
Abstract
Anastellin, a recombinant protein fragment from the first type III module of fibronectin, mimics a partially unfolded intermediate implicated in the assembly of fibronectin fibrils. Anastellin influences the structure of fibronectin and initiates in vitro fibrillation, yielding “superfibronectin”, a polymer with enhanced cell-adhesive properties. This ability is absent in an anastellin double mutant, L37AY40A. Here we demonstrate that both wild-type and L37AY40A anastellin affect fibronectin processing within the extracellular matrix (ECM) of smooth muscle cells. Fibronectin fibrils are diminished in the ECM from cells treated with anastellin, but are partially rescued by supplementation with plasma fibronectin in cell media. Proteomic analyses reveal that anastellin also impacts on the processing of other ECM proteins, with increased collagen and decreased laminin detected in media from cells exposed to wild-type anastellin. Moreover, both anastellin forms stimulate release of inflammatory cytokines, including interleukin 6. At the molecular level, L37AY40A does not exhibit major perturbations of structural features relative to wild-type anastellin, though the mutant showed differences in heparin binding characteristics. These findings indicate that wild-type and L37AY40A anastellin share similar molecular features but elicit slightly different, but partially overlapping, responses in smooth muscle cells resulting in altered secretion of cytokines and proteins involved in ECM processing.
Similar content being viewed by others

Introduction
Fibronectin (FN) is a major extracellular matrix (ECM) protein implicated in the regulation of cell adhesion, migration, differentiation, and proliferation. “Cell-derived” FN released from endothelial cells, smooth muscle cells and fibroblasts is assembled into insoluble, elastic fibrils that interact with other ECM proteins (e.g. collagens and proteoglycans) and integrins on cell surfaces1,2. On the other hand, soluble plasma FN synthesized by hepatocytes is released into the circulation and recruited to sites of injury where it plays a key role in blood clot formation3. Plasma FN is also deposited into ECM fibrils together with locally-synthesized, cell-derived FN4,5,6. The mechanism of FN polymerization in the ECM is not fully established, but there is strong evidence suggesting that cytoskeletal actins exert a pulling force on FN through interactions via membrane-bound integrin receptors, which expose hidden (“cryptic”) sites that stabilize interactions between individual protomers, thereby promoting polymerization2,7,8.
FN is a multimodular glycoprotein consisting of two subunits connected by two disulfide bonds near the C-termini9. Each subunit comprises 12 type I modules (FNI), 2 type II modules (FNII) and 15–17 type III modules (FNIII), that are organized into functional domains that enable macromolecule binding. Two of the FNIII modules (EDA and EDB) are uniquely expressed in cell-derived FN. Anastellin (AN), a small protein fragment derived from the C-terminal part of the first type III module of FN (FNIII1), initiates polymerization of plasma FN in vitro, resulting in the formation of ‘superfibronectin’, a polymer which resembles ECM-derived FN fibrils at the microscopic level. In addition to its influence on FN fibrillation, AN also inhibits cell proliferation, has anti-angiogenic properties, and suppresses tumor growth and metastasis10,11,12. AN also interacts with heparin-based polysaccharides13. These interactions are probably mediated by a cluster of positively-charged cryptic residues that are important for FN-cell interactions in the native FNIII1 module14.
Besides its ability to initiate FN polymerization in vitro, AN also modulates the structure of ECM-incorporated FN, and impacts on cell behavior15,16. With fibroblasts, AN activates p38 mitogen-activated protein kinase (MAPK) signaling with subsequent effects on cytoskeletal organization and cell cycle progression15,16. AN also modulates vascular endothelial growth factor (VEGF) signaling in endothelial cells, and inhibits lysophospholipid signaling via Ras/ERK17,18. Furthermore, AN stimulates the release of inflammatory cytokines from fibroblasts and mononuclear cells through activation of NF-κB dependent pathways19,20.
The structural features of AN that are responsible for these activities are incompletely resolved, but it has been demonstrated that mutation of Leu37 and Tyr40 to Ala (i.e. an L37AY40A mutant) removes the capacity of AN to induce FN polymerization21. However, the L37AY40A mutant retains the ability of wild-type (wt) AN to modulate FN structure in assembled ECM, and impact on MAPK signaling22. We therefore hypothesized that the activity of AN to induce plasma FN polymerization, and remodeling of FN in established ECM, operate through different mechanisms. We also hypothesized that AN may induce alterations in the processing of other extracellular proteins. This was examined using a proteomics approach to quantify the levels of ECM-related proteins secreted from human coronary artery smooth muscle cells (HCASMC) exposed to either wt or L37AY40A AN. In addition, we investigated the molecular features of the two forms of AN, and compared their interactions with isolated plasma FN and heparin, with the aim to provide new insights into the mechanisms of AN-mediated FN fibrillation and ECM remodeling.
Results
Impact of AN on the structure of FN fibrils generated by HCASMC
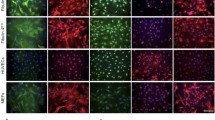
The influence of wt and L37AY40A AN on the assembly of the ECM laid down by HCASMC cultures was examined by seeding cells on chamber culture slides and incubating these in growth media without or with added AN. The resulting cultures were then probed for FN by immunofluorescence microscopy. A marked decrease in the extent of extracellular FN fibrils was observed for cells exposed to wt AN for 48 h, as detected by use of both a polyclonal FN antibody (FN 2413), and a monoclonal (FN 3E2) antibody directed against the EDA module of cell-derived FN (Fig. 1). In contrast, the FN detected in the cells treated with AN appears to be localized within the cell, or at the cell surface. With L37AY40A AN, a decrease in the levels of FN fibrils was also observed with the FN 3E2 mAb, which only detects FN containing the EDA module, whereas significant incorporation of FN into fibrils was using the pAb which recognizes both cell-derived FN species and plasma FN (Fig. 1).

AN influences FN structure in ECM from cells cultured in growth media. Primary HCASMC at an initial density of 2.25 × 104 cells per well in 8-well chamber slides were cultured overnight in growth media and then incubated in growth media containing 30 μM AN (wt or L37AY40A) for 48 h. The cells were then fixed and permeabilized followed by incubation with primary antibodies at 4 °C overnight. Total FN was visualized by an anti-FN pAb (2413, 1:500, green channel) and a mAb against the EDA module of cell-derived FN (3E2, 1:500, red channel). Nuclei were counterstained using DAPI (blue channel). Overlays of the three channels are shown on the right. Scale bars: 20 μm. Pixel intensity plots from three independent experiments are displayed in Supplementary Fig. S1.
A similar trend was observed when the HCASMC cultures were decellularized, to leave cell-free ECM, before incubation with the FN antibodies. Higher levels of FN in the ECM were detected from the cells incubated with the L37AY40A mutant compared to wt AN (Supplementary Fig. S2). These different effects of wt and L37AY40A AN imply that the L37 and Y40 residues of AN are important determinants for the observed effects on the structure of cell-derived FN. In contrast to the cell microscopy images (Fig. 1), staining of fibrillar FN was detected with the 3E2 antibody in the decellularized samples derived from cells exposed to AN, albeit at a lower level relative to control samples (Supplementary Fig. S2). This discrepancy may be explained by increased epitope accessibility when cells growing on top of the deposited matrix are removed.
To determine whether the FN incorporated into the ECM fibrils was associated with AN, decellularized ECM from cells exposed to AN was probed for both FN (using the pAb) and AN (using an antibody against the His-tag on the recombinant protein). The resulting data showed strong co-localization of FN with either wt or L37AY40A AN in the ECM, consistent with specific interactions between the two proteins (Supplementary Fig. S3a). The extent of AN incorporation was dependent on the concentration of AN added, and whether this was wt or L37AY40A AN, with the latter being incorporated to a greater extent than the former at modest, but not high AN concentrations (Supplementary Fig. S3b).
FN fibrils in cells exposed to AN are restored in the presence of plasma fibronectin
To examine the influence of the cell media on the extracellular FN fibrils, HCASMC were subjected to serum starvation, then incubated for 24 h with wt or L37AY40A AN in serum-free basal media. With both wt and L37AY40A AN, most of the FN detected with either the pAb or mAb appears to be mainly cell associated, and levels of fibrillar FN in the ECM were diminished relative to the control cells without AN (Fig. 2a). These findings indicate that the abundance of FN fibrils in cells exposed to L37AY40A AN is dependent on plasma-derived FN present in the growth (but not basal) media, or another component present in this media. In order to test this hypothesis, cells were incubated in basal media supplemented with different levels of plasma FN. These additions yielded significantly higher levels of fibrillar FN in cells exposed to AN (Fig. 2b,c vs 2a). The plasma FN-dependent increase in FN fibrils is most obvious for cells exposed to L37AY40A AN.

AN influences FN structure in ECM from cells cultured in basal media. Primary HCASMC at an initial density of 2.25 × 104 cells per well in 8-well chamber slides were cultured in basal media overnight and then incubated in basal media supplemented with 30 μM AN (wt or L37AY40A) and either 0 (a), 10 (b) or 100 (c) ug mL−1 FN for 24 h. HCASMCs on the slides were then fixed and permeabilized followed by incubation with primary antibodies at 4 °C overnight. Total FN was visualized by anti-FN pAb (2413, 1:500, green channel) and cell-derived FN detected using a mAb that recognizes the EDA epitope (3E2, 1:500, red channel); nuclei were counterstained using DAPI (blue channel). Overlays of images are shown on the right. Scale bars: 20 μm. Pixel intensity plots from three independent experiments are displayed in Supplementary Fig. S2.
Cells were also incubated in basal media for a shorter incubation time (2 h) and investigated by immunofluorescence microscopy. In contrast to the longer incubation time of 24 h (Fig. 2a), the differences in levels of fibrillar FN detected in the ECM from cells incubated in the absence or presence of AN were less pronounced after 2 h (Supplementary Fig. S5). These observations suggest that AN may influence the turnover of FN fibrils in the ECM from HCASMC.
AN modulates intracellular levels of FN to a moderate extent
To examine whether AN modulates the synthesis and processing of FN, HCASMC lysates were prepared from cells that had been cultured in growth media containing wt or L37AY40A AN for 48 h. Separation of the proteins present in the cell lysates (prepared using deoxycholate) by SDS-PAGE and subsequent immunoblotting using the FN pAb demonstrated that decreased cellular levels of FN were detected in cells exposed to either wt or L37AY40A AN, and there were no statistically-significant differences between the levels in the cells exposed to the two proteins (Supplementary Fig. S6a–c). In addition, quantitative real-time PCR (qPCR) analysis revealed that the levels of the FN-encoding transcript FN1 were not significantly different between the cells exposed to wt or L37AY40A AN for 2 h (Supplementary Fig. S6d). The modest changes at the transcriptional level suggest that the lack of extracellular FN fibrils in cells exposed to AN is not mainly due to suppression of FN expression.
AN induces modest alterations in cellular metabolic activity, viability and proliferation
To determine if some of the above differences in FN processing and matrix incorporation arise from other AN-mediated effects on the cells, cell viability was assayed by quantifying lactate dehydrogenase (LDH) release from the cells, relative to the total amount in the cells and media; these values showed no significant difference (Supplementary Fig. S7a). Furthermore, the mitochondrial reductive capacity (metabolic activity) of HCASMC co-incubated with wt or L37AY40A AN, at 15 or 30 μM, for 48 h was examined using the MTS assay. No significant changes, when compared to controls without AN, were detected with either wt or L37AY40A AN at 15 μM, though a small, but significant, enhancement was observed with wt AN, but not L37AY40A AN, at 30 μM (Supplementary Fig. S7b). However, at both concentrations, a significant increase in metabolic activity was detected for wt compared to L37AY40A AN. The total cell numbers (determined by trypan blue exclusion) did not show any significant decrease with either AN form, with a modest increase (relative to control cells) detected with wt AN at both concentrations (Supplementary Fig. S7c). No significant differences were detected between the two forms of AN. qPCR analysis of the mitosis-related genes PCNA (Proliferating Cell Nuclear Antigen), CCNA1 (Cyclin-A1) and CCNB1 (Cyclin-B1) indicated no significant differences in the expression of these genes between cells exposed to AN (wt or L37AY40A) and control cells incubated in the absence of AN (Supplementary Fig. S8). Together these data indicate that the changes in FN processing and matrix incorporation do not result from AN-mediated toxicity or major alterations in cell metabolic activity or proliferation.
AN stimulates release of cytokines and proteins involved in ECM processing
Culture supernatants from HCASMC were subjected to proteomics analysis by LC–MS/MS to evaluate if AN (wt or L37AY40A) induced changes in the levels of specific proteins secreted from the cells. ECM-associated proteins that are present at higher levels in conditioned media from cells exposed to wt and L37AY40A AN, relative to controls without AN exposure, are listed in Table 1 and include multiple collagens (I, III, IV, V, VI), enzymes involved in ECM processing (MMP2) as well as protease inhibitors (e.g. tissue factor pathway inhibitor 2). Some ECM-related proteins were more abundant in the conditioned media from cells exposed to wt AN, but not L37AY40A AN (e.g. cathepsin D and Z) or vice versa (e.g. peroxidasin and thrombospondin). Several cytokines, including interleukin 6 (IL-6) and CXC chemokines 1, 3, 5 and 6, showed the greatest increase in abundance in the cell conditioned media from cells exposed to wt or L37AY40A AN, relative to controls (Supplementary Fig. S9). In addition, CXC chemokines 2 and 10 were detected in supernatants from cells exposed to wt and L37AY40A AN, but not in supernatants from control cells (Supplementary Data S2). The level of IL-6 in the supernatants was also assessed by immunoblotting, with a dose-dependent increase detected from cells exposed to L37AY40A AN (Supplementary Fig. S10). Table 2 lists ECM-related proteins present at lower levels in the cell conditioned media from cells exposed to AN, with these including annexin, fillagrin, nidogen and versican. Some ECM proteins such as laminin and FN were detected at lower levels in cells exposed to wt AN, but not L37AY40A AN, relative to the controls.
The capacity of AN to modulate FN cell adhesiveness is not directly linked to superfibronectin formation
Incubation of plasma FN with wt AN, and subsequent coating onto plates, before addition of cells, resulted in an increase in adhesion of HCASMC, as detected by the use of cells preloaded with the fluorescent dye calcein-AM (Supplementary Fig. S11a). No significant differences were detected between the effects of wt or L37AY40A AN on cell adhesion. Formation of fibrillar superfibronectin by wt but not L37AY40A AN, was confirmed by turbidity measurements and sedimentation assays (Supplementary Fig. S12) indicating that superfibronectin formation and cell adhesion are distinct and separate events. Control experiments with AN alone (i.e. no FN) showed that neither form of AN exhibited direct cell adhesiveness (Supplementary Fig. S11a). In contrast, when the plasma FN was pre-coated on plates and then treated with AN, before addition of cells, a significant decrease in adhesion was detected with L37AY40A but not wt AN, when compared to controls (Supplementary Fig. S11b). These data suggest that L37AY40A AN may adversely affect the adhesiveness of FN by blocking interaction sites or inducing conformational changes in the material on the plates, with this being dependent on L37 and/or Y40.
Wt and L37AY40A AN induce conformational changes in plasma FN
The structural conformation of plasma FN exposed to wt and L37AY40A AN was probed in ELISA assays with mAb A32 that recognizes a conformation-sensitive epitope in the heparin 2 domain of FN23. Incubation of plasma FN in solution with increasing concentrations of AN gave a dose-dependent enhancement of antibody recognition, with a maximal signal detected at ≥ 1 μM AN (Fig. 3a). A similar trend was observed with L37AY40A AN, although higher values were detected with the latter at 1–10 μM when compared to wt AN (Fig. 3a). Corresponding assays performed with pAb 2413 gave only marginal increases in the ELISA signal with increasing AN concentrations (Fig. 3b), with these small increases possibly arising from recognition of AN by this pAb (Fig. 3b). Similar experiments performed with wt or L37AY40A AN added to FN pre-coated on plates, also yielded a significantly increased signal from mAb A32, although the magnitude of these changes was less than for the solution experiments (Figs. 3a vs 3c). These data suggest that both wt and L37AY40A AN induce conformational changes in plasma FN, with these changes being greater for FN in solution, than pre-coated on plates. The binding of AN to FN (either pre-coated or co-incubated) under these conditions was confirmed by an antibody that recognizes the His-tag of AN. These experiments showed a dose-dependent increase in AN binding to FN with a maximal signal detected at ≥ 1 μM AN (Supplementary Fig. S13). Thus, the increased recognition by mAb A32 correlates with AN binding to FN.

AN induces a conformational switch in plasma FN. Wt or L37AY40A AN were mixed with FN (1 μg mL−1) in a 96-well plate overnight at 21 °C then probed in an ELISA format using: (a) a mAb that recognizes the heparin-2 binding domain of FN (A32, 1:500), or (b) a pAb against FN (2413, 1:500). Alternatively, (c) plasma FN (20 μg mL−1) was pre-coated on plates, then incubated with wt or L37AY40A AN overnight, and probed by the A32 mAb. For controls without FN 20 μM AN was used. Data are presented as means ± SD from three independent experiments, analyzed by 2-way ANOVA with Tukey's multiple comparison test. * indicates significant difference from the control without AN at the p < 0.05 level. # indicates significant difference between the two AN forms at the p < 0.05 level. ‘ns’ indicates an absence of significant differences.
The observed increase in recognition of the heparin-binding 2 domain in FN prompted investigation of a possible increase in affinity of heparin for FN exposed to AN. When wt or L37AY40A AN was co-incubated with FN overnight, an increase in binding of fluorescein-tagged heparin was observed relative to a AN control without FN, but not relative to a FN control without AN (Supplementary Fig. S14a). Similar results were observed when AN was added to pre-coated FN, except that no significant increase was detected for wt AN relative to control without FN (Supplementary Fig. S14b).
Wt and L37AY40A AN display similar overall molecular structures but different heparin binding profiles
The influence of molecular structure on the observed effects of wt and L37AY40A AN was investigated by far-UV circular dichroism (CD) spectroscopy (Fig. 4a). Similar spectra were detected for both forms indicating that the L37A and Y40A mutations do not perturb the beta-sheet structure of the protein. Small-angle X-ray scattering (SAXS) analyses yielded radii of gyration (Rg) that were not significantly different (wt AN 18.2 ± 0.2 Å, L37AY40A AN 18.6 ± 0.1 Å), consistent with a similar size and molecular conformation (Fig. 4b). It is therefore concluded that the differences between wt and L37AY40A AN outlined above are not due to major perturbations in overall structure.

Molecular features of wt and L37AY40A AN. (a) Far-UV CD spectroscopy (190–260 nm) of wt and L37AY40A AN (0.1 mg mL−1) at 25 °C in 1 mm cells; for further details see Experimental procedues. (b) SAXS data for wt and L37AY40A AN (100 μM, 0.93 mg mL−1) measured in 10 mM phosphate buffer, pH 7.4, on an optimized NanoSTAR SAXS instrument. Data are displayed as a function of the scattering vector, q, with a scaling factor used to compare the data for wt and L37AY40A AN. (c) Separation of wt and L37AY40A AN on a Hitrap Heparin HP column. Samples were applied in 20 mM sodium phosphate buffer, pH 7.4, and eluted with a gradient of 0–500 mM NaCl in 20 mM phosphate buffer at 1 mL min−1. (d) Binding of fluorescein-tagged heparin (4 mg mL−1) to AN (wt or L37AY40A) coated on plates at 21 °C overnight, detected by fluorescence measurements (λex 495 nm and λem 525 nm). Data are presented as means ± SD from three independent experiments, analyzed by paired-sample T-tests. *Indicates statistically significant differences between wt and L37AY40A AN at the p < 0.05 level.
To examine other potential differences which might rationalize the observed alterations in functionality of L37AY40A AN, the heparin-binding profiles of wt and mutant AN were examined by chromatographic separation on a heparin-binding column (Fig. 4c). L37AY40A AN eluted as a single peak at high NaCl concentrations, whereas wt AN eluted as two peaks with one of these having a similar elution time to L37AY40A AN (Fig. 4c). These data suggest that wt AN binds to the heparin column in two different conformations, with one of these not adopted by L37AY40A. Isolation of the species that give rise to the two peaks with wt AN, and subsequent re-chromatography of the separate fractions, resulted again in the detection of two forms (data not shown) suggesting that the two wt AN conformations exist in a dynamic equilibrium. The heparin affinity of AN was also examined using a fluorescein-labelled heparin analogue in a binding assay format. Under these conditions a significant increase in heparin binding affinity was observed for L37AY40A AN (Fig. 4d), with the amount of bound label increasing with the AN concentration.
Discussion
The data obtained in this study demonstrate that AN has a major impact on the structure of FN in the ECM derived from HCASMC cultures, with a marked decrease in the extent of extracellular FN fibrils observed for cells exposed to this protein. FN fibrils in the presence of the inactive L37AY40A AN mutant were restored by supplementation of the basal cell media with plasma FN, which suggests that these events are independent of superfibronectin formation. These results are interesting in relation to previous data reporting that the antiangiogenic activity of AN in vivo is dependent on the presence of plasma FN24. Earlier work has reported different effects of AN on FN assembly and turnover with this being dependent on the experimental conditions and cell type. Thus, Bordoulous et al. observed that AN promoted disassembly of FN fibrils generated by fibroblast and endothelial cells15, whereas Klein et al. showed that AN induces a conformational switch in FN that decreases recognition of an epitope in the EDA module16. We also observed a diminished signal from the EDA module in assembled ECM, but no conclusive evidence was obtained for a conformational change of FN in the ECM, since a loss of signal from fibrillar FN was also observed with the pAb detecting total FN. Previous studies demonstrated that conformational changes in FN induced by AN alters cell signaling via the VEGF and Ras/ERK pathways17,18. Such alternations have been associated with the anti-proliferative effects of AN on endothelial and vascular smooth muscle cells10. In contrast, our data are consistent with an absence of significant changes in cell viability and proliferation, and a slight increase in metabolic activity observed on exposure of HCASMC to wt AN.
Since FN plays a central role as a scaffold and “master regulator” in the ECM, we reasoned that the observed gross changes in the morphology of FN fibrils would also be associated with changes in overall ECM composition. Proteomics analysis of conditioned media from cells exposed to wt and L37AY40A AN revealed strikingly different protein profiles relative to control cells incubated without AN. Some ECM proteins such as collagens were detected at higher levels in supernatants from cells exposed to either wt or L37AY40A AN. This may be due to inhibition of collagen fibril assembly or an increase in collagen turnover. In contrast, lower levels of both FN and laminin were detected from cells exposed to wt, but not L37AY40A AN. These results do not agree with the lower intracellular levels of FN detected in cells exposed to both wt and L37AY40A AN. This may be due to differences in the secretory or extracellular processing pathways for wt and L37AY40A AN. Peroxidasin was detected at higher levels in the conditioned media from cells exposed to L37AY40A AN, but not wt AN. Peroxidasin plays a key role in the intermolecular cross-linking of the NC1 domains of collagen, and increased levels of this enzyme may therefore influence ECM structure and particularly the extent of matrix crosslinking25. Several of the identified proteins are known to interact directly with FN, including alpha-2-macroglobulin, CCN2 and fibrillin26,27,28. Wt and L37AY40A AN also stimulated the release of proteases (e.g. collagenase and pappalysin) and protease inhibitors (e.g. TIMP1-2 and antithrombin) that influence ECM turnover. Some of these proteases (e.g. MMP2) are known to cleave FN, and may thus catalyze turnover of FN fibrils in the ECM29. Several of the upregulated proteins, including tissue factor pathway inhibitor 2 and thrombospondin (elevated only in L37AY40A AN) display antiangiogenic properties30,31. These observations may be of relevance to the antiangiogenic and antimetastatic properties of AN observed in mice with human tumor cell grafts11. Release of inflammatory cytokines such as IL-6 and CXC chemokines 3, 5 and 6 was also stimulated in cells exposed to wt and L37AY40A AN. IL-6 is known to be released from vascular smooth muscle cells after exposure to bacterial antigens, different pro-inflammatory molecules or mechanical stress, and has been associated with TLR4-dependent signaling32,33. A similar inflammatory response has previously been reported for fibroblasts and mononuclear cells exposed to AN19,20.
In addition to its ability to modulate ECM assembly, AN also promotes FN polymerization in vitro in the absence of cells. The resulting polymer is often referred to as “superfibronectin” on the assumption that “superadhesiveness” correlates with FN fibrillation34. We demonstrate here that AN-induced enhancement of FN adhesiveness is, at least partially, independent of FN polymerization, as no significant difference was observed between wt and L37AY40A AN that lacks the ability to form superfibronectin. An increase in AN-mediated cell adhesiveness was only seen when AN (wt or L37AY40A) was co-incubated with FN in solution; when AN was added to FN coated on plates decreased adhesiveness was seen. These data suggest that the increase in cell adhesiveness is mediated by direct interactions between AN and FN in solution, and that AN added to FN coated on plates may either block cell access to adhesion sites, or that the FN incorporated into ECM cannot undergo marked structural changes.
Our data also demonstrate that interactions of AN with plasma FN exposes an epitope in the heparin-2 binding domain. These alterations are detected at concentrations of AN where no superfibronectin formation is observed (i.e. < 1 μM). Furthermore, the exposure of the heparin-2 binding domain occurs primarily when AN and FN are co-incubated in solution, and not when AN is added to pre-coated FN. These observations suggest that the exposure of the epitope is due to conformational changes rather than polymerization of FN and that these changes occur primarily with the soluble, non-polymerized protein.
The structure of both soluble and fibrillar FN is dynamic and is influenced by interactions with various ligands35,36,37,38,39. Whether these functional alternations are dependent on large-scale conformational changes or more subtle structural rearrangements is unclear34,40,41,42. Previous data indicates that AN interacts with the FNIII1,2,3,11 modules46,47,48, but there are no reports of direct interactions between AN and the heparin-2 binding domain in FN that is localized in FNIII12-14. It is however possible that AN—FNIII1,2,3,11 interactions induce long-range conformational changes that enhance exposure of the heparin-2 binding domain. This process may drive superfibronectin formation, as previous data has demonstrated that the heparin-2 binding domain interacts with FNIII modules and plays a key role in FN assembly49. On the other hand, it is also possible that exposure of the heparin-2 binding domain contributes to the observed inhibition of FN matrix assembly in cells exposed to AN; similar effects have been demonstrated with cells exposed to an isolated heparin-2 binding domain fragment49.
The folded structure of the FNIII module that AN is derived from, is found in many proteins50. Even though FNIII modules display high sequence diversity, the FNIII fold is highly conserved and is composed of two anti-parallel beta sheets, comprised of 3 (A, B, E) and 4 (C, D, F, G) strands respectively. AN lacks the two first beta strands (A, B) of the FNIII fold, and is instead composed of a single beta sheet (C, D, F, G), with the remaining (E) strand present as a flexible loop containing L37 and Y4021. Stine et al. have reported that AN can engage in beta-strand exchange with FN, whereby the two first beta-strands from FNIII3 (A, B) form a beta-sheet with the E strand from AN46. It is therefore speculated that L37 and Y40 may stabilize contacts between the beta-strands of AN and FN and that these contacts are disrupted in the L37AY40A mutant and impact on fibril formation and matrix assembly of FN. Our data indicate that wt and L37AY40A AN have a similar molecular structure. It is therefore likely that the absence of fibrillation activity of L37AY40A results from local perturbations around the mutated residues, altered hydrophobicity, or disruption of hydrogen bonds involving the hydroxyl group of Y40. In this context it is interesting to reflect on the differences between wt and L37AY40A AN in terms of heparin binding. AN contains three clustered positively-charged residues in a 13RWRPK17 motif that binds negatively-charged heparin polysaccharides14. These residues constitute a “cryptic” heparin-binding motif that is exposed when FN is subjected to chemomechanical forces that partially unfold the FNIII1 module, or as a result of proteolytic processing. In AN, the 13RWRP16 portion of this motif is located on beta-strand C, while K17 is positioned in the loop between strands C and D. The distance between Y40 and R13, the closest neighbors between L37/Y40 and the 13RWRPK17 motif is ~ 14 Å, which rules out direct contacts. It therefore seems likely that the differences in heparin binding are due to changes in the local vicinity of the 13RWRPK17 motif.
In conclusion, the current data demonstrates that AN has a strong influence on the processing of FN in HCASMC and elicits release of inflammatory cytokines and proteins involved in ECM turnover. Further investigations are required to determine the detailed mechanisms responsible for these observations, but the data reported here indicate that L37 and Y40, play a key role. These findings may be of therapeutic importance, particularly in regard to wound healing, angiogenesis and cell dispersal in tumor metastasis, where FN-dependent matrices are of key importance.
Experimental procedures
Materials
All chemicals, including lyophilized human plasma FN (F1056) and anti-FN monoclonal antibody (mAb) 3E2, were purchased from Sigma-Aldrich (St Louis, Missouri, USA), unless stated otherwise. All solutions were prepared with Milli-Q grade water (Millipore Advantage A10; Merck-Millipore, Billerica, MA, USA). Anti-FN pAb (2413), anti-His tag mAb (18,184) and anti-IL 6 pAB (6672) were from Abcam. Anti-FN heparin-2 binding fragment mAb (A32) was from Life Technologies. Anti-β actin mAb (8929) was from R&D System. Horseradish peroxidase (HRP)-conjugated sheep anti-mouse/anti-rabbit whole immunoglobulin (IgG) secondary antibodies were purchased from Millipore. DAPI (4′,6-diamidino-2-phenylindole) and Alexa Fluor 488-conjugated anti-rabbit and 594-conjugated anti-mouse secondary antibodies were from Molecular Probes (Thermo Fisher Scientific). Fluorescein-conjugated heparin (~ 18 kDa; H7482) and RIPA Lysis Buffer (89,900) was purchased from Thermo Fisher Scientific. Recombinant AN was produced in Escherichia coli essentially as described previously51. The L37AY40A mutant of AN (translated sequence in Supplementary Fig. S15) was prepared using the QuickChange Lightning Site-Directed Mutagenesis kit (Agilent, USA) according to the manufacturer’s instructions. Primers used for engineering the mutant was purchased from TAG Copenhagen A/S, Denmark. The sequences of the primers are 5’-ACGCGAACAGCGCGACCATTAAGGGTCTGAAACCGGG-3’ (forward) and 5’-TCGCGCTGTTCGCGTGACCCGGGATGGTCGC-3’ (reverse). Correct mutation was verified by DNA sequencing.
Culture of primary human coronary artery smooth muscle cells
Primary human coronary artery smooth muscle cells (HCASMC, donor 1522) purchased from Cell Applications (San Diego, CA, USA) were used between passages 2 and 5. The cells were cultured in commercial HCASMC growth media or basal media (Cell Applications) in a humidified incubator under an atmosphere of 5% CO2 at 37 °C. Basal medium contains essential and non-essential amino acids, vitamins, inorganic salts, organic compounds, and trace elements, but does not contain the growth supplements necessary for cell proliferation such as specific growth factors, antibiotics and serum. For experiments, the cells were harvested with trypsin/EDTA solution (0.025% trypsin, 0.01% EDTA, in PBS), centrifuged at 220 g for 5 min and plated overnight at a density of 1 × 105 cells mL−1 in 6-, 12- or 96-well plates using volumes of 2 mL, 1 mL or 50 μL, respectively. For microscopy, cells were plated at a density of 7.5 × 104 cells mL−1 in eight-well chamber slides (734 2050, VWR) in a volume of 300 μL, or 96-well plates in 200 μL media. Before treatment, the cell media was removed, and the cells washed with warm (37 °C) Hanks’ buffered salt solution (HBSS).
Cell adhesion assays
Wt and L37AY40A AN (0.5 μM; 25 μL per well) was added to a 96-well black microplate (sterile, F-bottom, Costar, #3603, Corning, NY, USA) in the presence or absence of FN (2 μg mL−1; 25 μL per well) prior to washing with PBS followed by blocking with 1% (w/v) denatured BSA in PBS for 1 h and incubation overnight at 21 °C. Alternatively, 50 μL wt or L37AY40A AN (0.25 μM) was added to wells pre-coated with FN (1 μg mL−1; 50 μL per well) and blocked with 1% (w/v) denatured BSA in PBS, followed by incubation overnight at 21 °C. HCAMSC were added (0.5 × 104 cells per well) and the plates incubated for 1 h at 37 °C. After washing twice with HBSS, the plates were incubated with 50 μM calcein-AM (50 μL per well) for 30 min at 37 °C. Adhesion of dye-loaded cells to AN was determined by measuring fluorescence intensity with λex 490 nm and λem 520 nm, using a SpectraMax i3x microplate reader (Molecular Devices, San Jose, CA).
ELISA
Detection of AN in isolated ECM
HCASMC (1.5 × 104 cells, 96 well plate) were cultured in growth media for 1 week to establish native ECM. The plates were then rinsed twice with PBS followed by incubation with 1% sodium deoxycholate at 21 °C for 2 × 20 min to remove cells. After washing twice with PBS, the native ECM remaining on the plates was then blocked with 1% (w/v) BSA in PBS and treated with 0–20 μM wt and L37AY40A AN at 4 °C overnight. Residual materials were then removed from the plates by washing with PBS followed by blocking with 1% (w/v) BSA in PBS. Plates were then incubated with anti-His tag mAb 18,184 (1:500 dilution) antibody at 4 °C overnight. After that, plates were rinsed twice with PBS and incubated with anti-mouse IgG secondary antibody at 21 °C for 1 h. After washing twice with PBS, the plates were then incubated with ABTS (2,2'-azinobis-3-ethylbenzothiazoline-6-sulfonic acid; 2 mM) and H2O2 (30% w/v), mixed in 1000:1 ratio. Optical absorbance of the samples was measured at 405 nm using a microplate reader.
Detection of isolated plasma FN mixed with AN
Wt and L37AY40A AN (0–20 μM) were mixed with plasma FN (1 μg mL−1) in a 96-well cell culture plate followed by incubation at 21 °C overnight. Alternatively, wt and L37AY40A AN (0–20 μM) was added to wells pre-coated with FN (50 μL, 20 μg mL−1) followed by incubation at 21 °C overnight. In both cases wells were subsequently blocked with 1% (w/v) BSA in PBS for 1 h followed by incubation with primary antibodies (pAb 2413, mAb A32, or mAb 18,184; 1:500 dilutions) at 4 °C overnight. Plates were rinsed twice with PBS and incubated with IgG secondary antibodies at 21 °C for 1 h. After washing twice with PBS, the plates were then incubated with ABTS/H2O2 (as above), and the absorbance of the samples measured at 405 nm using a microplate reader.
Immunofluorescence microscopy
Detection of FN in HCASMC incubated with wt or L37AY40A AN
HCASMC (2.25 × 104 cells) were cultured in eight-well chamber slides overnight and then incubated with growth media containing 200 μL wt or L37AY40A AN (30 μM) for 48 h. Alternatively, cells were incubated with basal media containing 200 μL wt or L37AY40A AN (30 μM) for 24 h. In both cases, cells on slides were washed, fixed with 4% (v/v) formaldehyde at 37 °C for 15 min, permeabilized with 0.5% (v/v) Triton X-100 in PBS on ice for 5 min and blocked with 1% (v/v) BSA in PBS for 1 h. Primary antibodies (pAb 2413 or mAb 3E2) were added in 1:500 dilutions followed by incubation at 4 °C overnight. The slides were then rinsed three times with PBS and incubated with either anti-mouse or anti-rabbit IgG conjugated with Alexa Fluor 488 antibody or Alexa Fluor 594 antibody (1:500 dilution) in 1% (v/v) BSA in PBS at 21 °C for 1 h followed by three rinses with PBS and counterstaining with 1 μg mL−1 DAPI in PBS in the dark at 21 °C for 10 min. Cover slips were added after three washes with PBS, and samples imaged using a fluorescence microscope (Olympus, Japan) equipped with cellSense Entry v1.5 software. Exposure times were 100 ms for the FN 2413, 3E2, and His-tag antibodies and 10 ms for DAPI.
Detection of AN and FN in isolated ECM
HCASMC (2.25 × 104 cells) were cultured in eight-well chamber slide for 1 week to establish native ECM. The plates were then rinsed twice with PBS followed by incubation with two aliquots of 1% sodium deoxycholate at 21 °C for 20 min to induce cell lysis, each followed by two washes with PBS. The slides were then blocked with 1% (v/v) BSA in PBS at 21 °C for 1 h, washed twice with PBS, and then incubated with 0.1 μM wt or L37AY40A AN, followed by incubation with primary antibodies (pAb 2413 or mAb 18,184) at 4 °C overnight in 1:500 dilutions. The slides were rinsed twice with PBS then incubated with either anti-mouse or anti-rabbit IgG conjugated with Alexa Fluor 488 antibody or Alexa Fluor 594 antibody (1:500 dilution) at 21 °C for 1 h followed by three rinses with PBS. Cover slips were then added, and the samples imaged using a fluorescence microscope as above.
Immunoblot analysis
HCASMC (2 × 105 cells) were cultured in 6-well plates overnight (2 mL growth media per well) and then incubated in growth media containing 15–30 μM wt or L37AY40A AN for 48 h (1.5 mL per well). The plates were then rinsed with PBS followed by incubation with 150 μL RIPA Lysis Buffer (25 mM Tris HCl, pH 7.6; 150 mM NaCl; 1% NP-40; 1% sodium deoxycholate; 0.1% SDS) including protease inhibitor cocktail (P8340) in 1:100 dilution for 5 min. Samples were then transfered to 1.5 mL tubes, centrifuged at 14,000g for 10 min at 4 °C, and the protein supernatant collected. For detection of IL-6, supernatants from cells cultures in basal media containing 200 μL wt or L37AY40A AN (15 or 30 μM) for 24 h were harvested by centrifugation. Samples were analyzed (10 μg protein per lane) by SDS–PAGE using 3–8% NuPAGE Tris acetate gels according to the manufacturer's instructions. After separation, the proteins were transferred to PVDF membranes using an iBlot system. Membranes were then blocked with 1% (w/v) BSA in TBST and probed with primary antibodies: pAb 2413 (FN, 1:1000), mAb 8929 (β-actin, 1:1000), pAb 6672 (IL-6, 1:5000). The membranes were then washed twice with TBST, and incubated for 1 h with anti-mouse or anti-rabbit IgG secondary antibodies at 21 °C. Unbound secondary antibodies were removed by washing, before detection of immune complexes using Western Lightning Plus ECL reagent, and a Sapphire Biomolecular Imager (Azure Biosystems) to acquire images.
Proteomics analysis of conditioned media
Supernatants (300 μL) containing conditioned media from cells exposed ± 30 μM AN (wt or L37AY40A AN) for 24 h in basal media as described above were loaded on 1 mL HisTrap columns (Cytiva life science) equilibrated in 25 mM HEPES–NaOH pH 7, 500 mM NaCl, 0.25 mM EDTA, 0.25 mM EGTA, 20% v/v glycerol, 5 mM β mercaptoethanol to remove AN. The flow-through was concentrated to 300 μL and mixed with 200 μL 8 M urea (in 0.1 M Tris, pH 8.5) in a spin filter (10,000 kD MWCO, Vivacon 500, Sartorius), then centrifuged at 14,000g for 25 min at 20 °C. 400 μL 8 M urea in Tris buffer containing 50 mM DTT was added and incubated at 20 °C for 30 min. The filter was again centrifuged (14,000g for 25 min at 20 °C), before addition of 400 μL 8 M urea in Tris buffer containing 50 mM iodoacetamide, and incubated for another 30 min at 20 °C in the dark. Samples were centrifuged as above and 400 μL 1.6 M urea in 0.1 M Tris (pH 8.0) added. After repeating this centrifugation step, 100 μL 1.6 M urea in 0.1 M Tris (pH 8.0) and 1 μL trypsin (0.1 μg μL−1) were added followed by incubation overnight at 20 °C. Released peptides were pooled from the flow-through after centrifugation at 14,000g for 10 min, and the flow-through from a subsequent wash of the spin filter with 50 μL 0.5 M NaCl. Samples were subjected to stage-tip solid-phase extraction on C18 discs as described previously and analyzed on a Bruker TIMS-TOF PRO mass spectrometer (Bruker Daltonics) in the positive ion mode with a Captivespray ion source on-line connected to a Dionex Ultimate 3000RSnano chromatography systems (Thermo Fisher Scientific). Peptides were separated on a 25 cm × 75 µm Aurora column (Ion optics) at 60 °C with a solvent gradient over 42 min, using acetonitrile with 0.1% formic acid as eluent at a flow rate of 600 nL min−1. The mass spectrometer was operated in DIA PASEF mode with 0.53 s cycle time and TIMS ramp time of 100 ms. MS scan range was set to 100–1700 m/z. Database searches were performed using DIA-NN52 version 1.8 with a spectral library generated in silico from the human UniProt reference proteome (UP000000558) using the following parameters: trypsin with 1 missed cleavage, methionine oxidation and N-terminal acetylation (variable modifications), cysteine carbamidomethylation (fixed modification), MS1 accuracy ± 10 ppm, precursor false discovery rate (FDR) 1%. Post-processing and statistical analysis on triplicate samples of culture supernatants was performed using Perseus (version 1.6.15.0), with a permutation based t-test (FDR 0.05, S0 0.75) performed to determine up- and- downregulated proteins in supernatants from cell cultures treated with AN (wt or L37AY40A) relative to control cells (Supplementary data S1). Up/down regulated proteins included in the matrisome database53 are listed in Tables 1 and 2.
Metabolic activity
HCASMC (0.5 × 104 cells) were cultured in 96-well plates overnight and then incubated in 100 μL growth media containing 15–30 μM wt or L37AY40A AN for 48 h. Cells were then washed with HBSS and re-incubated with 100 μL growth media containing 10 μL MTS regent ((3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium)) for 4 h at 37 °C. The absorbance at 490 nm was then measured with a microplate reader.
Lactate dehydrogenase (LDH) release
HCASMC (1 × 105 cells) were cultured in 12-well plates overnight (1 mL growth media per well) and then incubated in 1 mL growth media containing 15–30 μM wt or L37AY40A AN for 48 h. After incubation, the media was collected and the cells were washed with HBSS and lysed with 1 mL npH2O. Both media and cell lysate solutions were centrifuged at 448g for 5 min at 4 °C to remove cell debris. Ten μL of supernatant were then mixed with 200 μL of reaction reagent containing 0.15 mg mL−1 NADH and 2.5 mM sodium pyruvate in PBS. LDH activity was measured by detecting the decrease in absorbance at 340 nm for 30 min at 5 min intervals with a microplate reader. Cell viability was calculated as intracellular LDH activity compared to the total intra- and extracellular LDH activity, expressed as a percentage.
Cell proliferation assay
HCASMC (1 × 105 cells) were plated into 12-well plates overnight and then incubated in 1 mL growth media containing 15–30 μM wt or L37AY40A AN for 48 h at 37 °C. Cells were then released from the plate using trypsin–EDTA, prior to cell counting using a hemocytometer with trypan blue staining to delineate non-viable cells. Total cell number was then calculated for each sample in triplicate.
qPCR analysis
HCASMC (1 × 105 cells in 1 mL/well) cultured overnight in a 12-well plate, and then incubated in growth media containing 30 μM wt or L37AY40A AN for 2 h. Total RNA was extracted with the RNeasy kit (Qiagen), and RNase free DNase (Qiagen) was used for DNase digestion. 600 ng RNA was used for cDNA synthesis using Quantinova Reverse Transcription Kit (Qiagen). For qPCR analysis, 20 μL reaction mixtures (containing 10 μL SYBR® GreenER™, 2 μL primer (10 pmol μL−1), 0.4 μL ROX reference dye and 4 μL cDNA) were incubated at 50 °C for 2 min followed by 95 °C for 10 min (hold stage); 95 °C for 15 s followed by 60 °C for 1 min (PCR stage, repeated 40 times); 95 °C for 15 s followed by gradual increase from 60 to 95 °C by 0.075 °C s−1 (melt curve stage). The reaction was performed on the QuantStudio™ 5 real-time PCR system (Applied Bio-systems). Primers used were (5ʹ–3ʹ): FN1 (F: GGTGACACTTATGAGCGTCCTAAA; R: CCCATCAGCAGGAACACCTT), PCNA (F: AGGCACTCAAGGACCTCATCA; R: GAGTCCATGCTCTGCAGGTTT), CCNB1 (F: AGCTGCTGCCTGGTGAAGAG; R: GCCATGTTGATCTTCGCCTTA), CCNA1 (F: GCACCCTGCTCGTCACTTG; R: CAGCCCCCAATAAAAGATCCA). Relative mRNA concentrations of the genes of interest were normalized to the expression of genes encoding 18S ribosomal RNA (18S rRNA) (F: GAGGATGAGGTGGAACGTGT; R: TCTTCAGTCGCTCCAGGTCT) and beta-2-microglobulin (B2M) (F: AGATGAGTATGCCTGCCGTG; R: GCGGCATCTTCAAACCTCCA) housekeeping genes. Experiments were performed in triplicates and data analysis was carried out using the 2−ΔΔCT method.
Heparin binding assay
Experiments were performed in three different ways: i) Plasma FN (20 μg mL−1) or wt or L37AY40A AN (0.25 μM and 1 μM) in 50 μL was added to a 96-well black microplate (sterile, F-bottom, Costar, 3603, Corning, NY, USA), which was incubated overnight, followed by blocking with 1% (w/v) BSA in PBS. ii) AN (wt or L37AY40A, 50 μL, 1 μM) was added to wells pre-coated with FN (20 μg mL−1) as outlined above, and then incubated at 21 °C overnight. iii) 50 μL plasma FN (1 μg mL−1) was co-incubated with 0.25 μM AN (wt or L37AY40A) in a 96-well black microplate overnight followed by blocking with 1% (w/v) BSA in PBS, pH 7.4. In all experiments plates were subsequently incubated with 50 μL fluorescein-labeled heparin (0.004 mg mL−1) overnight at 21 °C in the dark and analyzed using a microplate reader with λex 495 nm and λem 525 nm (9 nm slit width), with three spectra averaged.
Turbidity and sedimentation assays
Wt AN (2.5–40 μM) or L37AY40A AN (40 μM) was mixed with FN (0.4 mg mL−1; 1.4 μM) before measuring the turbidity of the solution at 550 nm at 1 min intervals for a period of 40 min at 21 °C, using a Spectra Max i3x microplate reader (Molecular Devices, San Jose, CA, USA). In the sedimentation assay, wt or L37AY40A AN (40 μM) and FN (1 μM) were mixed in a final volume of 20 μL in sodium phosphate buffer (10 mM, pH 7.4) containing 5 mM EDTA and incubated overnight at 21 °C. After incubation, the samples were centrifuged at 20,000 g for 10 min at 4 °C. The insoluble pellets were then washed with 20 μL sodium phosphate buffer (10 mM, pH 7.4). All samples were analyzed by SDS-PAGE under reducing conditions.
CD spectroscopy
CD spectra were acquired on a Jasco-815 CD Spectrometer (Jasco Corporation, Japan), as previously51. Far-UV CD spectra of 0.1 mg mL−1 wt and L37AY40A AN was recorded between 190–260 nm, in a 1 mm path length cell at 25 °C, at 1 nm intervals with a 1 nm bandwidth, and a scan speed of 50 nm min−1, with six scans averaged for each spectrum.
SAXS analysis
SAXS measurements were performed as previously51 on an optimized NanoSTAR SAXS instrument54, with wt and L37AY40A AN (100 μM; 0.93 mg mL−1) in 10 mM phosphate buffer (pH 7.4). The SAXS data is displayed as a function of the scattering vector, q. A scaling factor was used to compare data between wt and L37AY40A AN. The radius of gyration, Rg, was obtained by performing Guinier fits.
Heparin-affinity chromatography
Wt and L37AY40A AN (80 μM) in 100 μL 20 mM sodium phosphate buffer, pH 7.4) was loaded onto a Hitrap heparin high-performance column (GE healthcare, Boston, MA) equilibrated in 20 mM sodium phosphate buffer (pH 7.4) on an Äkta Prime chromatography system (GE Healthcare). The proteins were eluted with a gradient of 0–500 mM NaCl in 20 mM sodium phosphate buffer (pH 7.4) at a flow rate of 1 mL min−1. Protein elution was monitored by UV detection at 280 nm.
Errors and statistical analyses
Statistical analyses were performed using Graphpad Prism (version 9; GraphPad Software, San Diego, USA) by 2-way ANOVA with Tukey's multiple comparison test. Quantitative data are presented as mean ± SD from at least three independent experiments, with p < 0.05 (indicated by * or #) taken as being statistically significant, and ‘ns’ indicating an absence of statistical significance. Analysis of pixel intensities in microscopy images was carried out using R (version 4.1.3). For each condition, pixel intensities from three images (each derived from an independent experiment) were pooled. Each pixel was loaded into a numeric r vector using the readTIFF function from the tiff package. The median of all pixels was then calculated via the summary function, and statistical analysis was performed using one-way ANOVA with post-hoc Tukey's multiple comparison tests. The boxplots were calculated from a random subset of the pixel data (300,000 per sample) and the ggplot2 library.
Data availability
This article contains Supplementary information. Raw mass spectrometry data and associated database search engine output files have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD033732.
Abbreviations
- ABTS:
-
2,2'-Azino-bis(3-ethylbenzothiazoline-6-sulfonic acid)
- AN:
-
Anastellin
- CD:
-
Circular dichroism
- DAPI:
-
4′,6-Diamidino-2-phenylindole
- ECM:
-
Extracellular matrix
- FN:
-
Fibronectin
- HCASMC:
-
Human coronary artery smooth muscle cells
- IL-6:
-
Interleukin 6
- L37AY40A AN:
-
An isoform of anastellin with both Leu37 and Tyr40 mutated to Ala residues
- LDH:
-
Lactate dehydrogenase
- mAb:
-
Monoclonal antibody
- MTS:
-
(3-(4,5-Dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium)
- pAb:
-
Polyclonal antibody
- qPCR:
-
Quantitative real-time PCR
- SAXS:
-
Small-angle X-ray scattering
- VEGF:
-
Vascular endothelial growth factor
- wt:
-
Wild type
References
Mosher, D. F. Assembly of fibronectin into extracellular matrix. Curr. Opin. Struct. Biol. 3, 214–222 (1993).
Wu, C., Keivenst, V. M., O’Toole, T. E., McDonald, J. A. & Ginsberg, M. H. Integrin activation and cytoskeletal interaction are essential for the assembly of a fibronectin matrix. Cell 83, 715–724 (1995).
Allio, A. E. & McKeown-Longo, P. J. Extracellular matrix assembly of cell-derived and plasma-derived fibronectins by substrate-attached fibroblasts. J. Cell. Physiol. 135, 459–466 (1988).
Oh, E., Pierschbacher, M. & Ruoslahti, E. Deposition of plasma fibronectin in tissues. Proc. Natl. Acad. Sci. U.S.A. 78, 3218–3221 (1981).
Hayman, E. G. & Ruoslahti, E. Distribution of fetal bovine serum fibronectin and endogenous rat cell fibronectin in extracellular matrix. J. Cell Biol. 83, 255–259 (1979).
Allio, A. E. & McKeown-Longo, P. J. Extracellular matrix assembly of cell-derived and plasma-derived fibronectins by substrate-attached fibroblasts. J. Cell Physiol. 135, 459–466 (1988).
Zhong, C. et al. Rho-mediated contractility exposes a cryptic site in fibronectin and induces fibronectin matrix assembly. J. Cell Biol. 141, 539–551 (1998).
Smith, M. L. et al. Force-induced unfolding of fibronectin in the extracellular matrix of living cells. PLoS Biol. 5, e268 (2007).
Maurer, L. M., Ma, W. & Mosher, D. F. Dynamic structure of plasma fibronectin. Crit. Rev. Biochem. Mol. Biol. 51, 213–227 (2016).
Ambesi, A., Klein, R. M., Pumiglia, K. M. & McKeown-Longo, P. J. Anastellin, a fragment of the first type III repeat of fibronectin, inhibits extracellular signal-regulated kinase and causes G1 arrest in human microvessel endothelial cells. Cancer Res. 65, 148–156 (2005).
Yi, M. & Ruoslahti, E. A fibronectin fragment inhibits tumor growth, angiogenesis, and metastasis. Proc. Natl. Acad. Sci. U.S.A. 98, 620–624 (2001).
Pasqualini, R., Bourdoulous, S., Koivunen, E., Woods, V. L. & Ruoslahti, E. A polymeric form of fibronectin has antimetastatic effects against multiple tumor types. Nat. Med. 2, 1197–1203 (1996).
Litvinovich, S. V., Novokhatny, V. V., Brew, S. A. & Ingham, K. C. Reversible unfolding of an isolated heparin and DNA binding fragment, the first type III module from fibronectin. Biochim. Biophys. Acta 1119, 57–62. https://doi.org/10.1016/0167-4838(92)90234-5 (1992).
Gui, L., Wojciechowski, K., Gildner, C. D., Nedelkovska, H. & Hocking, D. C. Identification of the heparin-binding determinants within fibronectin repeat III1. J. Biol. Chem. 281, 34816–34825 (2006).
Bourdoulous, S., Orend, G., MacKenna, D. A., Pasqualini, R. & Ruoslahti, E. Fibronectin matrix regulates activation of RHO and CDC42 GTPases and cell cycle progression. J. Cell Biol. 143, 267–276 (1998).
Klein, R. M., Zheng, M., Ambesi, A., Van De Water, L. & McKeown-Longo, P. J. Stimulation of extracellular matrix remodeling by the first type III repeat in fibronectin. J. Cell Sci. 116, 4663–4674 (2003).
Ambesi, A. & McKeown-Longo, P. J. Anastellin, the angiostatic fibronectin peptide, is a selective inhibitor of lysophospholipid signaling. Mol. Cancer Res. 7, 255–265 (2009).
Ambesi, A. & McKeown-Longo, P. J. Conformational remodeling of the fibronectin matrix selectively regulates VEGF signaling. J. Cell Sci. 127, 3805–3816 (2014).
Valenty, L. M., Longo, C. M., Horzempa, C., Ambesi, A. & McKeown-Longo, P. J. TLR4 ligands selectively synergize to induce expression of IL-8. Adv. Wound Care (New Rochelle) 6, 309–319. https://doi.org/10.1089/wound.2017.0735 (2017).
You, R., Zheng, M. & McKeown-Longo, P. J. The first type III repeat in fibronectin activates an inflammatory pathway in dermal fibroblasts. J. Biol. Chem. 285, 36255–36259. https://doi.org/10.1074/jbc.C110.176990 (2010).
Briknarová, K., Åkerman, M. E., Hoyt, D. W., Ruoslahti, E. & Ely, K. R. Anastellin, an FN3 fragment with fibronectin polymerization activity, resembles amyloid fibril precursors. J. Mol. Biol. 332, 205–215 (2003).
You, R., Klein, R. M., Zheng, M. & McKeown-Longo, P. J. Regulation of p38 MAP kinase by anastellin is independent of anastellin’s effect on matrix fibronectin. Matrix Biol. 28, 101–109 (2009).
Hubbard, B., Buczek-Thomas, J. A., Nugent, M. A. & Smith, M. L. Heparin-dependent regulation of fibronectin matrix conformation. Matrix Biol. 34, 124–131 (2014).
Yi, M., Sakai, T., Fassler, R. & Ruoslahti, E. Antiangiogenic proteins require plasma fibronectin or vitronectin for in vivo activity. Proc. Natl. Acad. Sci. U. S. A. 100, 11435–11438. https://doi.org/10.1073/pnas.1635112100 (2003).
Bhave, G. et al. Peroxidasin forms sulfilimine chemical bonds using hypohalous acids in tissue genesis. Nat. Chem. Biol. 8, 784–790. https://doi.org/10.1038/nchembio.1038 (2012).
Chen, Y. et al. CCN2 (connective tissue growth factor) promotes fibroblast adhesion to fibronectin. Mol. Biol. Cell. 15, 5635–5646. https://doi.org/10.1091/mbc.e04-06-0490 (2004).
Falkenberg, C., Enghild, J. J., Thogersen, I. B., Salvesen, G. & Åkerström, B. Isolation and characterization of fibronectin-alpha 1-microglobulin complex in rat plasma. Biochem. J. 301(Pt 3), 745–751. https://doi.org/10.1042/bj3010745 (1994).
Sabatier, L. et al. Fibrillin assembly requires fibronectin. Mol. Biol. Cell. 20, 846–858. https://doi.org/10.1091/mbc.E08-08-0830 (2009).
Doucet, A. & Overall, C. M. Broad coverage identification of multiple proteolytic cleavage site sequences in complex high molecular weight proteins using quantitative proteomics as a complement to edman sequencing. Mol. Cell. Proteomics 10, 3533. https://doi.org/10.1074/mcp.M110.003533 (2011).
Bornstein, P. Thrombospondins function as regulators of angiogenesis. J. Cell Commun. Signal. 3, 189–200. https://doi.org/10.1007/s12079-009-0060-8 (2009).
Chand, H. S., Foster, D. C. & Kisiel, W. Structure, function and biology of tissue factor pathway inhibitor-2. Thromb. Haemost. 94, 1122–1130. https://doi.org/10.1160/TH05-07-0509 (2005).
Yang, X. et al. Proinflammatory phenotype of vascular smooth muscle cells: Role of efficient Toll-like receptor 4 signaling. Am. J. Physiol. Heart Circ. Physiol. 289, H1069-1076. https://doi.org/10.1152/ajpheart.00143.2005 (2005).
Zampetaki, A., Zhang, Z., Hu, Y. & Xu, Q. Biomechanical stress induces IL-6 expression in smooth muscle cells via Ras/Rac1-p38 MAPK-NF-kappaB signaling pathways. Am. J. Physiol. Heart Circ. Physiol. 288, H2946-2954. https://doi.org/10.1152/ajpheart.00919.2004 (2005).
Morla, A., Zhang, Z. & Ruoslahti, E. Superfibronectin is a functionally distinct form of fibronectin. Nature 367, 193–196 (1994).
Mitsi, M., Hong, Z., Costello, C. E. & Nugent, M. A. Heparin-mediated conformational changes in fibronectin expose vascular endothelial growth factor binding sites. Biochemistry 45, 10319–10328 (2006).
Little, W. C., Smith, M. L., Ebneter, U. & Vogel, V. Assay to mechanically tune and optically probe fibrillar fibronectin conformations from fully relaxed to breakage. Matrix Biol. 27, 451–461 (2008).
Klotzsch, E. et al. Fibronectin forms the most extensible biological fibers displaying switchable force-exposed cryptic binding sites. Proc. Natl. Acad. Sci. U.S.A. 106, 18267–18272 (2009).
Giamblanco, N., Yaseen, M., Zhavnerko, G., Lu, J. R. & Marletta, G. Fibronectin conformation switch induced by coadsorption with human serum albumin. Langmuir 27, 312–319 (2011).
Williams, E. C., Janmey, P., Ferry, J. & Mosher, D. Conformational states of fibronectin. Effects of pH, ionic strength, and collagen binding. J. Biol. Chem. 257, 14973–14978 (1982).
Johansson, S. Demonstration of high affinity fibronectin receptors on rat hepatocytes in suspension. J. Biol. Chem. 260, 1557–1561 (1985).
Morla, A. & Ruoslahti, E. A fibronectin self-assembly site involved in fibronectin matrix assembly: Reconstruction in a synthetic peptide. J. Cell Biol. 118, 421–429 (1992).
Ugarova, T. P. et al. Conformational transitions in the cell binding domain of fibronectin. Biochemistry 34, 4457–4466 (1995).
Harris, G., Ma, W., Maurer, L. M., Potts, J. R. & Mosher, D. F. Borrelia burgdorferi protein BBK32 binds to soluble fibronectin via the N-terminal 70-kDa region, causing fibronectin to undergo conformational extension. J. Biol. Chem. 289, 22490–22499. https://doi.org/10.1074/jbc.M114.578419 (2014).
Degendorfer, G. et al. Peroxynitrite-mediated oxidation of plasma fibronectin. Free Radic. Biol. Med. 97, 602–615 (2016).
Vanichkitrungruang, S. et al. Oxidation of human plasma fibronectin by inflammatory oxidants perturbs endothelial cell function. Free Radic. Biol. Med. 136, 118–134. https://doi.org/10.1016/j.freeradbiomed.2019.04.003 (2019).
Stine, J. M., Ahl, G. J., Schlenker, C., Rusnac, D.-V. & Briknarová, K. The interaction between the third type III domain from fibronectin and anastellin involves β-strand exchange. Biochemistry 56, 4667–4675 (2017).
Ohashi, T. & Erickson, H. P. Domain unfolding plays a role in superfibronectin formation. J. Biol. Chem. 280, 39143–39151 (2005).
Ohashi, T. & Erickson, H. P. Fibronectin aggregation and assembly: The unfolding of the second fibronectin type III domain. J. Biol. Chem. 286, 39188–39199 (2011).
Bultmann, H., Santas, A. J. & Peters, D. M. Fibronectin fibrillogenesis involves the heparin II binding domain of fibronectin. J. Biol. Chem. 273, 2601–2609. https://doi.org/10.1074/jbc.273.5.2601 (1998).
Little, E., Bork, P. & Doolittle, R. F. Tracing the spread of fibronectin type III domains in bacterial glycohydrolases. J. Mol. Evol. 39, 631–643. https://doi.org/10.1007/BF00160409 (1994).
He, J. et al. Peroxynitrous acid (ONOOH) modifies the structure of anastellin and influences its capacity to polymerize fibronectin. Redox Biol. 36, 101631 (2020).
Demichev, V., Messner, C. B., Vernardis, S. I., Lilley, K. S. & Ralser, M. DIA-NN: neural networks and interference correction enable deep proteome coverage in high throughput. Nat. Methods 17, 41–44. https://doi.org/10.1038/s41592-019-0638-x (2020).
Naba, A. et al. The matrisome: In silico definition and in vivo characterization by proteomics of normal and tumor extracellular matrices. Mol. Cell. Proteomics 11, 4647. https://doi.org/10.1074/mcp.M111.014647 (2012).
Lyngsø, J. & Pedersen, J. S. A high-flux automated laboratory small-angle X-ray scattering instrument optimized for solution scattering. J. Appl. Crystallogr. 54, 295–305 (2021).
Acknowledgements
We are grateful to Jan Skov Pedersen for access to the SAXS machine. This work was supported by the Novo Nordisk Foundation (Laureate grants: NNF13OC0004294 and NNF205A0064214 to MJD), the Danish Council for Independent Research|Natural Sciences (DFF-7014-00047 to MJD), the Carlsberg Foundation (CF19-0451 to PH), and the China Scholarship Council (scholarship to JH).
Author information
Authors and Affiliations
Contributions
J.H.: conceptualization, methodology, validation, formal analysis, investigation, writing, writing—review and editing. J.H.S.: resources, investigation. P.W.T.: formal analysis, validation, writing—reviewing and editing. J.N.P.: formal analysis, investigation, validation, writing—reviewing and editing. M.B.S.: formal analysis. D.E.O.: validation, writing—reviewing and editing. C.L.H.: supervision, writing—review and editing. P.G.: resources, writing—reviewing and editing. M.J.D.: conceptualization, methodology, validation, writing—review and editing, supervision, funding acquisition. P.H.: conceptualization, methodology, validation, formal analysis, data curation, writing, writing—review and editing, supervision, project administration.
Corresponding authors
Ethics declarations
Competing interests
Prof. Davies reports declares consultancy contracts with Novo Nordisk A/S, and is a founder and shareholder in Seleno Therapeutics plc. These funders had no role in the design of the study; in the collection, analyses or interpretation of data; in the writing of the manuscript, or in the decision to publish the results. All other authors declare no conflicts of interest.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
He, J., Steffen, J.H., Thulstrup, P.W. et al. Anastellin impacts on the processing of extracellular matrix fibronectin and stimulates release of cytokines from coronary artery smooth muscle cells. Sci Rep 12, 22051 (2022). https://doi.org/10.1038/s41598-022-26359-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-26359-9
- Springer Nature Limited