kg body weight has strong antifibrotic activities on bleomycin induced IPF in mice model157. An in-vitro study found that the drug molecule ‘simeprevir’ significantly decreases the SARS-CoV-2 viral load152. In a double-blind placebo-controlled trial, the drug ‘Hesperidin 1000 mg daily may help decrease fever, cough, shortness of breath, or anosmia symptoms of nonvaccinated COVID-19 infected patients169. In histopathological, biochemical, and micro-CT analyses on rat, Hesperidin successfully reduced the severity of lung injury caused by BLC, a model for IPF168. Therefore, the findings of this study might be useful resources for simultaneously diagnosis and therapies of the SARS-CoV-2 infections and IPF diseases.
Conclusion
In this article, we detected 10 common hub-genes (cHubGs) that can differentiate both COVID-19 and IPF diseases from the control samples based on their expression patterns by using the integrated bioinformatics approaches. We observed that our proposed cHubGs (CXCR4, TNFAIP3, VCAM1, NLRP3, TNFAIP6, SELE, MX2, IRF4, UBD and CH25H) were more representative of common differentially expressed genes (cDEGs) than the previously suggested cHubGs by others. The cHubGs regulatory network analysis commonly detected SARS-CoV-2 and IPF diseases causing two crucial transcriptional (JUN and NFKB1) and post-transcriptional (hsa-mir-155-5p and hsa-mir-21-3p) regulators of cHubGs based on three independent databases JASPAR, TarBase and RegNetwork. The cHubGs-set enrichment analysis commonly detected some crucial GO-terms (BPs, CCs, and MFs) and KEGG pathways that are significantly associated with the development mechanisms of SARS-CoV-2 and IPF diseases from three independent databases Enrichr, DAVID and GeneCodis. Then we recommended our proposed cHubGs-guided 10 candidate drugs for the treatment against the SAR-CoV-2 infections with IPF disease as co-existing risk factor. We observed that our suggested drugs are computationally more efficient compare to the previously suggested drug molecules for the treatment against the SAR-CoV-2 infections with IPF disease as comorbidity risk factor. Among our proposed ten drug molecules, seven molecules (Nilotinib, Digoxin, Simeprevir, Sorafenib, Rapamycin, Vancomycin and Hesperidin) are FDA approved for other diseases, where four molecules (Nilotinib, Digoxin, Sorafenib, and Hesperidin) are experimentally validated for the treatment against SARS-CoV-2 infections and IPF disease individually. Thus, our proposed molecular biomarkers and candidate drug molecules in this study has a merit for diagnosis and therapies of SAR-CoV-2 infections with IPF disease as comorbidity risk.
Data availability
The datasets of RNA-Seq profiles on SARS-CoV-2 infections and IPF diseases that were analyzed in this study are collected from the National Center for Biotechnology Information (NCBI) Gene Expression Omnibus (GEO) open access database, where datasets are freely available (http://www.ncbi.nlm.nih.gov/geo/). The RNA-Seq profiles dataset on SARS-CoV-2 infected patients were downloaded from GPL18573 platform of Illumina NextGen 500 (Homo sapiens) with the GEO accession numbers GSE147507 published by Blanco-Melo et al.21. On the hand, the RNA-Seq profiles dataset on IPF patients were downloaded from GPL11154 platform of Illumina HiSeq 2000 (Homo sapiens) with the GEO accession numbers GSE52463 published by Nance et al.22.
References
Gorbalenya, A. E. et al. The species severe acute respiratory syndrome-related coronavirus: Classifying 2019-nCoV and naming it SARS-CoV-2. Nat. Microbiol. 5, 536–544 (2020).
Zhang, R., Li, Y., Zhang, A. L., Wang, Y. & Molina, M. J. Identifying airborne transmission as the dominant route for the spread of COVID-19. Proc. Natl. Acad. Sci. U. S. A. 117, 14857–14863 (2020).
Wang, C., Horby, P. W., Hayden, F. G. & Gao, G. F. A novel coronavirus outbreak of global health concern. Lancet 395, 470–473 (2020).
Zhu, N. et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 382, 727–733 (2020).
Yuki, K., Fujiogi, M. & Koutsogiannaki, S. COVID-19 pathophysiology: A review. Clin. Immunol. 215, 108427(2020).
Cascella, M., Rajnik, M., Cuomo, A., Dulebohn, S. C. & Di Napoli, R. Features, Evaluation and Treatment Coronavirus (COVID-19) (StatPearls, 2020).
Bosch, B. J., van der Zee, R., de Haan, C. A. M. & Rottier, P. J. M. The coronavirus spike protein is a class I virus fusion protein: Structural and functional characterization of the fusion core complex. J. Virol. 77, 8801–8811 (2003).
Chen, Y., Guo, Y., Pan, Y. & Zhao, Z. J. Structure analysis of the receptor binding of 2019-nCoV. Biochem. Biophys. Res. Commun. 525, 135–140 (2020).
Walls, A. C. et al. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell 181, 281–292.e6 (2020).
Letko, M., Marzi, A. & Munster, V. Functional assessment of cell entry and receptor usage for SARS-CoV-2 and other lineage B betacoronaviruses. Nat. Microbiol. 5, 562–569(2020).
Zou, X. et al. Single-cell RNA-seq data analysis on the receptor ACE2 expression reveals the potential risk of different human organs vulnerable to 2019-nCoV infection. Front. Med. 14,185–192 (2020).
George, P. M., Wells, A. U. & Jenkins, R. G. Pulmonary fibrosis and COVID-19: The potential role for antifibrotic therapy. Lancet Respir. Med. 8, 807–815 (2020).
John Hopkins University of Medicine, Coronavirus Resource Center. 2022. https://coronavirus.jhu.edu/map.html.
Taz, T. A. et al. Network-based identification genetic effect of SARS-CoV-2 infections to Idiopathic pulmonary fibrosis (IPF) patients. Brief. Bioinform. 22, 1254–1266 (2021).
Raghu, G. et al. An Official ATS/ERS/JRS/ALAT Statement: Idiopathic pulmonary fibrosis: Evidence-based guidelines for diagnosis and management. Am. J. Respir. Crit. Care Med. 183, 788–824 (2011).
Akter, T., Silver, R. M. & Bogatkevich, G. S. Recent advances in understanding the pathogenesis of scleroderma-interstitial lung disease. Curr. Rheumatol. Rep. 16, 411 (2014).
King, T. E. et al. A Phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N. Engl. J. Med. 370, 2083–2092 (2014).
Brake, S. J. et al. Smoking upregulates angiotensin-converting enzyme-2 receptor: A potential adhesion site for novel coronavirus SARS-CoV-2 (Covid-19). J. Clin. Med. 9, 841 (2020).
Sohal, S. S., Hansbro, P. M., Shukla, S. D., Eapen, M. S. & Walters, E. H. Potential mechanisms of microbial pathogens in idiopathic interstitial lung disease. Chest 152, 899–900 (2017).
Zhao, S., Fung-Leung, W. P., Bittner, A., Ngo, K. & Liu, X. Comparison of RNA-Seq and microarray in transcriptome profiling of activated T cells. PLoS One 9, e78644 (2014).
Blanco-Melo, D. et al. Imbalanced Host Response to SARS-CoV-2 Drives Development of COVID-19. Cell. 181,1036–1045.e9 (2020)
Nance, T. et al. Transcriptome analysis reveals differential splicing events in IPF lung tissue. PLoS One 9, e92111 (2014).
Beck, B. R., Shin, B., Choi, Y., Park, S. & Kang, K. Predicting commercially available antiviral drugs that may act on the novel coronavirus (SARS-CoV-2) through a drug-target interaction deep learning model. Comput. Struct. Biotechnol. J. 18, 784–790 (2020).
Robinson, M. D., McCarthy, D. J. & Smyth, G. K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2009).
Robinson, M. D., McCarthy, D. J. & Smyth, G. K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data | Bioinformatics | Oxford Academic. Bioinformatics 26, 139–140 (2010).
Benjamini, Y. & Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B 57, 289–300 (1995).
Szklarczyk, D. et al. The STRING database in 2011: Functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res. 39, D561–D568 (2011).
Shannon, P. et al. Cytoscape: A software Environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504 (2003).
Chin, C. H. et al. cytoHubba: Identifying hub objects and sub-networks from complex interactome. BMC Syst. Biol. 8, S11 (2014).
Jeong, H., Mason, S. P., Barabási, A. L. & Oltvai, Z. N. Lethality and centrality in protein networks. Nature 411, 41–42 (2001).
Pržulj, N., Wigle, D. A. & Jurisica, I. Functional topology in a network of protein interactions. Bioinformatics 20, 340–348 (2004).
Freeman, L. C. A Set of measures of centrality based on betweenness. Sociometry 40, 35–41 (1977).
Shimbel, A. Structural parameters of communication networks. Bull. Math. Biophys. 15, 501–507 (1953).
Sabidussi, G. The centrality index of a graph. Psychometrika 31, 581–603 (1966).
Khan, A. et al. JASPAR 2018: Update of the open-access database of transcription factor binding profiles and its web framework. Nucleic Acids Res. 46, D260–D266 (2018).
Karagkouni, D. et al. DIANA-TarBase v8: A decade-long collection of experimentally supported miRNA-gene interactions. Nucleic Acids Res. 46, D239–D245 (2018).
Liu, Z. P., Wu, C., Miao, H. & Wu, H. RegNetwork: An integrated database of transcriptional and post-transcriptional regulatory networks in human and mouse. Database 2015, bav095 (2015).
Zhou, G. et al. NetworkAnalyst 3.0: A visual analytics platform for comprehensive gene expression profiling and meta-analysis. Nucleic Acids Res. 47, W234–W241 (2019).
Boyle, E. I. et al. GO::TermFinder-open source software for accessing Gene Ontology information and finding significantly enriched Gene Ontology terms associated with a list of genes introduction: Motivation and design. Bioinforma. Appl. NOTE 20, 3710–3715 (2004).
Kanehisa, M. & Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30 (2000).
Chen, E. Y. et al. Enrichr: Interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinform 14, 128 (2013).
Huang, D. W., Sherman, B. T. & Lempicki, R. A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57 (2009).
Huang, D. W., Sherman, B. T. & Lempicki, R. A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 37, 1–13 (2009).
Nogales-Cadenas, R. et al. GeneCodis: Interpreting gene lists through enrichment analysis and integration of diverse biological information. Nucleic Acids Res. 37, W317–W322 (2009).
Kuleshov, M. V. et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 44, W90–W97 (2016).
Piñero, J. et al. The DisGeNET knowledge platform for disease genomics: 2019 update. Nucleic Acids Res. 48, D845–D855 (2020).
Piñero, J. et al. DisGeNET: A discovery platform for the dynamical exploration of human diseases and their genes. Database 2015, bav028 (2015).
Aguirre-Gamboa, R. et al. SurvExpress: An online biomarker validation tool and database for cancer gene expression data using survival analysis. PLoS One 8, e74250 (2013).
Ray, W. D. & Collett, D. Modelling survival data in medical research. J. R. Stat. Soc. Ser. A Stat. Soc. 158, 188 (1995).
Bøvelstad, H. M. & Borgan, Ø. Assessment of evaluation criteria for survival prediction from genomic data. Biometrical J. 53, 202–216 (2011).
Berman, H. M. et al. The protein data bank. Nucleic Acids Res. 28, 235–242 (2000).
Varadi, M. et al. AlphaFold protein structure database: Massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 50, D439–D444 (2022).
Waterhouse, A. et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 46, W296–W303 (2018).
Kim, S. et al. PubChem 2019 update: Improved access to chemical data. Nucleic Acids Res. 47, D1102–D1109 (2019).
Visualizer, D. S. v4. 0. 100. 13345. Accelrys Sof tware Inc (2005).
Morris, G. M. et al. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 30, 2785–2791 (2009).
Trott, O. & Olson, A. J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 31, 455–461(2010).
Delano, W. L. & Bromberg, S. PyMOL User’s Guide. DeLano Scientific LLC (2004).
Deng, X. et al. Structure-guided mutagenesis alters deubiquitinating activity and attenuates pathogenesis of a murine coronavirus. J. Virol. 94, 11 (2020).
Huang, C. et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 395, 497–506 (2020).
Ruan, Q., Yang, K., Wang, W., Jiang, L. & Song, J. Clinical predictors of mortality due to COVID-19 based on an analysis of data of 150 patients from Wuhan, China. Intensive Care Med. 46, 846–848 (2020).
Chen, L. et al. [Analysis of clinical features of 29 patients with 2019 novel coronavirus pneumonia]. Zhonghua Jie He He Hu Xi Za Zhi 43, 203–208 (2020).
Sun, D. et al. Clinical features of severe pediatric patients with coronavirus disease 2019 in Wuhan: A single center’s observational study. World J. Pediatr. 16, 251–259 (2020).
Vishnubalaji, R., Shaath, H. & Alajez, N. M. Protein coding and long noncoding RNA (lncRNA)) transcriptional landscape in SARS-CoV-2 infected bronchial epithelial cells highlight a role for interferon and inflammatory response. Genes (Basel) 11, 760 (2020).
Racanelli, A. C., Kikkers, S. A., Choi, A. M. K. & Cloonan, S. M. Autophagy and inflammation in chronic respiratory disease. Autophagy 14, 221–232 (2018).
Chen, Q. et al. Identification of hub genes associated with COVID-19 and idiopathic pulmonary fibrosis by integrated bioinformatics analysis. PLoS One 17, e0262737 (2022).
De Almeida, R. M. C., Thomas, G. L. & Glazier, J. A. Transcriptogram analysis reveals relationship between viral titer and gene sets responses during Corona-virus infection. NAR Genom. Bioinform. 4, lqac020 (2022).
Taz, T. A. et al. Identification of biomarkers and pathways for the SARS-CoV-2 infections that make complexities in pulmonary arterial hypertension patients. Brief. Bioinform. 22, 1451–1465 (2021).
Xia, Y., Lei, C., Yang, D. & Luo, H. Identification of key modules and hub genes associated with lung function in idiopathic pulmonary fibrosis. PeerJ 8, e9848(2020).
Hasankhani, A. et al. Differential co-expression network analysis reveals key hub-high traffic genes as potential therapeutic targets for COVID-19 pandemic. Front. Immunol. 12, 789317 (2021).
Singh, S. & Yang, Y. F. Pharmacological mechanism of NRICM101 for COVID-19 treatments by combined network pharmacology and pharmacodynamics. Int. J. Mol. Sci. 23, 15385 (2022).
Sohn, E. J. Functional roles and targets of COVID-19 in blood cells determined using bioinformatics analyses. Bioinform. Biol. Insights 16, 11779322221080266 (2022).
Xia, J. et al. Immune response is key to genetic mechanisms of SARS-CoV-2 infection with psychiatric disorders based on differential gene expression pattern analysis. Front. Immunol. 13, 79853 (2022).
Morenikeji, O. B., Bernard, K., Strutton, E., Wallace, M. & Thomas, B. N. Evolutionarily conserved long non-coding RNA regulates gene expression in cytokine storm during COVID-19. Front. Bioeng. Biotechnol. 8, 582953 (2021).
Agrawal, P., Sambaturu, N., Olgun, G. & Hannenhalli, S. A path-based analysis of infected cell line and COVID-19 patient transcriptome reveals novel potential targets and drugs against SARS-CoV-2. Front. Immunol. 13, 918817 (2022).
More, S. A., Patil, A. S., Sakle, N. S. & Mokale, S. N. Network analysis and molecular mapping for SARS-CoV-2 to reveal drug targets and repurposing of clinically developed drugs. Virology 555, 10–18 (2021).
Fang, K. Y. et al. Exploration and validation of related hub gene expression during SARS-CoV-2 infection of human bronchial organoids. Hum. Genom. 15, 18 (2021).
Xu, Z., Mo, L., Feng, X., Huang, M. & Li, L. Using bioinformatics approach identifies key genes and pathways in idiopathic pulmonary fibrosis. Medicine (Baltimore) 99, e22099 (2020).
Zolfaghari Emameh, R., Nosrati, H., Eftekhari, M., Falak, R. & Khoshmirsafa, M. Expansion of single cell transcriptomics data of SARS-CoV infection in human bronchial epithelial cells to COVID-19. Biol. Proceed. Online 22, 16 (2020).
Dey, L., Chakraborty, S. & Mukhopadhyay, A. Machine learning techniques for sequence-based prediction of viral–host interactions between SARS-CoV-2 and human proteins. Biomed. J. 43, 438–450 (2020).
Liu, W. et al. Exploring the potential targets and mechanisms of Huang Lian Jie Du decoction in the treatment of coronavirus disease 2019 based on network pharmacology. Int. J. Gen. Med. 14, 9873–9885 (2021).
Lin, H. et al. Exploring the treatment of COVID-19 with Yinqiao powder based on network pharmacology. Phyther. Res. 35, 2651–2664 (2021).
Tao, Q. et al. Network pharmacology and molecular docking analysis on molecular targets and mechanisms of Huashi Baidu formula in the treatment of COVID-19. Drug Dev. Ind. Pharm. 46, 1345–1353 (2020).
Feldmann, M. et al. Trials of anti-tumour necrosis factor therapy for COVID-19 are urgently needed. Lancet 395, 1407–1409 (2020).
Orlov, M., Wander, P. L., Morrell, E. D., Mikacenic, C. & Wurfel, M. M. A case for targeting Th17 cells and IL-17A in SARS-CoV-2 infections. J. Immunol. 205, 892–898 (2020).
Sodhi, C. P. et al. A dynamic variation of pulmonary ACE2 is required to modulate neutrophilic inflammation in response to pseudomonas aeruginosa lung infection in mice. J. Immunol. 203, 3000–3012 (2019).
Jiang, Y., Tian, M., Lin, W., Wang, X. & Wang, X. Protein kinase serine/threonine kinase 24 positively regulates interleukin 17-induced inflammation by promoting IKK complex activation. Front. Immunol. 9, 921 (2018).
Leng, L. et al. Pathological features of COVID-19-associated lung injury: A preliminary proteomics report based on clinical samples. Signal Transduct. Target. Ther. 5, 240 (2020).
Bawage, S. S. et al. Gold nanorods inhibit respiratory syncytial virus by stimulating the innate immune response. Nanomed. Nanotechnol. Biol. Med. 12, 2299–2310 (2016).
Wang, H., Xie, Q., Ou-Yang, W. & Zhang, M. Integrative analyses of genes associated with idiopathic pulmonary fibrosis. J. Cell. Biochem. 120, 8648–8660 (2019).
Monteleone, G., Sarzi-Puttini, P. C. & Ardizzone, S. Preventing COVID-19-induced pneumonia with anticytokine therapy. Lancet Rheumatol. 2, e255–e256 (2020).
The Uniprot Consortium. UniProt: A worldwide hub of protein knowledge The UniProt Consortium. Nucleic Acids Res 47, D506–D515 (2019).
Ackermann, M., Mentzer, S. J., Kolb, M. & Jonigk, D. Inflammation and intussusceptive angiogenesis in COVID-19: Everything in and out of flow. Eur. Respir. J. 56, 2003147 (2020).
Dimova, I. et al. SDF-1/CXCR4 signalling is involved in blood vessel growth and remodelling by intussusception. J. Cell. Mol. Med. 23, 3916–3926 (2019).
Phillips, R. J. et al. Circulating fibrocytes traffic to the lungs in response to CXCL12 and mediate fibrosis. J. Clin. Invest. 114, 438–46 (2004).
Mehrad, B., Burdick, M. D. & Strieter, R. M. Fibrocyte CXCR4 regulation as a therapeutic target in pulmonary fibrosis. Int. J. Biochem. Cell Biol. 41, 1708–1718 (2009).
Andersson-Sjöland, A. et al. Fibrocytes are a potential source of lung fibroblasts in idiopathic pulmonary fibrosis. Int. J. Biochem. Cell Biol. 40, 2129–2140 (2008).
Vereecke, L., Beyaert, R. & van Loo, G. The ubiquitin-editing enzyme A20 (TNFAIP3) is a central regulator of immunopathology. Trends Immunol. 30, 383–391 (2009).
Liu, S. shan et al. Targeting degradation of the transcription factor C/EBPβ reduces lung fibrosis by restoring activity of the ubiquitin-editing enzyme A20 in macrophages. Immunity 51, 522–534.e7 (2019).
Tong, M. et al. Elevated expression of serum endothelial cell adhesion molecules in COVID-19 patients. J. Infect. Dis. 222, 894–898 (2020).
Agassandian, M. et al. VCAM-1 is a TGF-β1 inducible gene upregulated in idiopathic pulmonary fibrosis. Cell. Signal. 27, 2467–2473 (2015).
Ge, C. & He, Y. In silico prediction of molecular targets of astragaloside IV for alleviation of COVID-19 hyperinflammation by systems network pharmacology and bioinformatic gene expression analysis. Front. Pharmacol. 11, 556984 (2020).
Gasse, P. et al. Uric acid is a danger signal activating NALP3 inflammasome in lung injury inflammation and fibrosis. Am. J. Respir. Crit. Care Med. 179, 903–913 (2009).
Smith, R. E., Strieter, R. M., Phan, S. H. & Kunkel, S. L. C-C Chemokines: Novel mediators of the profibrotic inflammatory response to bleomycin challenge. Am. J. Respir. Cell Mol. Biol. 15, 693–702 (1996).
De Nardo, D., De Nardo, C. M. & Latz, E. New insights into mechanisms controlling the NLRP3 inflammasome and its role in lung disease. Am. J. Pathol. 184, 42–54 (2014).
Kavianpour, M., Saleh, M. & Verdi, J. The role of mesenchymal stromal cells in immune modulation of COVID-19: Focus on cytokine storm. Stem Cell Res. Ther. 11, 404 (2020).
Xu, Z et al. Current status of cell-based therapies for COVID-19: Evidence from mesenchymal stromal cells in sepsis and ARDS. Front Immunol. 12, 738697 (2021).
Ni, K. et al. Rapid clearance of heavy chain-modified hyaluronan during resolving acute lung injury. Respir. Res. 19, 107 (2018).
Balta, S. Endothelial dysfunction and inflammatory markers of vascular disease. Curr. Vasc. Pharmacol. 19, 243–249 (2020).
Smadja, D. M. et al. Angiopoietin-2 as a marker of endothelial activation is a good predictor factor for intensive care unit admission of COVID-19 patients. Angiogenesis 23, 611–620 (2020).
Umesh, Kundu, D., Selvaraj, C., Singh, S. K. & Dubey, V. K. Identification of new anti-nCoV drug chemical compounds from Indian spices exploiting SARS-CoV-2 main protease as target. J. Biomol. Struct. Dyn. 39, 3428–3434 (2021).
Bizzotto, J. et al. SARS-CoV-2 infection boosts MX1 antiviral effector in COVID-19 patients. iScience 23, 101585 (2020).
Hadjadj, J. et al. Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science. 369, 718–724 (2020).
Fraser, E. et al. Multi-modal characterization of monocytes in idiopathic pulmonary fibrosis reveals a primed type I interferon immune phenotype. Front. Immunol. 12, 623430 (2021).
Laha, S. et al. In silico analysis of altered expression of long non-coding RNA in SARS-CoV-2 infected cells and their possible regulation by STAT1, STAT3 and interferon regulatory factors. Heliyon 7, e06395 (2021).
Satoh, T. et al. The Jmjd3-Irf4 axis regulates M2 macrophage polarization and host responses against helminth infection. Nat. Immunol. 11, 936–944 (2010).
Zhang, L. et al. Macrophages: Friend or foe in idiopathic pulmonary fibrosis? Respir. Res. 19, 170 (2018).
McCarron, S. et al. Functional characterization of organoids derived from irreversibly damaged NASH patient liver. Hepatology. 74, 1825–1844 (2021).
Wang, S. et al. Cholesterol 25‐hydroxylase inhibits SARS ‐CoV‐2 and other coronaviruses by depleting membrane cholesterol. EMBO J. 39, e106057 (2020).
Satu, M. S. et al. Diseasome and comorbidities complexities of SARS-CoV-2 infection with common malignant diseases. Brief. Bioinform. 74, 1825–1844 (2021).
Chen, T., Song, J., Liu, H., Zheng, H. & Chen, C. Positive Epstein–Barr virus detection in coronavirus disease 2019 (COVID-19) patients. Sci. Rep. 11, 10902 (2021).
Páramo, J. A. Inflammatory response in relation to COVID-19 and other prothrombotic phenotypes. Reumatol clínica (English Ed). 18, 1–4 (2020).
Zhou, Y. et al. Network-based drug repurposing for novel coronavirus 2019-nCoV/SARS-CoV-2. Cell Discov. 6,14 (2020).
Wernig, G. et al. Unifying mechanism for different fibrotic diseases. Proc. Natl. Acad. Sci. U. S. A. 114, 4757–4762 (2017).
Nain, Z. et al. Pathogenetic profiling of COVID-19 and SARS-like viruses. Brief. Bioinform. 22, 1175–1196 (2021).
Jonigk, D. et al. Obliterative airway remodelling in transplanted and non-transplanted lungs. Virchows Arch. 457, 369–380 (2010).
Ruan, Z. et al. SARS-CoV-2 and SARS-CoV: Virtual screening of potential inhibitors targeting RNA-dependent RNA polymerase activity (NSP12). J. Med. Virol. 93, 389–400 (2021).
Chandel, V. et al. Structure-based drug repurposing for targeting Nsp9 replicase and spike proteins of severe acute respiratory syndrome coronavirus 2. J. Biomol. Struct. Dyn. 40, 249–262 (2022).
Encinar, J. A. & Menendez, J. A. Potential drugs targeting early innate immune evasion of SARS-coronavirus 2 via 2′-O-Methylation of Viral RNA. Viruses 12, 525 (2020).
Sahoo, B. M. et al. Drug repurposing strategy (DRS): Emerging approach to identify potential therapeutics for treatment of novel coronavirus infection. Front. Mol. Biosci. 8, 628144 (2021).
Zhou, Y. W. et al. Therapeutic targets and interventional strategies in COVID-19: Mechanisms and clinical studies. Signal Transduct. Target. Ther. 6, 317 (2021).
Murugan, N. A., Kumar, S., Jeyakanthan, J. & Srivastava, V. Searching for target-specific and multi-targeting organics for Covid-19 in the Drugbank database with a double scoring approach. Sci. Rep. 10, 19125 (2020).
de Oliveira, O. V., Rocha, G. B., Paluch, A. S. & Costa, L. T. Repurposing approved drugs as inhibitors of SARS-CoV-2 S-protein from molecular modeling and virtual screening. J. Biomol. Struct. Dyn. 39, 3924–3933 (2021).
Cagno, V., Magliocco, G., Tapparel, C. & Daali, Y. The tyrosine kinase inhibitor nilotinib inhibits SARS-CoV-2 in vitro. Basic Clin. Pharmacol. Toxicol. 128, 621–624 (2021).
Bouchlarhem, A. et al. Multiple cranial nerve palsies revealing blast crisis in patient with chronic myeloid leukemia in the accelerated phase under nilotinib during severe infection with SARS-COV-19 virus: Case report and review of literature. Radiol. Case Rep. 16 , 3602–3609 (2021).
Banerjee, S. et al. Drug repurposing to identify nilotinib as a potential SARS-CoV-2 main protease inhibitor: Insights from a computational and in vitro study. J. Chem. Inf. Model. 61, 5469–5483 (2021).
Heidari, A. et al. Recent new results and achievements of California South University (CSU) BioSpectroscopy core research laboratory for COVID-19 or 2019-nCoV treatment: Diagnosis and treatment methodologies of “coronavirus”. J. Curr. Viruses Treat. Methodol. 1, 3–41 (2020).
Rhee, C. K. et al. Effect of nilotinib on bleomycin-induced acute lung injury and pulmonary fibrosis in mice. Respiration 82, 273–287 (2011).
Cho, J. et al. Antiviral activity of digoxin and ouabain against SARS-CoV-2 infection and its implication for COVID-19. Sci. Rep. 10, 16200 (2020).
Sekhar, T. Virtual Screening based prediction of potential drugs for COVID-19. Comb. Chem. High Throughput Screen. 23, 1–25 (2020).
Rattanawong, P. et al. Guidance on short-term management of atrial fibrillation in coronavirus disease 2019. J. Am. Heart Assoc. 9, e017529 (2020).
Talluri, S. Molecular docking and virtual screening based prediction of drugs for COVID-19. Comb. Chem. High Throughput Screen. 24, 716–728 (2020).
Xing, Y. et al. Therapeutic monitoring of plasma digoxin for COVID-19 patients using a simple UPLC-MS/MS method. Curr. Pharm. Anal. 17, 1308–1316 (2021).
Peltzer, B., Lerman, B. B., Goyal, P. & Cheung, J. W. Role for digoxin in patients hospitalized with COVID-19 and atrial arrhythmias. J. Cardiovasc. Electrophysiol. 32, 880–881 (2021).
Jia, L., Yang, M., Tian, X., Zhao, P. & Mei, X. B. Y. Digoxin alleviates pulmonary fibrosis by regulating phosphatidylinositol-3-kinase/Akt signaling through inhibiting the activation of fibroblast: An in vivo and in vitro experiment. Zhonghua Wei Zhong Bing Ji Jiu Yi Xue 34, 116 (2022).
Aishwarya, S., Gunasekaran, K. & Margret, A. A. Computational gene expression profiling in the exploration of biomarkers, non-coding functional RNAs and drug perturbagens for COVID-19. J. Biomol. Struct. Dyn. 40, 3681–3696 (2022).
Xu, J. et al. Drug repurposing approach to combating coronavirus: Potential drugs and drug targets. Med. Res. Rev. 41, 1375–1426 (2021).
Jeon, S. et al. Identification of antiviral drug candidates against SARS-CoV-2 from FDA-approved drugs. Antimicrob. Agents Chemother. 64, e00819-20 (2020).
Mosharaf, M. P. et al. Meta-data analysis to explore the hub of the hub-genes that influence SARS-CoV-2 infections highlighting their pathogenetic processes and drugs repurposing. Vaccines 10, 1248 (2022).
Feng, Z. et al. Virus-CKB: An integrated bioinformatics platform and analysis resource for COVID-19 research. Brief. Bioinform. 22, 882–895 (2021).
Feng, Z. et al. MCCS: A novel recognition pattern-based method for fast track discovery of anti-SARS-CoV-2 drugs. Brief. Bioinform. 22, 946–962 (2021).
Lo, H. S. et al. Simeprevir potently suppresses SARS-CoV-2 replication and synergizes with remdesivir. ACS Cent. Sci. 7, 792–802 (2021).
Abhithaj, J. et al. Repurposing simeprevir, calpain inhibitor IV and a cathepsin F inhibitor against SARS-CoV-2 and insights into their interactions with Mpro. J. Biomol. Struct. Dyn. 40, 325–336 (2022).
Kadioglu, O., Saeed, M., Greten, H. J. & Efferth, T. Identification of novel compounds against three targets of SARS CoV-2 coronavirus by combined virtual screening and supervised machine learning. Comput. Biol. Med. 133,104359 (2021).
Khan, R. J. et al. Identification of promising antiviral drug candidates against non-structural protein 15 (NSP15) from SARS-CoV-2: An in silico assisted drug-repurposing study. J. Biomol. Struct. Dyn. 40, 438–448 (2022).
Weisberg, E. et al. Repurposing of Kinase Inhibitors for Treatment of COVID-19. Pharm. Res. 37, 167 (2020).
Chen, Y. L. et al. Sorafenib ameliorates bleomycin-induced pulmonary fibrosis: Potential roles in the inhibition of epithelial-mesenchymal transition and fibroblast activation. Cell Death Dis. 4, e665 (2013).
Klann, K. et al. Growth Factor Receptor Signaling Inhibition Prevents SARS-CoV-2 Replication. Mol. Cell 80, 164–174.e4 (2020).
Garcia, G. et al. Antiviral drug screen identifies DNA-damage response inhibitor as potent blocker of SARS-CoV-2 replication. Cell Rep. 35, 108940 (2021).
Husain, A. & Byrareddy, S. N. Rapamycin as a potential repurpose drug candidate for the treatment of COVID-19. Chemico Biol. Interact. 331, 109282 (2020).
Molina-Molina, M. et al. Anti-fibrotic effects of pirfenidone and rapamycin in primary IPF fibroblasts and human alveolar epithelial cells. BMC Pulm. Med. 18, 63 (2018).
Qiao, Z., Zhang, H., Ji, H. F. & Chen, Q. Computational view toward the inhibition of SARS-CoV-2 spike glycoprotein and the 3CL protease. Computation 8, 53 (2020).
Cheng, F. J. et al. Hesperidin is a potential inhibitor against sars-cov-2 infection. Nutrients 13, 2800 (2021).
Bellavite, P. & Donzelli, A. Hesperidin and SARS-CoV-2: New light on the healthy function of citrus fruits. Antioxidants 9, 742 (2020).
Das, S., Sarmah, S., Lyndem, S. & Singha Roy, A. An investigation into the identification of potential inhibitors of SARS-CoV-2 main protease using molecular docking study. J. Biomol. Struct. Dyn. 39, 3347–3357 (2021).
Wu, C. et al. Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharm. Sin. B 10, 766–788 (2020).
Balmeh, N., Mahmoudi, S., Mohammadi, N. & Karabedianhajiabadi, A. Predicted therapeutic targets for COVID-19 disease by inhibiting SARS-CoV-2 and its related receptors. Informatics Med. Unlocked 20, 100407 (2020).
Görmeli, C. A., Saraç, K., Çiftçi, O., Timurkaan, N. & Malkoç, S. The effects of hesperidin on idiopathic pulmonary fibrosis evaluated by histopathologial-biochemical and micro-computed tomography examinations in a bleomycin-rat model. Biomed. Res. 27, 737–742 (2016).
Dupuis, J. et al. Fourteen-day evolution of COVID-19 symptoms during the third wave in nonvaccinated subjects and effects of hesperidin therapy: A randomized, double-blinded, placebo-controlled study. Evid. Based Complement. Altern. Med. 2022, 3125662 (2022).
Acknowledgements
We would like to acknowledge Ministry of Science and Technology (MOST) Research Project (Sl#569, ID#SRG-224569, 2022–2023), Government of Bangladesh, for supporting this study.
Author information
Authors and Affiliations
Contributions
M.A.I and M.N.H.M. conceived the idea of the study. M.A.I analyzed RNA-Seq profiles and drafted the manuscript. M.K.K and M.S.R performed molecular docking for drug screening. S.A.T., K.F.T. and M.P.M. helped during the data preparation, M.A.I and M.B.H revised the manuscript, S.R.K. and H.K. edited the manuscript and provided suggestions. M.N.H.M edited the manuscript and supervised the project.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Islam, M.A., Kibria, M.K., Hossen, M.B. et al. Bioinformatics-based investigation on the genetic influence between SARS-CoV-2 infections and idiopathic pulmonary fibrosis (IPF) diseases, and drug repurposing. Sci Rep 13, 4685 (2023). https://doi.org/10.1038/s41598-023-31276-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-31276-6
- Springer Nature Limited