
Abstract
Endangered species with small population sizes are susceptible to genetic erosion, which can be detrimental to long-term persistence. Consequently, monitoring and mitigating the loss of genetic diversity are essential for conservation. The Peninsular pronghorn (Antilocapra americana peninsularis) is an endangered pronghorn subspecies that is almost entirely held in captivity. Captive breeding has increased the number of pronghorns from 25 founders in 1997 to around 700 individuals today, but it is unclear how the genetic diversity of the captive herd may have changed over time. We therefore generated and analysed data for 16 microsatellites spanning 2009–2021. We detected a decline in heterozygosity and an increase in the proportion of inbred individuals over time. However, these trends appear to have been partially mitigated by a genetically informed breeding management attempt that was implemented in 2018. We also reconstructed the recent demographic history of the Peninsular pronghorn, revealing two sequential population declines putatively linked to the desertification of the Baja California peninsula around 6000 years ago, and hunting and habitat loss around 500 years ago, respectively. Our results provide insights into the genetic diversity of an endangered antelope and indicate the potential for genetically informed management to have positive conservation outcomes.
Similar content being viewed by others


Introduction
Many species have experienced severe declines over the past two centuries as a result of growing anthropogenic pressures including direct exploitation, habitat destruction and climate change1,2,3. Some authors have even argued that Earth’s biodiversity is entering a sixth mass extinction event, characterised by the unprecedented loss of diversity at all levels4,5,6. Consequently, nowadays the persistence of many species is critically dependent on intensive management actions such as captive breeding, habitat restoration and reintroduction programs.
For many species, captive management has been the only option for persistence7. For example, species like the Kakapo (Strigops habroptilus), Przewalski’s horse (Equus przewalskii) and giant Galapagos tortoise (Chelonoidis niger), among many others, would have gone extinct without human intervention and ex situ management8,9,10. Captive breeding is frequently used for the preservation of threatened species and, in some cases, for the rehabilitation of declining populations11,12,13,14. However, it can sometimes inadvertently lead to genetic or behavioural changes that are not always beneficial15. For example, when selective pressures in captivity differ to those that are usually encountered by a species in the wild, maladaptive alleles or behaviours can rise to high frequency in captive populations, which can compromise the survival of individuals after they are reintroduced into the wild16,17. Furthermore, in small captive populations, strong genetic drift and the increased probability of mating between close relatives can decrease genome-wide heterozygosity and lead to inbreeding depression18,19,20,21,22. The fitness costs associated with inbreeding have been documented across taxonomic groups and include negative effects on litter size, longevity, female reproduction, male fertility and weight, in addition to hereditary defects23,24,25,26, all of which can have a strong impact on population viability.
Given that conserving genetic diversity and minimising inbreeding are important goals of most if not all captive breeding programmes27,28 and reduced genetic diversity has been associated with increased extinction risk and reduced adaptive potential29,30,31, knowledge of the effects of captive breeding on genetic diversity is crucial. In this regard, time-series genetic data from captive populations can be particularly useful32,33,34,35, as they can shed light on changes in key genetic characteristics of a population such as allelic richness, heterozygosity and the effective population size (Ne); measures that reflect a combination of the speed of allele frequency change through genetic drift, the efficacy of selection and expected genetic diversity levels for selectively neutral loci36,37.
The pronghorn (Antilocapra americana) is the only extant species of the North American family Antilocapridae38,39. Pronghorns are thought to have been historically abundant, with documents from the 1800s suggesting that roughly 30–40 million individuals inhabited North America prior to the westward settlement of humans on the continent40,41. Nevertheless, current pronghorn numbers have been severely affected by habitat fragmentation and overhunting, with many populations having declined or disappeared entirely42,43,44. Nowadays, four pronghorn subspecies are recognized: the American pronghorn (A. a. americana), the Sonoran pronghorn (A. a. sonoriensis), the Peninsular pronghorn (A. a. peninsularis) and the Mexican pronghorn (A. a. mexicana)45. The American pronghorn is the most widespread subspecies, with the Sonoran, Peninsular and Mexican subspecies occupying more peripheral southern areas40,44,45. Of these subspecies, the Peninsular and Sonoran are currently under national and international protection46,47,48. Overall, the pronghorn is one of the many species currently undergoing captive breeding and translocation, with independent breeding programs active in the USA and Mexico49,50,51.
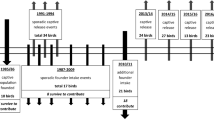
As with all of the pronghorn subspecies, wild populations of the Peninsular pronghorn have declined substantially since the arrival of the fist Spanish settlers51. By the beginning of the twentieth century, the Peninsular pronghorn was thought to number fewer than 1000 individuals40. These numbers have since fallen to fewer than a hundred individuals in the 1980s42,51,52,53. In the face of imminent extinction, a captive breeding program was established by the Peninsular Pronghorn Species Recovery Programme51. This commenced in 1997 at the Vizcaino Biosphere Reserve, Mexico, with 25 wild-caught adults and fawns being introduced to the breeding facilities during the first six years of the programme49. Since then, the captive herd has experienced steady growth, with around 60–100 young individuals being incorporated every year. Although systematic censuses have not been performed, the herd is known to have grown to around 198 individuals in 2006 and 250 individuals in 2010. Furthermore, some additional individuals were translocated to the USA and a number of animals also escaped captivity, with an aerial survey documenting a wild herd of 133 individuals in 2020. Therefore, the Peninsula pronghorn conservation programme represents a good example of a successful ongoing species recovery initiative in Mexico.
Currently, the animals are held in three management stations, with an additional six small populations held by a consortium of zoos in the Southwestern USA53. The main conservation area encompasses over 54,000 ha located in two protected natural areas: the El Vizcaíno Biosphere Reserve and the Valle de los Cirios Flora and Fauna Protection Area. Some of the individuals are allowed to roam freely over the protected areas and are provided only with supplementary feeding and water during the dry season53. Other animals, mainly the breeding herd and pregnant females, are managed in four smaller pens with year-round supplemental food and water, which are protected from predators by anti-coyote fencing.
In 2018, a genetically informed breeding management attempt was undertaken. A random selection of young but sexually mature males and females was microsatellite genotyped (2018 cohort, this paper) and a breeding plan was developed that focused on minimizing the relatedness of potential partners. Group-based management was implemented at one of the pens. At this pen, which consisted of only breeding females, two sexually mature males, selected on the basis of molecular estimates of kinship (and the possession of rare alleles), were introduced and kept there until the following year35. As not all of the individuals in the pen were sampled, some females with unknown relationships to the introduced males were also allowed to breed. Currently, the management team is looking to expand this strategy to include other management units and additional pens.
Previous population genetic studies of pronghorns reported moderate to high levels of genetic diversity in the American subspecies45,54,55,56,57, while genetic diversity appears to be somewhat lower for the Sonoran54,58 and Peninsular pronghorn subspecies58,59. Moreover, the American subspecies shows little evidence of population genetic structure57 while population genetic differentiation at the subspecies level is more pronounced, revealing clear genetic discontinuities between geographically isolated populations35,58,59.
The reasons for the relatively low genetic diversity of the Peninsular pronghorn subspecies are unknown, with two (non-mutually exclusive) explanations being possible. The first of these is that human induced habitat loss, competition with domestic animals and uncontrolled hunting may have caused the Peninsular pronghorn to decline over the past three centuries44,51, which may have been further exacerbated by small population sizes and inbreeding over the past few decades of captivity. Alternatively, or additionally, dramatic ecological changes during the last glacial maximum (LGM; ca. 12,000 years ago) resulted in the desertification of most of the Baja California peninsula, reducing water availability60,61,62 and likely contributing to a gradual reduction in pronghorn numbers over thousands of years.
Here, we generated a time series dataset of multilocus microsatellite data for the captive Peninsular pronghorn spanning the period 2009–2021 inclusive. We first evaluated changes in genetic diversity, heterozygosity and inbreeding over the past 13 years. We then used approximate Bayesian computation63 to evaluate support for alternative demographic scenarios that could explain the low genetic diversity of the Peninsular pronghorn, and to estimate relevant parameters such as the current Ne and the strength and timing of historical declines. We hypothesised that the collapse of the Peninsular pronghorn may have been driven by a combination of historical ecological changes and more recent anthropogenic pressures. We furthermore hypothesised that, although the captive breeding programme has been successful in increasing the number of individuals, there may have been some unavoidable loss of genetic diversity and an increase in inbreeding over time, although we expected that some of these changes might have been mitigated by the breeding management attempt.
Results
Summary statistics
We genotyped 144 pronghorn individuals at 16 microsatellite loci. Our genotyping error rate, estimated from 12 samples genotyped at eight loci, was 0.03 per locus. The overall rate of missing data was 6.1%, which fell to 4.6% when seven individuals with missing data at four loci were excluded. All of the multilocus genotypes were unique, indicating that no individuals had been inadvertently sampled more than once (e.g. initially as fawns and later as adults). Analysing the full dataset of 124 individuals (only fawns were included for 2012, 2016 and 2021; only adults were included for 2009 and 2018, Supplementary Table S1 online), we found significant deviations from Hardy–Weinberg equilibrium (HWE) at six loci (Supplementary Table S2 online). However, when the dataset was partitioned by year, we did not detect any consistent patterns of deviation from HWE across loci (Supplementary Table S2 online). Similar results were obtained for null alleles, with two loci (Aam1 and Anam6) showing indications of the presence of null alleles when all of the data were analysed together, while no consistent patterns were obtained when the years were analysed separately (Supplementary Table S3 online). Significant linkage disequilibrium (LD) was also detected for the full dataset (p = 0.02) but when the cohorts were analysed separately this was only present in 2009 (p = 0.01, Supplementary Table S4 online). Consequently, we retained all of the microsatellite loci for subsequent analyses.
Genetic diversity
Among 124 captive Peninsular pronghorn individuals genotyped at 16 microsatellite loci, we detected a total of 88 alleles, with the mean number of alleles per locus being 5.5 (Table 1; Supplementary Table S5 online). Observed heterozygosity (Ho) was slightly but not significantly (Bartlett’s K-squared = 0.071, p = 0.79) lower than expected heterozygosity (He, Table 1; Supplementary Table S5 online). No significant differences among years were found for the basic diversity estimates (Supplementary Fig. S1 online), although Ho and He showed a weak tendency to decline over time, with the highest values being observed in 2009 and the lowest values being observed in 2018 (Supplementary Fig. S1).
Heterozygosity and inbreeding
Three frequency-weighted microsatellite-based measures of individual heterozygosity—standardized multilocus heterozygosity (sMLH), internal relatedness (IR) and homozygosity weighted by locus (HL)—showed consistent trends of declining heterozygosity over time (all significant at p < 0.05, Table 2, Fig. 1a–c). Based on the TrioML inbreeding index, we found that the captive herd of the Peninsula pronghorn is moderately inbred, with f averaging 0.12 (95% SE = 0.01) and ranging from zero to 0.53 (Fig. 1d, Table 1). We also detected a significant increase in inbreeding over time (Table 2, Fig. 1e), which was mainly attributable to an increase in the proportion of moderately to highly inbred individuals (f > 0.125) from 16.6% in 2009 to 55.5% in 2021 (Fig. 1, Supplementary Table S6 online).

Violin plots showing temporal changes in heterozygosity and inbreeding in the captive peninsular pronghorn. Panels (a–c) show changes in three frequency-weighted measures of individual heterozygosity: standardized multilocus heterozygosity (sMLH), internal relatedness (IR) and homozygosity weighted by locus (HL) respectively. Panel (d) shows bar charts depicting the proportion (%) of individuals falling within different inbreeding classes, from none (f = 0), through low (f = < 0.125) and moderate (0.125 < f < 0.25) to high (f > 0.25), as estimated using TrioML. Panel (e) shows violin plots of changes in the inbreeding coefficient, estimated using TrioML. In panels (a–c) and (e), the boxplots span the first to third quartiles, with horizontal lines inside the boxes representing the medians. The raw data are plotted as black points and the lines connecting the boxplots correspond to regression lines smoothed and fitted with the “glm” function separately for the years 2009–2018 (dashed lines) and 2018–2021 (solid lines).
Temporal change in heterozygosity and inbreeding
To investigate whether the genetically informed breeding management attempt in 2018 could have helped to slow down the loss of heterozygosity, we implemented regressions of diversity estimates on time for the periods 2009–2021, 2009–2018 and 2018–2021 (Fig. 1, Table 2 and Supplementary Table S7 online). Almost all of the frequency-weighted measures of individual heterozygosity and inbreeding (sMLH, IR, HL and TrioML) showed significant temporal trends at p < 0.05 for the periods 2009–2018 and 2009–2021. The only exception was the TrioML inbreeding index over the period 2009–2021 (p = 0.06). Nevertheless, for the marker-based estimates (Ar, Ho and He) only Ho was significant and only for the period 2009–2018. None of the estimates were significant for the period 2018–2021 (Table 2 and Supplementary Table S7 online). Although we do not have a sufficiently long time series after the breeding management attempt to allow us to formally test for differences in the slopes, we did observe a tendency for the slopes to decrease after 2018, at least for sMLH and HL (Fig. 1a–c). Furthermore, the proportion of highly inbred offspring (f > 0.25) declined from 26.3% in 2018 to 11.1% in 2021 (Fig. 1d, Supplementary Table S6 online). Accordingly, the predicted values from the GLMs based on data from 2009 to 2018 projected a greater amount of genetic erosion than was actually observed (Table 3).
Historical demography
We used approximate Bayesian computation (ABC) to evaluate four alternative historical demographic scenarios (Supplementary Fig. S2 online and Methods section for details). The best supported model (59%, CI = 58–60%) contained both a historical and a recent demographic reduction, while the second-best supported model (~ 33%) contained only a recent demographic reduction. The prior predictive error (i.e. the proportion of wrongly identified scenarios over 1000 test datasets drawn from a random sample of the chosen scenario) was high (logistic approach, 0.56) but the posterior error (i.e. the proportion of wrongly identified scenarios over 1000 test datasets drawn from the simulated datasets closest to the observed dataset) was lower at 0.39. Five demographic parameters were estimated for the best supported model (Fig. 2). Although posterior estimates for the contemporary and historical Ne were broad, we observed a large, over 200-fold decrease in the current Ne in comparison to the historical estimate (Fig. 2a–c). Assuming a pronghorn generation time of approximately two years35, we inferred that the first historical decline occurred approximately 6000 (95% CI 2060–17,720) years ago, whereas second decline appears to date back to around 554 (95% CI 7–2420) years ago (Fig. 2d,e).

Posterior density curves and numerical estimates of demographic parameters for the best supported demographic scenario, which contains both a historical and a more recent reduction. (a) Historical effective population size; (b) effective population size before the recent demographic reduction; (c) current effective size of the captive peninsular pronghorn herd; (d) the number of generations since the historical demographic reduction; and (e) the number of generations since the recent demographic reduction. Panel (f) shows the mean, mode and 95% confidence intervals of each estimated demographic parameter.
Discussion
Our time-series genetic dataset represents a new resource for the conservation management of the Peninsular pronghorn and has produced at least two significant discoveries. First, we uncovered a gradual erosion of the genetic diversity of the captive herd over time, although this trend appears to have partially abated in response to a genetically informed breeding management. Second, we could show that the genetic diversity of the Peninsular pronghorn has been shaped by a combination of historical and recent demographic changes driven by the ecological transformation of the Baja California peninsula and by anthropogenic pressures including hunting and habitat destruction. Below, we discuss the relevance of these findings to the Peninsula pronghorn conservation.
Temporal changes in the genetic diversity of the captive herd
The Peninsular pronghorn experienced a severe decline over the last two centuries, from once being present across much of the Baja California peninsula to being functionally extinct in the wild51,52,59. This decline motivated the captive breeding program initiative44,53. In spite of early difficulties related to management, health problems and juvenile mortality64, by the end of 2021 and with approximately 700 individuals, the Peninsular Pronghorn Conservation Programme achieved this goal53. Until now, however, we lacked an understanding of how the last 13 years of captivity may have shaped the genetic composition of the captive herd.
Although we did not find any statistically significant temporal changes in several marker-based estimates of genetic diversity (Ar, He and Ho), a tendency for reduction was observed for He (9.8%, from 0.51 in 2009 to 0.46 in 2021) and Ho (16.3%, from 0.55 in 2009 to 0.46 in 2021). Furthermore, all three frequency-based estimates of individual heterozygosity (sMLH, IR and HL) revealed significant decreases in heterozygosity during the course of the study with, for example, sMLH falling by around 27% over the past 13 years. This pattern was mirrored by the TrioML based inbreeding coefficient, which showed a gradual reduction in the number of non-inbred and weakly inbred individuals and a concurrent increase in the number of moderately to highly inbred individuals over time. These findings are consistent with the theoretical expectation of zygosity being a function of the breeding system65. Thus, inbreeding directly reduces heterozygosity by increasing the proportion of homozygotes relative to random expectations, but only indirectly affects allelic richness. By contrast, genetic drift directly affects allelic diversity but only indirectly impacts heterozygosity66,67. Therefore, our results suggest that inbreeding is currently the predominant force shaping the genetic diversity of the Peninsular pronghorn.
The genetic management of captive populations has proven to be extremely effective in preventing the loss of genetic diversity and ameliorating the negative effects of inbreeding68,69,70. Accordingly, we found that the implementation of a genetically informed breeding management attempt that have begun in 2018 was associated with a slight reduction in the slope of the relationship between heterozygosity and time, as well as with a reduction in the proportion of highly inbred individuals. Furthermore, model-based predictions suggest that the 2021 offspring cohort is significantly more outbred than would be expected if no genetic management had been undertaken. Consequently, although it is still rather early to tell, our preliminary results suggest that this strategy might be beneficial in terms of mitigating inbreeding and the loss of heterozygosity.
Limitations of our study
While our results provide grounds for cautious optimism, a number of caveats should be born in mind. First, in our study, comparisons among different years were not always based on the same age class, with only fawns being sampled in 2012, 2016 and 2021, and adults being sampled in 2009 and 2018. However, the difficulty of handling captive pronghorn meant that it was not possible to exhaustively sample both age classes across all years. We therefore focused on sampling adults at the beginning of the study and in the year of the breeding management attempt in order to provide reference populations against which subsequent generations of offspring could be compared. Furthermore, estimates of inbreeding increased between adults sampled in 2009 and 2018, while the offspring cohorts sampled in 2012, 2016 and 2021 also showed trends of decreasing heterozygosity and increasing inbreeding over time.
A second important caveat is that our sample size of individuals was modest (n = 144) in comparison to the total size of the captive herd (approximately 700 individuals). Incomplete sampling may be particularly important regarding the outcomes of management actions, as the effect of breeding recommendations will not be fully realised if animals with unknown relationships are allowed to mate as well as animals of known kinship. Consequently, the reduction in the loss of diversity that we observed after the implementation of the genetically informed breeding attempt may be conservative in the sense that more thorough sampling might have produced an even better outcome. Regardless, given that it is not possible to provide breeding recommendations for unsampled individuals, a strong case can be made for increasing sampling rates into the future. This would help to further optimise partner selection and thereby ensure the best possible retention of genetic diversity and reduction of inbreeding. Larger sample sizes of genotyped individuals would also be beneficial for the ongoing monitoring of genetic changes within the captive population.
Third, previous studies of the American pronghorn subspecies have documented inbreeding depression for multiple traits from birth mass through fawn survival to body condition71. However, fitness data have not yet been systematically collected for the Peninsular pronghorn, precluding an analysis of the potential negative effects of inbreeding in this subspecies. Consequently, future studies should aim to quantify the magnitude of inbreeding depression in the captive Peninsula pronghorn population, as well as to evaluate whether temporal changes in the amount of inbreeding are associated with changes in the mean fitness of the population. The simplest approach for this would be to test for associations between heterozygosity and fitness components such as fawn body mass and survival, although it would be preferable to estimate inbreeding more reliably using population genomic approaches such as reduced representation sequencing72.
Fourth, over longer timescales, a handful of microsatellites cannot tell us very much about the nature or magnitude of functional genetic variation, although neutral genetic diversity should in general provide a rough indication of the adaptive potential of a given species73. Nonetheless, we believe that the recent sequencing of the pronghorn genome74 will facilitate more detailed investigation of how unintended selection and drift may have impacted the genetic composition of the Peninsula pronghorn. In particular, dense single nucleotide polymorphisms mapped to the reference genome could be used to characterize selective sweeps as well as runs of homozygosity, identical by descent haplotypes that are informative both about inbreeding and population history75. Furthermore, computational approaches have been developed to infer the presence of putatively deleterious alleles from whole genome resequencing data76,77. Application of these approaches to pronghorn would shed light on the mutation load and its relationship to historical demography both within and across species.
Finally, it should be born in mind that developing a larger breeding programme that extends to the entire captive pronghorn herd would bring significant challenges. Currently, the Peninsular pronghorn population is kept in large enclosures, with the smallest of these housing the pregnant females, spanning a total of around 100 ha. Therefore, the size of the pens places limits on the scope of intensive management actions. The captive handling of pronghorns also carries an increased risk of serious injury to the animals, especially as this species is susceptible to capture myopathy78,79,80. Finally, captive breeding and genetic management require access to financial resources81. Consequently, pronghorn managers will need to weigh all of the pros and cons to design and implement a genetic management programme that is optimized for this species and which is feasible given financial and logistical constraints. For example, group-based management actions may be a good alternative to individual-based management actions. Specifically, breeding females could be distributed among management units and among smaller pens within each unit. Small numbers of breeding males could subsequently be moved between pens to maintain gene flow. A single breeding male could potentially breed for several seasons in different pens and management units before being replaced35.
Historical demography
Characterizing the strength and timing of historical declines can provide insights into the causes of those declines and thereby help conservation practitioners to create conditions that promote population recovery82. Based on our demographic reconstruction of the Peninsular pronghorn, we inferred that the onset of the decline may have been linked to climatic changes at the end of the LGM and the ensuing desertification of the Baja California peninsula. This is not surprising given that the contraction of open woodlands and expansion of desert scrub after the last glaciation are believed to be responsible for multiple extinction events as well as shifts in the geographical distributions of many animal species on the Baja California peninsula83,84,85. Furthermore, droughts have been recognized as one of the most important factors affecting the recruitment, mortality and abundance of pronghorns in arid and semi-arid areas86,87,88. For example, a devastating drought in 2002 reduced the number of Sonoran pronghorns in the USA to just 21 animals, motivating a captive breeding initiative as well as the introduction of animals from Mexico48,89. Consequently, our findings are consistent with the argument that precipitation is one of the most important factors limiting the abundance and geographical distribution of pronghorn62.
Our demographic analysis also uncovered evidence for a more recent demographic decline dating back around five hundred years ago. This is supported by recent studies showing that Peninsular pronghorn numbers have decreased to fewer than 100–150 individuals over the past hundred years or so44,51. Our results therefore point towards a scenario involving two consecutive declines, the first mediated by climate related vegetational changes on the Baja California peninsula and the second driven by increasing anthropogenic pressures such as hunting, fencing and cattle ranching. Both climatic and anthropogenic stressors will likely continue to be significant threats to the Peninsular pronghorn over the coming decades44,53,62. In this regard, species abundance models could be a useful tool for identifying suitable areas for future reintroductions based on a combination of human threats and climatic projections. Moreover, genetic information could be used to optimally select individuals for release in such a way as to minimize inbreeding and maximise genetic diversity7,90.
To conclude, we investigated changes in heterozygosity and inbreeding over time in the captive Peninsular pronghorn herd and used demographic reconstruction to evaluate alternative hypotheses relating to the decline of this subspecies. We found that, although the captive population has become progressively more inbred over time, genetically informed management appears to have partially counteracted this trend. We could also show that the Peninsular pronghorn likely experienced a gradual, protracted decline with two consecutive phases linked respectively to environmental change and anthropogenic impacts. Although the Peninsular pronghorn still faces multiple threats, the success of the captive breeding programme at building a large and demographically stable population may hint at unexpected resilience, and is a clear testament to the success of ongoing protection measures.
Methods
Research permissions and ethical considerations
All samples were collected by the management team of the Peninsular Pronghorn Conservation Programme under the registration key DGVS‐UMA‐VL‐3755‐BC given to the management unit by the Mexican Secretariat of Environment and Natural Resources. All procedures were approved by the authorized personal of the Valle de los Cirios Flora and Fauna Protection Area and followed the guidelines of the American Society of Mammalogists (www.mammalsociety.org/uploads/committee_files/CurrentGuidelines.pdf, accessed 7 January 2022). This work did not require any approval from the ethical committee since no experiments on live animals were performed, aside from the routine tagging that was performed by trained personnel and according to the conservation programme internal schedule. All procedures were in compliance with the ARRIVE guidelines for how to report animal experiments91.
Sample collection
Tissue samples were collected from 144 peninsular pronghorn individuals by trained personnel during 2009–2021 from the Vizcaino Biosphere Reserve and Valle de los Cirios Flora and Fauna Protection Area (Supplementary Table S1 online). Small pieces of ear tissue were taken during the tagging and from deceased animals whenever those were found by the management team. Tissue samples were preserved in 100% ethanol at room temperature until processing. Samples were mainly taken from young individuals (newborns to animals up to 6 months of age) and occasionally from adults in 2012, 2016 and 2021, while in 2009 adults were sampled as a reference group. We additionally sampled adults in 2018 as part the breeding management attempt described above. Those individuals formed part of the breeding herd, with unrelated individuals preferentially selected as mating partners. Consequently, our sampling scheme spans 13 consecutive years out of the 23 years of the captive breeding programme.
Molecular techniques
DNA extractions were performed using DNeasy Blood and Tissue Kit (Qiagen Inc., Valencia CA, USA) following the manufacturer’s protocol. DNA concentration was determined using a NanoDrop2000 (Thermo Scientific™) and each extract was adjusted to a concentration of approximately 100 ng/µl. We amplified 16 microsatellite loci previously described for pronghorn92,93,94 using the M13 genotyping approach95. Additionally, 12 samples were independently re-genotyped at eight microsatellites in order to estimate our genotyping error rate. Polymerase chain reactions were carried out in an 11.5 µL volume containing 1 µL of the DNA template, 1 × buffer (Invitrogen, Carlsbad, California), 1 mM MgCl2, 0.2 mM deoxynucleoside triphosphates, 0.05% bovine serum albumin, 0.5 U of Taq DNA Polymerase (Invitrogen), and 0.5 µM of each primer. The polymerase chain reaction profile consisted of an initial denaturalization step at 95 °C for 5 min, followed by 35 cycles of 60 s each at 95 °C, annealing by ramping from 55 to 60 °C, followed by 60 s extension at 72 °C. Cycles were terminated with a final extension stage of 10 min at 72 °C. PCR products were resolved on an Applied Biosystems 3730XL capillary sequencer at the University of Arizona Gene Core Facility and alleles were scored using PeakScanner v1.0 (Applied Biosystems).
Data analysis
Genotypes were binned to size classes using FlexiBin96. After that, we imported the binned microsatellite data into the R environment97 (R version 4.1.2) and converted it into a GENIND object using adegenet98. We quantified the amount of missing data per locus and per individual using the R package poppr99 and removed all individuals that failed to genotype at four or more loci. We also used package PopGenReport100 to estimate null allele frequencies. To test for linkage disequilibrium (LD) between pairs of loci, we used poppr. For this analysis, we used the standardized index of association (rbarD)101 and the number of permutations was specified using the ‘sample = 999’ argument. We further tested for Hardy–Weinberg equilibrium using the pegas package102. Finally, in order to determine the uniqueness of the genotypes we used “mlg” function as implemented in poppr. All of the above analyses were performed on the complete dataset and separately for each year. Whenever multiple tests we used, the resulting p-values were adjusted for the false discovery rate (FDR) using the R package stats97.
Genetic diversity and summary statistics
The number of alleles (A), allelic richness (Ar), expected heterozygosity (He) and observed heterozygosity (Ho) were calculated for the full dataset and separately for each year using the R packages adegenet98 and hierfstat103. Multilocus heterozygosity was quantified as standardized multilocus heterozygosity (sMLH), internal relatedness (IR) and homozygosity weighted by locus (HL)104 using R packages Rhh105 and inbreedR105,106. We used COANCESTRY v. 1.0107 to calculate individual inbreeding coefficient using the TrioML method108, using 10,000 reference individuals and bootstrapping on 10,000 samples. Following Marshall et al.109, we designated inbreeding coefficients (f) of zero as ‘none’, below 0.125 as ‘low’, 0.125 ≥ f < 0.25 as ‘moderate’, and f ≥ 0.25 as ‘high’. Wilcox tests were then used to test for significant differences in the diversity indices using the R package stats. We also used generalized linear models (GLMs) to quantify the strength of diversity decline over the years (2009–2021, 2009–2018 and 2018–2021) using the R package lme4110. Finally, using the “predict” function in the R package stats and the slope of the GLM spanning 2009–2018, we determined the modeled value for each diversity estimate assuming that no genetically based breeding management attempt had been implemented.
Demographic reconstruction
To investigate the demographic history of the Peninsular pronghorn, we used approximate Bayesian computation (ABC) as implemented in DIYABC v. 2.063,111,112. For modeling alternative demographic histories and reconstructing demographic trajectories, we used data from 58 samples (fawns and adults) from 2016, which was the year represented by the largest number of individuals.
ABC allows the evaluation of alternative demographic scenarios, expressed as a stepwise series of population size changes, and then uses summary statistics from the observed and simulated datasets to estimate parameter values and to assess the relative support for each scenario. We first developed four alternative demographic models intended to describe plausible patterns of effective population size (Ne) change over time. The mutation rate was set to range between 1e−2 and 1e−5. Priors for the timing of events and the magnitude of changes of Ne (Supplementary Fig. S2 online and Supplementary Table S8 online) were based on prior knowledge of the factors likely shaping the demographic history of the species, including environmental change on the Baja California peninsula after the LGM, anthropogenically induced population reduction and the captive breeding programme42,60,61. The first scenario represented the null hypothesis of (a) constant Ne over time; the alternative scenarios invoked: (b) a recent reduction caused by overexploitation and habitat loss, (c) a historical reduction caused by the desertification of the Baja California peninsula, and (d) a combination of recent and historical reductions, expressed as a two-step model (Supplementary Fig. S2 online). After simulating one million datasets for each scenario, we used a polychotomous logistic regression procedure113 to estimate the posterior probability of each scenario based on the 1% of simulated data sets for each model that produced summary statistics closest to the observed values. The error rate was estimated using prior data space and the posterior distributions. The posterior error rate represents the proportion of wrongly identified scenarios over the 1000 test datasets63. Based on the best supported scenario, local linear regression was used to estimate the posterior distributions of the parameters. Specifically, a logit transformation of parameter values was performed and the 1% closest simulated datasets to the observed were used for regression and posterior parameter estimation113.
Data availability
Our microsatellite dataset is available from the corresponding author on request or from Zenodo repository, https://doi.org/10.5281/zenodo.6014746.
References
Butchart, S. H. M. et al. Global biodiversity: Indicators of recent declines. Science 328(5982), 1164–1168. https://doi.org/10.1126/science.1187512 (2010).
Dirzo, R. et al. Defaunation in the anthropocene. Science 345(6195), 401–406. https://doi.org/10.1126/science.1251817 (2014).
Bradshaw, C. J. A. et al. Underestimating the challenges of avoiding a ghastly future. Front. Conserv. Sci. https://doi.org/10.3389/fcosc.2020.615419 (2021).
Barnosky, A. D. et al. Has the Earth’s sixth mass extinction already arrived?. Nature 471(7336), 51–57. https://doi.org/10.1038/nature09678 (2011).
Ceballos, G. et al. Accelerated modern human-induced species losses: Entering the sixth mass extinction. Sci. Adv. https://doi.org/10.1126/sciadv.1400253 (2015).
Ceballos, G., Ehrlich, P. R. & Raven, P. H. Vertebrates on the brink as indicators of biological annihilation and the sixth mass extinction. Proc. Natl. Acad. Sci. U.S.A. 117(24), 13596–13602. https://doi.org/10.1073/pnas.1922686117 (2020).
McGowan, P. J., Traylor-Holzer, K. & Leus, K. IUCN guidelines for determining when and how ex situ management should be used in species conservation. Conserv. Lett. 10(3), 361–366. https://doi.org/10.1111/conl.12285 (2016).
Clout, M. N. & Merton, D. V. Saving the Kakapo: The conservation of the world’s most peculiar parrot. Bird Conserv. Int. 8(3), 281–296. https://doi.org/10.1017/s0959270900001933 (1998).
Milinkovitch, M. C. et al. Genetic analysis of a successful repatriation programme: Giant Galápagos tortoises. Proc. R. Soc. B Biol. Sci. 271(1537), 341–345. https://doi.org/10.1098/rspb.2003.2607 (2004).
Ryder, O. A. & Wedemeyer, E. A. A cooperative breeding programme for the Mongolian wild horse Equus przewalskii in the United States. Biol. Conserv. 22(4), 259–271. https://doi.org/10.1016/0006-3207(82)90021-0 (1982).
Mallinson, J. J. C. Conservation breeding programmes: An important ingredient for species survival. Biodivers. Conserv. 4(6), 617–635. https://doi.org/10.1007/bf00222518 (1995).
Seddon, P. J., Armstrong, D. P. & Maloney, R. F. Developing the science of reintroduction biology. Conserv. Biol. 21(2), 303–312. https://doi.org/10.1111/j.1523-1739.2006.00627.x (2007).
Bowkett, A. E. Recent captive-breeding proposals and the return of the ark concept to global species conservation. Conserv. Biol. 23(3), 773–776. https://doi.org/10.1111/j.1523-1739.2008.01157.x (2009).
Shan, L. et al. Large-scale genetic survey provides insights into the captive management and reintroduction of giant pandas. Mol. Biol. Evol. 31(10), 2663–2671. https://doi.org/10.1093/molbev/msu210 (2014).
Fischer, J. & Lindenmayer, D. An assessment of the published results of animal relocations. Biol. Conserv. 96(1), 1–11. https://doi.org/10.1016/s0006-3207(00)00048-3 (2014).
Christie, M. R., Marine, M. L., French, R. A. & Blouin, M. S. Genetic adaptation to captivity can occur in a single generation. Proc. Natl. Acad. Sci. U.S.A. 109(1), 238–242. https://doi.org/10.1073/pnas.1111073109 (2011).
Fraser, D. J. et al. Population correlates of rapid captive-induced maladaptation in a wild fish. Evol. Appl. 12(7), 1305–1317. https://doi.org/10.1111/eva.12649 (2018).
Ralls, K., Brugger, K. & Ballou, J. Inbreeding and juvenile mortality in small populations of ungulates. Science 206(4422), 1101–1103. https://doi.org/10.1126/science.493997 (1979).
Charlesworth, D. & Charlesworth, B. Inbreeding depression and its evolutionary consequences. Annu. Rev. Ecol. Evol. Syst. 18(1), 237–268. https://doi.org/10.1146/annurev.es.18.110187.001321 (1987).
Ralls, K., Ballou, J. D. & Templeton, A. Estimates of lethal equivalents and the cost of inbreeding in mammals. Conserv. Biol. 2(2), 185–193. https://doi.org/10.1111/j.1523-1739.1988.tb00169.x (1988).
Hedrick, P. W. & Kalinowski, S. T. Inbreeding depression in conservation biology. Annu. Rev. Ecol. Evol. Syst. 31(1), 139–162. https://doi.org/10.1146/annurev.ecolsys.31.1.139 (2000).
Frankham, R. Introduction to Conservation Genetics 2nd edn. (Cambridge University Press, 2010).
Laikre, L. Conservation genetics of Nordic carnivores: Lessons from zoos. Hereditas 130(3), 203–216. https://doi.org/10.1111/j.1601-5223.1999.00203.x (2004).
Gomendio, M., Cassinello, J. & Roldan, E. R. S. A comparative study of ejaculate traits in three endangered ungulates with different levels of inbreeding: Fluctuating asymmetry as an indicator of reproductive and genetic stress. Proc. R. Soc. B Biol. Sci. 267(1446), 875–882. https://doi.org/10.1098/rspb.2000.1084 (2000).
Swinnerton, K. J., Groombridge, J. J., Jones, C. G., Burn, R. W. & Mungroo, Y. Inbreeding depression and founder diversity among captive and free-living populations of the endangered pink pigeon Columba mayeri. Anim. Conserv. 7(4), 353–364. https://doi.org/10.1017/s1367943004001556 (2004).
Farquharson, K. A., Hogg, C. J. & Grueber, C. E. Offspring survival changes over generations of captive breeding. Nat. Commun. https://doi.org/10.1038/s41467-021-22631-0 (2021).
Kleiman, D. G., Thompson, K. V. & Baer, C. K. Wild Mammals in Captivity: Principles and Techniques for Zoo Management 2nd edn. (University of Chicago Press, 2021).
Ralls, K. & Ballou, J. D. Captive breeding and reintroduction. In Encyclopedia of Biodiversity (ed. Levin, S. A.) 662–667 (Academic Press, 2013). https://doi.org/10.1016/b978-0-12-384719-5.00268-9.
Reed, D. H. & Frankham, R. Correlation between fitness and genetic diversity. Conserv. Biol. 17(1), 230–237. https://doi.org/10.1046/j.1523-1739.2003.01236.x (2003).
Spielman, D., Brook, B. W. & Frankham, R. Most species are not driven to extinction before genetic factors impact them. Proc. Natl. Acad. Sci. U.S.A. 101(42), 15261–15264. https://doi.org/10.1073/pnas.0403809101 (2003).
Willi, Y., van Buskirk, J. & Hoffmann, A. A. Limits to the adaptive potential of small populations. Annu. Rev. Ecol. Evol. Syst. 37(1), 433–458. https://doi.org/10.1146/annurev.ecolsys.37.091305.110145 (2006).
Habel, J. C., Husemann, M., Finger, A., Danley, P. D. & Zachos, F. E. The relevance of time series in molecular ecology and conservation biology. Biol. Rev. 89(2), 484–492. https://doi.org/10.1111/brv.12068 (2013).
Araki, H., Cooper, B. & Blouin, M. S. Genetic effects of captive breeding cause a rapid, cumulative fitness decline in the wild. Science 318(5847), 100–103. https://doi.org/10.1126/science.1145621 (2007).
Purohit, D. et al. Genetic effects of long-term captive breeding on the endangered pygmy hog. PeerJ 9, e12212. https://doi.org/10.7717/peerj.12212 (2021).
Hahn, E. E. & Culver, M. Genetic diversity and structure in Arizona pronghorn following conservation efforts. Conserv. Sci. Pract. https://doi.org/10.1111/csp2.498 (2021).
Charlesworth, B. Effective population size and patterns of molecular evolution and variation. Nat. Rev. Gen. 10(3), 195–205. https://doi.org/10.1038/nrg2526 (2009).
Wang, J., Santiago, E. & Caballero, A. Prediction and estimation of effective population size. Heredity 117(4), 193–206. https://doi.org/10.1038/hdy.2016.43 (2016).
O’Gara, W., Yoakum, J. D. & McCabe, R. E. Pronghorn: Ecology and Managment (University Press of Colorado, 2004).
Janis, C. M., Scott, K. M. & Jacobs, L. L. Evolution of Tertiary Mammals of North America: Terrestrial Carnivores, Ungulates, and Ungulate like Mammals Vol. 1 (Cambridge University Press, 2005).
Nelson, E. W. Status of the Pronghorn Antelope, 1922–1924 (U.S Department Agriculture Bulletin, 1925).
O’Gara, B. W. & McCabe, R. E. From exploitation to conservation. In Pronghorn: Ecology and Management (eds O’Gara, B. W. & Yoakum, J. D.) 41–73 (University Press Colorado, 2004).
Cancino, J., Ortega-Rubio, A. & Sanchez-Pacheco, J. A. Status of an endangered subspecies: The peninsular pronghorn at Baja California. J. Arid Environ. 32(4), 463–467. https://doi.org/10.1006/jare.1996.0039 (1996).
Laliberte, A. S. & Ripple, W. J. Range contractions of North American carnivores and ungulates. Bioscience 54(2), 123–138. https://doi.org/10.1641/0006-3568 (2004).
Medellín, R. A. et al. History, ecology, and conservation of the pronghorn antelope, bighorn sheep, and black bear in Mexico. In Biodiversity, Ecosystems, and Conservation in Northern Mexico (eds Cartron, J.-L. et al.) 387–405 (Oxford University Press, 2005).
Lee, T. E., Bickham, J. W. & Scott, M. D. Mitochondrial DNA and allozyme analysis of North American pronghorn populations. J. Wildl. Manag. 58(2), 307–318. https://doi.org/10.2307/3809396 (1994).
IUCN SSC Antelope Specialist Group. Antilocapra americana ssp. peninsularis. The IUCN Red List of Threatened Species 2021: e.T1679A200726719. https://doi.org/10.2305/IUCN.UK.2021-2.RLTS.T1679A200726719.en (2021).
SEMARNAT. Norma Oficial Mexicana NOM-059-SEMARNAT-2010, Protección ambiental– Especies nativas de México de flora y fauna silvestres– Categorías de riesgo y especificaciones para su inclusión, exclusión o cambio– Lista de especies en riesgo. Diario Oficial de la Federación 30 diciembre (2010).
U. S. Fish and Wildlife Service. Recovery Plan for the Sonoran pronghorn (Antilocapra americana sonoriensis), Second Revision. (U.S. Fish and Wildlife Service, Southwest Region, Albuquerque, 2016).
Cancino, J., Sanchez-Sotomayor, V. & Castellanos, R. From the field: Capture, hand-raising, and captive management of peninsular pronghorn. Wildl. Soc. Bull. 33(1), 61–65. https://doi.org/10.2193/0091-7648 (2005).
Horne, J. S., Hervert, J. J., Woodruff, S. P. & Mills, L. S. Evaluating the benefit of captive breeding and reintroductions to endangered Sonoran pronghorn. Biol. Conserv. 196, 133–146. https://doi.org/10.1016/j.biocon.2016.02.005 (2016).
CONANP. Programa de Acción para la Conservación de la Especie: Berrendo (Antilocapra americana), 2009 año del berrendo. Secretaria del Medio Ambiente y Recursos Naturales (SEMARNAT). www.conanp.gob.mx (2009).
Cancino, J., Rodríguez-Estrella, R. & Miller, P. Using population viability analysis for management recommendations of the endangered endemic peninsular pronghorn. Acta Zool. Mex. 26(1), 173–189 (2010).
Danoff-Burg, J. A. & Mulroe, K. Peninsular Pronghorn Species Action Plan (2021) (in press).
Stephen, C. L. et al. Population genetic analysis of sonoran pronghorn (Antilocapra americana sonoriensis). J. Mammal. 86(4), 782–792. https://doi.org/10.1644/1545-1542 (2005).
Stephen, C. L., Whittaker, D. G., Gillis, D., Cox, L. L. & Rhodes, O. E. Genetic consequences of reintroductions: An example from oregon pronghorn antelope (Antilocapra americana). J. Wildl. Manag. 69(4), 1463–1474. https://doi.org/10.2193/0022-541x (2005).
Barnow-Meyer, K. & Byers, J. Genetic diversity and gene flow in Yellowstone Basin pronghorn (Antilocapra americana). UW Natl. Parks Serv. Res. Station Annu. Rep. 31, 65–72. https://doi.org/10.13001/uwnpsrc.2008.3705 (2008).
LaCava, M. E. F. et al. Pronghorn population genomics show connectivity in the core of their range. J. Mammal. 101(4), 1061–1071. https://doi.org/10.1093/jmammal/gyaa054 (2020).
Klimova, A., Munguia-Vega, A., Hoffman, J. I. & Culver, M. Genetic diversity and demography of two endangered captive pronghorn subspecies from the Sonoran Desert. J. Mammal. 95(6), 1263–1277. https://doi.org/10.1644/13-mamm-a-321 (2014).
Hahn, E. E., Klimova, A., Munguía-Vega, A., Clark, K. B. & Culver, M. Use of museum specimens to refine historical pronghorn subspecies boundaries. J. Wildl. Manag. 84(3), 524–533. https://doi.org/10.1002/jwmg.21810 (2020).
Axelrod, D. I. The evolution of desert vegetation in western North America. Carnegie Instit. Wash. Publ. 590, 215–306 (1950).
Dolby, G. A., Bennett, S. E. K., Lira-Noriega, A., Wilder, B. T. & Munguía-Vega, A. Assessing the geological and climatic forcing of biodiversity and evolution Surrounding the Gulf of California. J. Southwest. 57, 391–455. https://doi.org/10.1353/jsw.2015.0005 (2015).
Gedir, J. V., Cain, J. W., Harris, G. & Turnbull, T. T. Effects of climate change on long-term population growth of pronghorn in an arid environment. Ecosphere 6(10), art189. https://doi.org/10.1890/es15-00266.1 (2015).
Cornuet, J. M. et al. DIYABC v2.0: A software to make approximate Bayesian computation inferences about population history using single nucleotide polymorphism, DNA sequence and microsatellite data. Bioinformatics 30(8), 1187–1189. https://doi.org/10.1093/bioinformatics/btt763 (2014).
Islas-Espinoza, M. & de las Heras, A. Peninsular pronghorn conservation: Too many paradigms, too few indicators. In Sustainability Indicators in Practice (eds Latawiec, A. & Agol, D.) 126–145 (De Gruyter Open Poland, 2015). https://doi.org/10.1515/9783110450507-012.
Willoughby, J. R. et al. The impacts of inbreeding, drift and selection on genetic diversity in captive breeding populations. Mol. Ecol. 24(1), 98–110. https://doi.org/10.1111/mec.13020 (2014).
Crow, J. F. & Kimura, M. An Introduction in Population Genetics Theory (Harper and Row, 1970).
Falconer, D. S. Introduction to Quantitative Genetics 3rd edn. (Longman Scientific and Technical, 1989).
Ballou, J. D. Strategies for maintaining genetic diversity in captive populations through reproductive technology. Zoo Biol. 3(4), 311–323. https://doi.org/10.1002/zoo.1430030404 (1984).
Ballou, J. D. & Lacy, R. C. Identifying genetically important individuals for management of genetic diversity in pedigreed populations. In Population Management for Survival and Recovery (eds Ballou, J. D. et al.) 76–111 (Columbia Press, 1995).
Montgomery, M. E. et al. Minimizing kinship in captive breeding programs. Zoo Biol. 16(5), 377–389. https://doi.org/10.1002/(sici)1098-2361 (1997).
Dunn, S. J., Clancey, E., Waits, L. P. & Byers, J. A. Inbreeding depression in pronghorn (Antilocapra americana) fawns. Mol. Ecol. 20(23), 4889–4898. https://doi.org/10.1111/j.1365-294x.2011.05327.x (2011).
Hoffman, J. I. et al. High-throughput sequencing reveals inbreeding depression in a natural population. Proc. Natl. Acad. Sci. U.S.A. 111(10), 3775–3780. https://doi.org/10.1073/pnas.1318945111 (2014).
Kardos, M. et al. The crucial role of genome-wide genetic variation in conservation. Proc. Natl. Acad. Sci. U.S.A. 118(48), e2104642118. https://doi.org/10.1073/pnas.2104642118 (2021).
Zoonomia Consortium. A comparative genomics multitool for scientific discovery and conservation. Nature 587(7833), 240–245. https://doi.org/10.1038/s41586-020-2876-6 (2020).
Ceballos, F. C., Joshi, P. K., Clark, D. W., Ramsay, M. & Wilson, J. F. Runs of homozygosity: Windows into population history and trait architecture. Nat. Rev. Genet. 19(4), 220–234. https://doi.org/10.1038/nrg.2017.109 (2018).
Supple, M. A. & Shapiro, B. Conservation of biodiversity in the genomics era. Genome Biol. https://doi.org/10.1186/s13059-018-1520-3 (2018).
Hohenlohe, P. A. & Rajora, O. P. Population Genomics: Wildlife (Springer, 2020).
Chalmers, G. A. & Barrett, M. W. Capture myopathy in pronghorns in Alberta, Canada. J. Am. Vet. Med. Assoc. 171(9), 918–923 (1977).
Sotelo-Gallardo, H., Contreras Balderas, A. J. & Espinosa Treviño, A. Comparación de dos métodos de liberación del berrendo, Antilocapra americana (Artiodactyla: Antilocapridae) en Coahuila, México. Rev. Biol. Trop. 65(3), 1208. https://doi.org/10.15517/rbt.v65i3.29447 (2017).
Breed, D. et al. Conserving wildlife in a changing world: Understanding capture myopathy—A malignant outcome of stress during capture and translocation. Conserv. Physiol. https://doi.org/10.1093/conphys/coz027 (2019).
Snyder, N. F. et al. Limitations of captive breeding in endangered species recovery. Conserv. Biol. 10(2), 338–348. https://doi.org/10.1046/j.1523-1739.1996.10020338.x (1996).
Bonebrake, T. C., Christensen, J., Boggs, C. L. & Ehrlich, P. R. Population decline assessment, historical baselines, and conservation. Conserv. Lett. 3(6), 371–378. https://doi.org/10.1111/j.1755-263x.2010.00139.x (2010).
Grismer, L. L. & McGuire, J. A. The oases of central Baja California, Mexico. Part I. A preliminary account of the relict mesophilic herpetofauna and the status of the oases. Bull. South. Calif. Acad. Sci. 92, 2–24 (1993).
Welsh, H. H., Clark, W. H., Franco-Vizcaíno, E. & Valdéz-Villavicencio, J. H. Herpetofauna associated with palm oases across the Californian-Sonoran transition in Northern Baja California, Mexico. Southwest. Nat. 55(4), 581–585. https://doi.org/10.1894/pas-15.1 (2010).
Mann, D. H., Groves, P., Gaglioti, B. V. & Shapiro, B. A. Climate-driven ecological stability as a globally shared cause of Late Quaternary megafaunal extinctions: The Plaids and Stripes Hypothesis. Biol. Rev. 94(1), 328–352. https://doi.org/10.1111/brv.12456 (2018).
Brown, D. E., Warnecke, D. & McKinney, T. Effects of midsummer drought on mortality of doe pronghorn (Antilocapra americana). Southwest. Nat. 51(2), 220–225. https://doi.org/10.1894/0038-4909 (2006).
Simpson, D. C., Harveson, L. A., Brewer, C. E., Walser, R. E. & Sides, A. R. Influence of precipitation on pronghorn demography in Texas. J. Wildl. Manag. 71(3), 906–910. https://doi.org/10.2193/2005-753 (2007).
McKinney, T., Brown, D. E. & Allison, L. Winter precipitation and recruitment of pronghorns in Arizona. Southwest. Nat. 53(3), 319–325. https://doi.org/10.1894/cj-147.1 (2008).
Otte, A. Partners save the Sonoran pronghorn. Endang. Species Bull. 31, 22–23 (2006).
McCullough, D. R. & Barrett, R. H. Wildlife 2001: Populations (Springer, 1992).
Percie Du Sert, N. et al. Reporting animal research: Explanation and elaboration for the ARRIVE guidelines 20. PLOS Biol. 18(7), e3000411. https://doi.org/10.1371/journal.pbio.3000411 (2020).
Carling, M. D., Passavant, C. W. & Byers, J. A. DNA microsatellites of pronghorn (Antilocapra americana). Mol. Ecol. Not. 3(1), 10–11. https://doi.org/10.1046/j.1471-8286.2003.00334.x (2002).
Dunn, S. J. et al. Ten polymorphic microsatellite markers for pronghorn (Antilocapra americana). Conserv. Genet. Resour. 2(1), 81–84. https://doi.org/10.1007/s12686-009-9166-9 (2010).
Munguia-Vega, A., Klimova, A. & Culver, M. New microsatellite loci isolated via next-generation sequencing for two endangered pronghorn from the Sonoran Desert. Conserv. Genet. Resour. 5(1), 125–127. https://doi.org/10.1007/s12686-012-9749-8 (2012).
Boutin-Ganache, I., Raposo, M., Raymond, M. & Deschepper, C. F. M13-Tailed primers improve the readability and usability of microsatellite analyses performed with two different allele-sizing methods. Biotechniques 31(1), 25–28. https://doi.org/10.2144/01311bm02 (2001).
Amos, W. et al. Automated binning of microsatellite alleles: Problems and solutions. Mol. Ecol. Not. 7(1), 10–14. https://doi.org/10.1111/j.1471-8286.2006.01560.x (2006).
R Core Team. R: A Language and Environment for Statistical Computing (R Foundation for Statistical Computing, 2021). https://www.R-project.org/.
Jombart, T. & Ahmed, I. Adegenet 1.3–1: New tools for the analysis of genome-wide SNP data. Bioinformatics 27(21), 3070–3071. https://doi.org/10.1093/bioinformatics/btr521 (2011).
Kamvar, Z. N., Tabima, J. F. & Grünwald, N. J. Poppr: An R package for genetic analysis of populations with clonal, partially clonal, and/or sexual reproduction. PeerJ 2, e281. https://doi.org/10.7717/peerj.281 (2014).
Adamack, A. T. & Gruber, B. PopGenReport: Simplifying basic population genetic analyses in R. Methods Ecol. Evol. 5(4), 384–387. https://doi.org/10.1111/2041-210x.12158 (2014).
Agapow, P. M. & Burt, A. Indices of multilocus linkage disequilibrium. Mol. Ecol. Not. 1(1–2), 101–102. https://doi.org/10.1046/j.1471-8278.2000.00014.x (2001).
Paradis, E. pegas: An R package for population genetics with an integrated-modular approach. Bioinformatics 26(3), 419–420. https://doi.org/10.1093/bioinformatics/btp696 (2010).
Goudet, J. hierfstat, a package for r to compute and test hierarchical F-statistics. Mol. Ecol. Not. 5(1), 184–186. https://doi.org/10.1111/j.1471-8286.2004.00828.x (2005).
Aparicio, J. M., Ortego, J. & Cordero, P. J. What should we weigh to estimate heterozygosity, alleles or loci?. Mol. Ecol. 15(14), 4659–4665. https://doi.org/10.1111/j.1365-294x.2006.03111.x (2006).
Alho, J. S., Välimäki, K. & Merilä, J. Rhh: An R extension for estimating multilocus heterozygosity and heterozygosity–heterozygosity correlation. Mol. Ecol. Res. 10(4), 720–722. https://doi.org/10.1111/j.1755-0998.2010.02830.x (2010).
Stoffel, M. A. et al. inbreedR: An R package for the analysis of inbreeding based on genetic markers. Methods Ecol. Evol. 7(11), 1331–1339. https://doi.org/10.1111/2041-210x.12588 (2016).
Wang, J. Coancestry: A program for simulating, estimating and analysing relatedness and inbreeding coefficients. Mol. Ecol. Res. 11(1), 141–145. https://doi.org/10.1111/j.1755-0998.2010.02885.x (2010).
Wang, J. Triadic IBD coefficients and applications to estimating pairwise relatedness. Genet. Res. 89(3), 135–153. https://doi.org/10.1017/s0016672307008798 (2007).
Marshall, T. C. et al. Estimating the prevalence of inbreeding from incomplete pedigrees. Proc. R. Soc. B Biol. Sci. 269(1500), 1533–1539. https://doi.org/10.1098/rspb.2002.2035 (2002).
Bates, D., Mächler, M., Bolker, B. & Walker, S. Fitting linear mixed-effects models using lme4. J. Stat. Softw. https://doi.org/10.18637/jss.v067.i01 (2015).
Beaumont, M. A., Zhang, W. & Balding, D. J. Approximate Bayesian computation in population genetics. Genetics 162(4), 2025–2035. https://doi.org/10.1093/genetics/162.4.2025 (2002).
Bertorelle, G., Benazzo, A. & Mona, S. ABC as a flexible framework to estimate demography over space and time: Some cons, many pros. Mol. Ecol. 19(13), 2609–2625. https://doi.org/10.1111/j.1365-294x.2010.04690.x (2010).
Fagundes, N. J. R. et al. Statistical evaluation of alternative models of human evolution. Proc. Natl. Acad. Sci. U.S.A. 104(45), 17614–17619. https://doi.org/10.1073/pnas.0708280104 (2007).
Acknowledgements
This work was partially funded by the Espacios Naturales y Desarrollo Sustentable A.C. (ENDESU). Support for the Article Processing Charge was granted by the Deutsche Forschungsgemeinschaft and the Open Access Publication Fund of Bielefeld University. We are thankful to the management team of Valle de los Cirios Flora and Fauna Protected Area, in particular to José Martín Gutiérrez Perea and Fernando Escoto Rodríguez. We are also thankful to Dr. Santiago Sanchez-Ramirez for his valuable comments that helped improve this manuscript.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
A.K. and J.I.H. designed the study; A.K and J.N.G.R. conducted the lab work and data analyses, A.K. and J.I.H. wrote the manuscript; A.K, J.N.G.R. and V.S.S. commented on and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Klimova, A., Gutiérrez-Rivera, J., Sánchez-Sotomayor, V. et al. The genetic consequences of captive breeding, environmental change and human exploitation in the endangered peninsular pronghorn. Sci Rep 12, 11253 (2022). https://doi.org/10.1038/s41598-022-14468-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-14468-4
- Springer Nature Limited