
Abstract
In previous study, ectopic expression of GhSAMDC1 improved vegetative growth and early flowering in tobacco, which had been explained through changes of polyamine content, polyamines and flowering relate genes expression. To further disclose the transcript changes of ectopic expression of GhSAMDC1 in tobacco, the leaves from wild type and two transgenic lines at seedling (30 days old), bolting (60 days old) and flowering (90 days old) stages were performed for transcriptome analysis. Compared to wild type, a total of 938 differentially expressed genes (DEGs) were found to be up- or down-regulated in the two transgenic plants. GO and KEGG analysis revealed that tobacco of wild-type and transgenic lines were controlled by a complex gene network, which regulated multiple metabolic pathways. Phytohormone detection indicate GhSAMDC1 affect endogenous phytohormone content, ABA and JA content are remarkably increased in transgenic plants. Furthermore, transcript factor analysis indicated 18 transcript factor families, including stress response, development and flowering related transcript factor families, especially AP2-EREBP, WRKY, HSF and Tify are the most over-represented in those transcript factor families. In conclusion, transcriptome analysis provides insights into the molecular mechanism of GhSAMDC1 involving rapid vegetative growth and early flowering in tobacco.
Similar content being viewed by others


Introduction
Polyamines (PAs) are low molecular weight aliphatic nitrogenous bases with biological activity, which contain two or more amino groups1, and mainly include putrescine (Put), spermidine (Spd), spermine (Spm) and thermospermine (T-Spm) in plants2,3. PAs showed tissue and organ specific distribution patterns4,5, and played crucial roles in various physiological processes in plants, some reports had been showed PAs involved in abiotic/biotic stress response6,7,8,9,10,11,12, leaf development and senescence13,14,15, fruit development and ripening16,17,18 and floral initiation and development19,20,21,22. Beside, many reports have indicated PAs as signaling molecules or mediated through the products of their catabolism together with plant hormone23, such as jasmonic acid (JA)24, abscisic acid (ABA)25,26, salicylic acid (SA)27 and ethylene28 were involved in various physiological processes.
In plants, Put is synthesized through arginine decarboxylase (ADC) and/or ornithine decarboxylase (ODC), and conversion of Put to Spd and Spm requires Spd synthase and Spm synthase, respectively7. S-adenosylmethionine decarboxylase (SAMDC) involve in the biosynthesis of decarboxylated SAM, which donates the aminopropyl moiety for the biosynthesis of these PAs. On the other hand, catabolism of PAs involves diamine oxidases (CuAOs) and polyamine oxidases (PAOs). The homeostasis of cellular PAs levels, being well regulated by a dynamic balance of biosynthesis and catabolism, and it is important for maintaining normal growth and development in plants. The functions of SAMDC genes had been reported in many plants, which indicate it play important roles in stress tolerance and plant development through regulating in polyamine synthesis2,10,13,22.
In previous study, an upland cotton S-adenosylmethionine decarboxylase (GhSAMDC1, GenBank NO. JN020148) gene was transformed into tobacco and resulted in rapid vegetative growth and early flowering. To further disclose the potential molecular mechanism of GhSAMDC1, a transcriptome analysis was performed in wild type and transgenic plants at different stages.
Results
Phenotype identification of transgenic plants
In previous study, transgenic plants (3–1, 3–2, and 4–3) from three independent transformation events had been analyzed and identified22. Compared to wild type, transgenic plants showed rapid vegetative growth in early bolting and flowering stage (Fig. 1A–C). Based on the previous expression analysis, we selected seedling stage (30 days, S) (Fig. 1A), bolting stage (60 days, B) (Fig. 1B) and flowering stage (90 days, F) (Fig. 1C) of wild type (WT) and transgenic (3–2 and 4–3) lines for transcription sequencing.

Phenotype of wild type and transgenic plants. (a–c) Phenotype of wild type and transgenic plants at seedling, bolting and flowering stages, respectively.
Primary analysis of transcriptome data
Transcriptome analysis indicated sequencing produced libraries averaging 57.83 million clean reads per library, and average of Q20 and Q30 value were 97.24% and 92.11%, respectively (Table S1). Heat map of Pearson correlation coefficient between different samples was shown in Fig. 2A, the coefficient were 0.915–0.982 between twice replicates, and details were described in supplementary. Principal Component Analysis (PCA) show that the clusters in different periods have large differences (Figure S1). Fragments Per Kilobase of transcript sequence per Millions base pairs sequenced (FPKM) distribution analysis suggested the percentage of FPKM interval 0–1, 1–3, 3–15, 15–60, > 60 are 54.52%, 14.96%, 21.76%, 6.69% and 2.08%. The violin diagram of gene expression level is shown in Fig. 2B, and details were described in supplementary Table S2. Based on the above data analysis, a cluster analysis of the differentially expressed transcripts was described in Fig. 2C.

Primary analysis of transcriptome datas. (a) Heat map of Pearson correlation coefficient between samples; (b) Violin diagram of gene expression level in wild type and transgenic lines at different stages; (c) Heat map for cluster analysis of the differentially expressed transcripts. The color scale corresponds to the log2 (FPKM) values of genes in various samples, red represented up-regulated expression and blue represented down-regulated expression.
Gene expression analysis of WT and transgenic lines at different developmental stages
Comparisons of gene expression between WT and the two transgenic lines were performed at different stages. Overall, there were 938 genes identified that were differentially expressed in the transgenic lines compared to the WT (|log 2 (fold change) |≥ 2 and padj < 0.05). Among these genes, 82, 241 and 615 DEGs were acquired at seedling, bolting and flowering stages, respectively (Table S3). Compared to WT, transgenic line 3–2 had 181 DEGs (131 up- and 50 down- regulated), and transgenic line 4–3 had 757 DEGs (438 up- and 319 down-regulated), the details are described in Fig. 3A. As was shown in Fig. 3B, two three-way Venn diagrams of 3–2 vs WT and 4–3 vs WT at different stages were displayed, respectively. 120 were unique ones with 15, 23 and 41 unique to seedling, bolting and flowering stage in the transgenic line 3–2 and WT comparison, respectively. Of the 676 non-redundant DEGs in the transgenic line 4–3 compared to WT, there were 31 DEGs shared at different stages, there were 6, 133 and 487 genes to seedling, bolting and flowering stage in the transgenic line 4–3 and WT.

Analysis of gene expression differences between wild type and transgenic lines. (a) Diagram showing the number of genes up- and down-regulated genes between wild type and transgenic lines at different stages; (b) Venn diagrams for differentially expressed genes between wild type and transgenic lines.
GO and KEGG pathway analysis of DEGs between 4–3 and WT at different stages
To gain a better understanding of the molecular mechanism of GhSAMDC1 involving in rapid vegetative growth and early flowering in tobacco, GO annotations of the DEGs and enrichment of KEGG pathway were performed in comparison 3–2 vs WT and 4–3 vs WT at different stages, it found that due to the small number of DEGs between 3 and 2 and WT, GO were not enriched at different stages. Therefore, we only compared between 4 and 3 and WT at different stages. GO annotations of the DEGs between WT plants and transgenic line 4–3 at different stages were presented in Fig. 4A–C, Using the criterion of corrected PValue ≤ 0.05, a total of 49 GO terms were enriched at bolting stage, There were 41 GO terms involved in biological process level, mainly including regulation of cellular process, regulation of biological process, primary metabolic process, transcription, DNA-templated, nucleic acid-templated transcription, RNA biosynthetic process, cellular macromolecule biosynthetic process, macromolecule metabolic process, RNA metabolic process, nitrogen compound metabolic process, nucleobase-containing compound metabolic process. In cellular component level, DEGs were over-represented in proteinaceous extracellular matrix, extracellular matrix, extracellular region part, extracellular region. For molecular function level, over-represented DEGs were associated with nucleic acid binding transcription factor activity, sequence-specific DNA binding, inositol-3-phosphate synthase activity, intramolecular lyase activity (Fig. 4B, Table S4). 15 GO terms were enriched at flowering stage, 8 biological process terms were enriched were related to coenzyme M biosynthetic process, coenzyme M metabolic process, cell morphogenesis, cellular component morphogenesis, cellular developmental process, protein folding, oxidation–reduction process, anatomical structure morphogenesis, in molecular function level mainly involved terpene synthase activity, carbon–oxygen lyase activity, magnesium ion binding, lyase activity, oxidoreductase activity, unfolded protein binding. (Fig. 4C, Table S5). In order to further analyze the DEGs, we performed Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways analysis (Fig. 4D-F), using the criterion of corrected P Value ≤ 0.05, KEGG enrichment analysis indicated histidine metabolism between 3 and 2 and WT at bolting stage (Table S6). At the bolting stage in 4–3 vs WT, pathways related to plant-pathogen interaction (Fig. 4E, Table S6), at the flowering stage between 4 and 3 and WT, they were involved protein processing in endoplasmic reticulum, alpha-Linolenic acid metabolism, spliceosome, sesquiterpenoid and triterpenoid biosynthesis, linoleic acid metabolism (Fig. 4F, Table S6). These results indicate that tobacco of wild-type and transgenic are controlled by a complex gene network, which regulates multiple metabolic pathways.

GO annotations and KEGG enrichmentanalysis of the DEGs between wild type and transgenic line 4–3 at different stages. (a–c) GO annotations of the DEGs between wild type and transgenic line 4–3 at different stages; (d–f) KEGG enrichment analysis of the DEGs between wild type and transgenic line 4–3 at different stages.
Protein–protein interaction (PPI) analysis
To further understand the relationship of DEGs, PPI network analyses of DEGs with FDR ≤ 0.05 and |Log2FC|≥ 2 was performed using STRING database. Through BlastX comparison, the interaction relationship among these DEGs was found and visualized in the String database. A total of 105 interaction relationships were identified among 77 DEGs between transgenic line 4–3 and WT at flowering stage (Fig. 5), which indicated average node degree was 2.73, average local clustering coefficient was 0.354, expected number of edges was 12 and PPI enrichment P value was less than 1.0 × e16.The DEGs including Hsp90-1, hsc70 (Heat shock cognate 70 kDa protein 1), Solyc03g007890.2.1 (Heat shock protein 90), 101,252,822(Heat shock protein 70 family protein), 101,243,963 (Heat shock transcription factor B2A), 101,255,185 (Heat shock protein 70B), 101,267,632 (Winged-helix DNA-binding transcription factor family protein), 101,255,164 (Heat shock protein 70) and 101,265,819 (Chaperone protein htpG family protein) were found to play important roles in maintaining the tight connection of the whole network. The result indicated ectopic expression of GhSAMDC1 might involve in early flowering through activating heat shock protein in transgenic tobacco29,30.

Protein–protein interaction network analysis of DEGs between transgenic 4–3 and wild type at flowering stage. The STRING database (http://string-db.org/) was used to analyze the protein–protein interaction network based on the proteins corresponding to all DEGs.
Quantitative RT-PCR validation of RNA-sequencing data
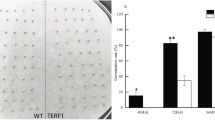
To confirm the accuracy of the transcriptome analysis results, real-time quantitative reverse transcription-PCR (qRT-PCR) was used to analyze DEGs identified in the RNA-Seq results. 6 DEGs between wild type and transgenic lines at different stages were selected for qRT-PCR analysis. There was a general agreement between the RNA-seq (Table S7) and qRT- PCR (Fig. 6) results for all genes analyzed validating the RNA-seq library.

Relative expression analysis of 6 DEGs by qRT-PCR.
Discussion
Interaction of polyamine and hormone affected plant growth and development
Based on our previous study, ectopic expression of GhSAMDC1 resulted in Spd content decrease and then increase accompany with Put decrease at flowering stage in transgenic lines, which indicates Spd plays an important role in floral induction22. Changes of PA content could alter gene expression level of genes associated with plant hormone signaling transduction in Arabidopsis31, and Spd involved floral induction by decreasing GA3 and increasing MdNCED1 and MdNCED3 through ABA enrichment in apple32. Recently, plant CuAOs involving in polyamine terminal oxidation were induced by stress-related hormones, including methyl-jasmonate (MeJA), abscisic acid (ABA) and salicylic acid (SA), which indicated polyamine homeostasis was affected by plant hormone33. In thus, crosstalk between polyamine and hormone plays a crucial role in plant growth and development.
Phytohormone detection was performed in wild type and transgenic lines at different stages (Fig. 7). IAA, BR and ZT were not detected in all samples, GA3 was only detected in transgenic line 4–3 at flowering stage. GA are closely associated with DELLA involving in flowering pathways34,35, and a highly conserved family of R2R3 MYB transcription factors had been reported involving in the GA signaling pathway and flowering36,37,38. SA content was detected in all stages and decreased accompany with growth in wild type plants, whereas only was detected in later stages and showed reverse tendency in transgenic lines. Increase of Spd enhanced SA content in the leaves had been reported in wheat39, and vice versa SA treatment affects the PAs metabolism in plants27,40,41,42. In Arabidopsis, exogenous SA appears to be a repressor of the expression of flowering related gene FLC43. Remarkably, ABA and JA were detected in all samples, and the content of them in transgenic lines were higher than that in wild type plants, it was consistent with exogenous Spm induced JA accumulation in lima bean24. Furthermore, a positive feedback loop has also been found between ABA and PAs, ABA activates the PA metabolism as well as PA induces ABA synthesis25,44,45, and various miRNAs have also been documented to be involved in flowering by means of ABA signaling and regulation46,47,48. Further indicated a complicated relationship exist between PAs and plant hormones, and which play crucial role in ectopic expression of GhSAMDC1 involving rapid vegetative growth and early flowering in tobacco.

Plant hormone content of wild type and transgenic lines at different stages.
Transcription factor genes affect rapid vegetative growth and early flowering in transgenic plants overexpressing GhSAMDC 1
Analyzing GO annotation of the DEGs between 4 and 3 and WT at bolting stage found that there were enriched transcription factor activity, therefore, the transcription factor was analyzed. As is shown in Table S8, 18 transcription factor families were identified in those DEGs, and AP2-EREBP, WRKY, HSF and Tify were the most over represented families. In these families, AP2/EREBP including 14 genes was the most abundant transcription factor family. As we known, AP2/EREBP comprise AP2-like genes (with two AP2 domains) and EREBP-like or ERF-like genes (with one AP2 domain)49, and they are involved in plant development, especially reproductive growth through regulating ABA content50,51. In this study, compared to wild type, ABA content was remarkably increased in transgenic lines at different stages, which indicated increase of ABA content might be regulated by those AP2/EREBP genes. Furthermore, 9 WRKY transcription factor genes were identified and over-represented. Except for responding to biotic and abiotic stress52,53, WRKY genes are also involved in several physiological and developmental processes54,55. Especially, many studies have demonstrated that WRKY genes play crucial roles in flowering56,57,58,59. In addition, both HSF and Tify transcription factor family contained 6 genes, and they were also over-represented in those DEGs. HSF had been reported that they not only involve in abiotic stress response60,61,62, but also regulate plant development63 and flowering29,30. Tify are plant-specific transcription factors characterized by the presence of a highly conserved motif (TIF(F/Y)XG) in the TIFY domain64. Many studies had been reported they encodes proteins involve in multiple biological processes through modulating jasmonate signaling pathway65, including leaf development66, abiotic stress resistance67,68,69 and flowering70,71. Altogether, identification of abundant plant development and flowering related transcription factor genes indicated these genes at least partly participate regulating rapid vegetative growth and early flowering in ectopic expression of GhSAMDC1 in tobacco.
Methods
Plant materials and sampling
The tobacco variety used in this study was SR1 (Nicotiana tabacum pv SR1), and the plant was preserved and planted in the Hubei Key Laboratory of Economic Forest Germplasm Improvement and Resources Comprehensive Utilization. Wild type and homozygous T3 transgenic tobacco seeds (including 3–2 and 4–3) were surface sterilized with 2% NaClO and washed three times with sterile water. The sterile seeds were then suspended in 0.2% agar and plated on 1⁄2 Murashige and Skoog (MS) medium plus 1.5% sucrose72. 50 seedlings were selected from each variety and planted in artificial climate chamber (16 h light and 8 h dark cycle at 25 °C), observe the phenotypic changes of seedlings and leaves from the same position on 30 days (Seedling stage, S), 60 days (Bolting stage, B), 90 days (Flowering stage, F) and wild type (WT), transgenic lines 3–2 (3–2) and 4–3 (4–3) plants were sampled for transcriptome analysis (Table S9). Twice biological replicates were performed.
Library preparation and Transcriptome sequencing
Total RNA was extracted using Trizol reagent following the manufacturer’s protocol. RNA purity was checked using the NanoPhotometer® spectrophotometer (IMPLEN). RNA concentration was measured using Qubit® RNA Assay Kit in Qubit® 2.0 Flurometer. RNA integrity was assessed using the RNA Nano 6000 Assay Kit with the Bioanalyzer 2100 system. A total amount of 1 μg RNA per sample was used as input material for the RNA sample preparations. Sequencing libraries were generated using NEBNext® UltraTM RNA Library Prep Kit for Illumina® (NEB, USA) following manufacturer’s recommendations. The clustering of the index-coded samples was performed on a cBot Cluster Generation System using TruSeq PE Cluster Kit v3-cBot-HS (Illumia) according to the manufacturer’s instructions. After cluster generation, the library preparations were sequenced on an Illumina Hiseq platform and 125 bp/150 bp paired-end reads were generated.
Data analysis
The datasets used and analysed have been deposited in the National Center for Biotechnology Information (NCBI). The accession number is PRJNA759726 (http://www.ncbi.nlm.nih.gov/bioproject/759726), which includes 18 accession items (SRR15709784—SRR15709801). Raw data of fastq format were firstly processed through in-house perl scripts. Nicotiana tabacum reference genome and gene model annotation files were downloaded from genome website directly (https://www.ncbi.nlm.nih.gov/genome/425?genome_assembly_id=274804). HTSeq v0.9.1 was used to count the reads numbers mapped to each gene. And then FPKM of each gene was calculated based on the length of the gene and reads count mapped to this gene, The DESeq software was used to determine differential gene expression by negative binomial distribution and calculation of false discovery rate (FDR) by the Benjamini and Hochberg method, as well as the adjusted Pvalue. The adjusted Pvalue < 0.05 was used as the threshold for identification of DEGs among samples. Differential expression analysis of two conditions/groups (two biological replicates per condition) was performed using the DESeq R package (1.18.0) (https://bioconductor.org/packages/release/bioc/html/DESeq2.html). Genes with an adjusted Pvalue < 0.05 found by DESeq were assigned as differentially expressed. Gene Ontology (GO) enrichment analysis of differentially expressed genes was implemented by the clusterProfler R package (http://www.bioconductor.org/packages/release/bioc/html/clusterProfiler.html). GO terms with corrected Pvalue less than 0.05 were considered significantly enriched by differential expressed genes. KOBAS software was used to test the statistical enrichment of differential expression genes in KEGG pathways (www.kegg.jp/kegg/kegg1.html). PPI analysis of differentially expressed genes was based on the STRING database (https://cn.string-db.org/), which known and predicted Protein–Protein Interactions73, Nodes represent the DEGs enriched in the STRING database, while edges reflect the interactions between differentially expressed genes, the interaction data was imported into Cytoscape software v3.2.0 (http://cytoscape.org) to realize the visualization of the interaction network. Transcript factors analysis was performed using iTAK software online (http://itak.feilab.net/cgi-bin/itak/index.cgi).
RNA isolation and quantitative reverse transcriptase-polymerase chain reaction (qRT-PCR) analysis
Total RNA was extracted from the samples using the modified CTAB method22. qRT-PCR was performed using Power SYBR Green Master (Roche, Basel, Switzerland) on a Roche Light Cycler 480 system (Roche), as described previously. The reaction was run as follows: pre-incubation at 94 °C for 2 min, 40 cycles of 94 °C for 20 s, 58 °C for 20 s and 72 °C for 20 s. The actin gene (Tac9; X69885) was used as the reference gene, and the relative 2−∆ct quantification method was used to evaluate quantitative variation. Three biological replicates and three technical repeats were run. The qRT-PCR primers are listed in Table S10.
Phytohormone detection
Leaves from wild type and transgenic lines 3–2 and 4–3 were sampled for phytohormone detection at seedling (30 days old), bolting (60 days old) and flowering (90 days old) stages, Three biological replicates were performed for each sample. Samples were rapidly frozen by liquid nitrogen and transported with dry ice for analysis. Phytohormone detection was performed by Shiseido SP HPLC-Thermo TSQ Quantum Ulta MS/Ms in sci-tech innovation company in Qingdao. All images are combined through Photoshop.
Ethics statement
The tobacco varieties were supplied by Molecular Breeding Laboratory of Shihezi University (Tobacco, Xinjiang), including SR1 (Nicotiana tabacum cv. Petit Habana SR1) varieties. Experimental research and field studies on plants in this work, including the collection of plant material, comply with relevant institutional, national, and international guidelines and legislation.
Data availability
The data and material that support the fndings of this study are available from the corresponding author on reasonable request.
References
Tabor, C. W. & Tabor, H. Polyamines. Annu. Rev. Biochem. 53, 749–790 (1984).
Kusano, T., Berberich, T., Tateda, C. & Takahashi, Y. Polyamines: Essential factors for growth and survival. Planta 228, 367–381 (2008).
Takano, A., Kakehi, J. & Takahashi, T. Thermospermine is not a minor polyamine in the plant kingdom. Plant Cell Physiol. 53, 606–616 (2012).
Paschalidis, K. A. & Roubelakis-Angelakis, K. A. Spatial and temporal distribution of polyamine levels and polyamine anabolism in different organs/tissues of the tobacco plant. Correlations with age, cell division/expansion, and differentiation. Plant Physiol. 138, 142–152 (2005).
Takahashi, Y., Tahara, M., Yamada, Y., Mitsudomi, Y. & Koga, K. Characterization of the polyamine biosynthetic pathways and salt stress response in brachypodium distachyon. J. Plant Growth Regul. 37, 625–634 (2017).
Moschou, P. N., Paschalidis, K. A. & Roubelakis-Angelakis, K. A. Plant polyamine catabolism: The state of the art. Plant Signal Behav. 3, 1061–1066 (2008).
Alcazar, R. et al. Polyamines: Molecules with regulatory functions in plant abiotic stress tolerance. Planta 231, 1237–1249 (2010).
Minocha, R., Majumdar, R. & Minocha, S. C. Polyamines and abiotic stress in plants: A complex relationship. Front. Plant Sci. 5, 175 (2014).
Hatmi, S. et al. Drought stress tolerance in grapevine involves activation of polyamine oxidation contributing to improved immune response and low susceptibility to Botrytis cinerea. J. Exp. Bot. 66, 775–787 (2015).
Liu, J. H., Wang, W., Wu, H., Gong, X. & Moriguchi, T. Polyamines function in stress tolerance: From synthesis to regulation. Front. Plant Sci. 6, 827 (2015).
Pal, M., Szalai, G. & Janda, T. Speculation: Polyamines are important in abiotic stress signaling. Plant Sci. 237, 16–23 (2015).
Park, J. Y., Kang, B. R., Ryu, C. M., Anderson, A. J. & Kim, Y. C. Polyamine is a critical determinant of Pseudomonas chlororaphis O6 for GacS-dependent bacterial cell growth and biocontrol capacity. Mol. Plant Pathol. 19, 1257–1266 (2018).
Moschou, P. N. & Roubelakis-Angelakis, K. A. Polyamines and programmed cell death. J. Exp. Bot. 65, 1285–1296 (2014).
Cai, G. et al. Polyamines are common players in different facets of plant programmed cell death. Amino Acids 47, 27–44 (2015).
Sobieszczuk-Nowicka, E. Polyamine catabolism adds fuel to leaf senescence. Amino Acids 49, 49–56 (2017).
Agudelo-Romero, P., Bortolloti, C., Pais, M. S., Tiburcio, A. F. & Fortes, A. M. Study of polyamines during grape ripening indicate an important role of polyamine catabolism. Plant Physiol. Biochem. 67, 105–119 (2013).
Tsaniklidis, G. et al. Spatial and temporal distribution of genes involved in polyamine metabolism during tomato fruit development. Plant Physiol. Biochem. 100, 27–36 (2016).
Fortes, A. M. & Agudelo-Romero, P. Polyamine metabolism in climacteric and Non-Climacteric fruit ripening. Methods Mol. Biol. 1694, 433–447 (2018).
Kakehi, J., Kuwashiro, Y., Niitsu, M. & Takahashi, T. Thermospermine is required for stem elongation in Arabidopsis thaliana. Plant Cell Physiol. 49, 1342–1349 (2008).
Applewhite, P. B., Awhney, R. K. & Galston, A. W. A role for spermidine in the bolting and flowering of Arabidopsis. Physiol. Plantarum 108, 314–320 (2010).
Ahmed, S. et al. Altered expression of polyamine transporters reveals a role for spermidine in the timing of flowering and other developmental response pathways. Plant Sci. 258, 146–155 (2017).
Zhu, H. et al. Ectopic expression of GhSAMDC1 improved plant vegetative growth and early flowering through conversion of spermidine to spermine in tobacco. Sci. Rep. 10, 14418 (2020).
Anwar, R., Mattoo, A. K. & Handa, A. K. Polyamine interactions with plant hormones: Crosstalk at several levels. Int. J. Hydrogen Energ. 38, 1039–1051 (2015).
Ozawa, R. et al. Exogenous polyamines elicit herbivore-induced volatiles in lima bean leaves: Involvement of calcium, H2O2 and Jasmonic acid. Plant Cell Physiol. 50, 2183–2199 (2015).
Pal, M. et al. Interaction of polyamines, abscisic acid and proline under osmotic stress in the leaves of wheat plants. Sci. Rep. 8, 12839 (2018).
Bai, Q., Huang, Y. & Shen, Y. The physiological and molecular mechanism of abscisic acid in regulation of fleshy fruit ripening. Front. Plant Sci. 11, 619953 (2020).
Németh, M., Janda, T., Horváth, E., Páldi, E. & Szalai, G. Exogenous salicylic acid increases polyamine content but may decrease drought tolerance in maize. Plant Sci. 162, 569–574 (2020).
Yang, W. et al. Involvement of ethylene and polyamines biosynthesis and abdominal phloem tissues characters of wheat caryopsis during grain filling under stress conditions. Sci. Rep. 7, 46020 (2017).
Margaritopoulou, T. et al. HSP90 canonical content organizes a molecular scaffold mechanism to progress flowering. Plant J. 87, 174–187 (2016).
Ye, J. et al. Genome-Wide investigation of heat shock transcription factor family in Wheat (Triticum aestivum L.) and possible roles in anther development. Int. J. Mol. Sci. 21, 608 (2020).
Marco, F., Alcazar, R., Tiburcio, A. F. & Carrasco, P. Interactions between polyamines and abiotic stress pathway responses unraveled by transcriptome analysis of polyamine overproducers. OMICS 15, 775–781 (2011).
Qin, L. et al. Effect of exogenous spermidine on floral induction, endogenous polyamine and hormone production, and expression of related genes in “Fuji” apple (Malus domestica Borkh). Sci. Rep. 9, 12777 (2019).
Fraudentali, I. et al. Developmental, hormone- and stress-modulated expression profiles of four members of the Arabidopsis copper-amine oxidase gene family. Plant Physiol. Biochem. 147, 141–160 (2020).
Teotia, S. & Tang, G. To bloom or not to bloom: Role of microRNAs in plant flowering. Mol. Plant 8, 359–377 (2015).
Park, J. et al. Gibberellin signaling requires chromatin remodeler PICKLE to promote vegetative growth and phase transitions. Plant Physiol. 173, 1463–1474 (2017).
Gocal, G. F. et al. GAMYB-like genes, flowering, and gibberellin signaling in Arabidopsis. Plant Physiol. 127, 1682–1693 (2001).
Woodger, F. J., Millar, A., Murray, F., Jacobsen, J. V. & Gubler, F. The role of GAMYB transcription factors in GA-Regulated gene expression. J. Plant Growth Regul. 22, 176–184 (2003).
Pal, M. et al. Role of polyamines in plant growth regulation of Rht wheat mutants. Plant Physiol. Biochem. 137, 189–202 (2019).
Rahdari, P. & Hoseini, S. M. Roll of poly amines (Spermidine and Putrescine) on Protein, Chlorophyll and Phenolic Compounds in Wheat (Triticum aestivum L.) under Salinity Stress. J. Nov. Appl. Sci. 2, 746–751 (2013).
Szepesi, A. et al. Interaction between salicylic acid and polyamines and their possible roles in tomato hardening processes. Acta Biol. Szeged. 55, 165–166 (2011).
Hassannejad, S., Bernard, F., Mirzajani, F. & Gholami, M. SA improvement of hyperhydricity reversion in Thymus daenensis shoots culture may be associated with polyamines changes. Plant Physiol. Biochem. 51, 40–46 (2012).
Wang, X. & Zhang, Y. Regulation of Salicylic Acid on Polyamine Synthesize under NaCl Stress in Leaves of the Yali Pear. Res. J. Appl. Sci. Eng. Technol. 4, 3704–3708 (2012).
Martinez, C., Pons, E., Prats, G. & Leon, J. Salicylic acid regulates flowering time and links defence responses and reproductive development. Plant J. 37, 209–217 (2004).
Cuevas, J. C. et al. Putrescine is involved in Arabidopsis freezing tolerance and cold acclimation by regulating abscisic acid levels in response to low temperature. Plant Physiol. 148, 1094–1105 (2008).
Diao, Q., Song, Y., Shi, D. & Qi, H. Interaction of Polyamines, Abscisic Acid, Nitric Oxide, and Hydrogen Peroxide under Chilling Stress in Tomato (Lycopersicon esculentum Mill.) Seedlings. Front. Plant Sci. 8, 203 (2017).
Achard, P., Herr, A., Baulcombe, D. C. & Harberd, N. P. Modulation of floral development by a gibberellin-regulated microRNA. Development 131, 3357–3365 (2004).
Reyes, J. L. & Chua, N. H. ABA induction of miR159 controls transcript levels of two MYB factors during Arabidopsis seed germination. Plant J. 49, 592–606 (2007).
Li, W., Wang, T., Zhang, Y. & Li, Y. Overexpression of soybean miR172c confers tolerance to water deficit and salt stress, but increases ABA sensitivity in transgenic Arabidopsis thaliana. J. Exp. Bot. 67, 175–194 (2016).
Riechmann, J. L. & Meyerowitz, E. M. The AP2/EREBP family of plant transcription factors. Biol. Chem. 379, 633–646 (1998).
Yant, L. et al. Orchestration of the floral transition and floral development in Arabidopsis by the bifunctional transcription factor APETALA2. Plant Cell 22, 2156–2170 (2010).
Zhang, B., Wang, L., Zeng, L., Zhang, C. & Ma, H. Arabidopsis TOE proteins convey a photoperiodic signal to antagonize CONSTANS and regulate flowering time. Genes Dev. 29, 975–987 (2015).
Rushton, P. J., Somssich, I. E., Ringler, P. & Shen, Q. J. WRKY transcription factors. Trends Plant Sci. 15, 247–258 (2010).
Chen, L., Zhang, L., Li, D., Wang, F. & Yu, D. WRKY8 transcription factor functions in the TMV-cg defense response by mediating both abscisic acid and ethylene signaling in Arabidopsis. Proc. Natl. Acad. Sci. USA 110, E1963–E1971 (2013).
Jiang, Y., Liang, G., Yang, S. & Yu, D. Arabidopsis WRKY57 functions as a node of convergence for jasmonic acid- and auxin-mediated signaling in jasmonic acid-induced leaf senescence. Plant Cell 26, 230–245 (2014).
Chen, L., Xiang, S., Chen, Y., Li, D. & Yu, D. Arabidopsis WRKY45 interacts with the DELLA protein RGL1 to positively regulate Age-Triggered leaf senescence. Mol. Plant. 10, 1174–1189 (2017).
Li, W., Wang, H. & Yu, D. Arabidopsis WRKY transcription factors WRKY12 and WRKY13 oppositely regulate flowering under Short-Day conditions. Mol. Plant. 9, 1492–1503 (2016).
Yu, Y. et al. WRKY71 accelerates flowering via the direct activation of flowering locust and leafy in Arabidopsis thaliana. Plant J. 85, 96–106 (2016).
Zhang, L., Chen, L. & Yu, D. Transcription factor WRKY75 interacts with DELLA proteins to affect flowering. Plant Physiol. 176, 790–803 (2018).
Ma, Z., Li, W., Wang, H. & Yu, D. WRKY transcription factors WRKY12 and WRKY13 interact with SPL10 to modulate age-mediated flowering. J. Integr. Plant Biol. 62, 1659–1673 (2020).
Mishra, S. K. et al. In the complex family of heat stress transcription factors, HsfA1 has a unique role as master regulator of thermotolerance in tomato. Genes Dev. 16, 1555–1567 (2002).
Banti, V., Mafessoni, F., Loreti, E., Alpi, A. & Perata, P. The heat-inducible transcription factor HsfA2 enhances anoxia tolerance in Arabidopsis. Plant Physiol. 152, 1471–1483 (2010).
Ma, J. et al. Genome-wide analysis of HSF family transcription factors and their responses to abiotic stresses in two Chinese cabbage varieties. Acta Physiol. Plant. 36, 513–523 (2014).
Chidambaranathan, P. et al. Genome-wide analysis identifies chickpea (Cicer arietinum) heat stress transcription factors (Hsfs) responsive to heat stress at the pod development stage. J. Plant Res. 131, 525–542 (2018).
Bai, Y., Meng, Y., Huang, D., Qi, Y. & Chen, M. Origin and evolutionary analysis of the plant-specific TIFY transcription factor family. Genomics 98, 128–136 (2011).
Monte, I. et al. A single JAZ repressor controls the jasmonate pathway in marchantia polymorpha. Mol. Plant. 12, 185–198 (2019).
Gonzalez, N. et al. A repressor protein complex regulates leaf growth in arabidopsis. Plant Cell 27, 2273–2287 (2015).
Wu, H., Ye, H., Yao, R., Zhang, T. & Xiong, L. OsJAZ9 acts as a transcriptional regulator in jasmonate signaling and modulates salt stress tolerance in rice. Plant Sci. 232, 1–12 (2015).
Fu, J. et al. OsJAZ1 attenuates drought resistance by regulating JA and ABA signaling in rice. Front Plant Sci. 8, 2108 (2017).
Liu, S., Zhang, P., Li, C. & Xia, G. The moss jasmonate ZIM-domain protein PnJAZ1 confers salinity tolerance via crosstalk with the abscisic acid signalling pathway. Plant Sci. 280, 1–11 (2019).
Yu, X. et al. The Jasmonate ZIM-domain protein gene SlJAZ2 regulates plant morphology and accelerates flower initiation in Solanum lycopersicum plants. Plant Sci. 267, 65–73 (2018).
Liu, H. et al. TaZIM-A1 negatively regulates flowering time in common wheat (Triticum aestivum L.). J. Integr. Plant Biol. 61, 359–376 (2019).
Murashige, T. & Skoog, F. A revised medium for rapid growth and bio assays with tobacco tissue cultures. Physiol. Plant 15, 473–497 (1962).
Mering, C. V. STRING: A database of predicted functional associations between proteins. Nucleic Acids Res. 31, 258–261 (2003).
Acknowledgements
This work was supported by the grant from the National Natural Science Foundation of China (Grant no. 31660427). Doctor Fund of Huanggang Normal University (No. 2042019014), and High-level Training Program of Huanggang Normal University (202001003).
Author information
Authors and Affiliations
Contributions
H.Z. conceived and designed research. W.T. and X.C. conducted experiments. H.Z. and X.T. contributed new reagents or analytical tools. X.C. and F.P. analyzed data. H.Z. and X.C. wrote the manuscript. L.W. and X.H. helped with editing the manuscript. All authors read and approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Cheng, X., Pang, F., Tian, W. et al. Transcriptome analysis provides insights into the molecular mechanism of GhSAMDC1 involving in rapid vegetative growth and early flowering in tobacco. Sci Rep 12, 13612 (2022). https://doi.org/10.1038/s41598-022-18064-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-18064-4
- Springer Nature Limited
This article is cited by
-
Comparative analysis of POD genes and their expression under multiple hormones in Pyrus bretschenedri
BMC Genomic Data (2024)
-
Polyamines metabolism and their biological role in plant cells: what do we really know?
Phytochemistry Reviews (2024)