
Abstract
Tobacco use is the leading preventable cause of cancer, and affects the respiratory, oral, fecal, and duodenal mucosa-associated microbiota. However, the effects of smoking on the duodenal luminal microbiome have not been studied directly. We aimed to compare the duodenal luminal microbiome in never-smokers, current smokers, and ex-smokers who quit ≥ 10 years ago. In a cross-sectional study, current smokers (CS, n = 24) were identified and matched to never-smokers (NS, n = 27) and ex-smokers (XS, n = 27) by age (± 5 years), body mass index (BMI, ± 3 kg/m2), and sex. Current antibiotic users were excluded. The duodenal luminal microbiome was analysed in 1 aspirate sample per subject by 16S rRNA gene sequencing. Relative abundances (RA) of families associated with increased duodenal microbial diversity, Prevotellaceae, Neisseriaceae, and Porphyromonadaceae, were significantly lower in CS vs. NS. This was driven by lower RA of unknown Prevotella and Porphyromonas species, and Neisseria subflava and N. cinerea, in CS. In contrast, RA of Enterobacteriaceae and Lactobacillaceae (associated with decreased diversity), were significantly higher in CS, due to higher RA of Escherichia-Shigella, Klebsiella and Lactobacillus species. Many of these changes were absent or less pronounced in XS, who exhibited a duodenal luminal microbiome more similar to NS. RA of taxa previously found to be increased in the oral and respiratory microbiota of smokers were also higher in the duodenal luminal microbiome, including Bulledia extructa and an unknown Filifactor species. In conclusion, smoking is associated with an altered duodenal luminal microbiome. However, ex-smokers have a duodenal luminal microbiome that is similar to never-smokers.
Similar content being viewed by others


Introduction
Tobacco use is the leading preventable cause of cancer and cancer deaths1,2,3. Despite increased awareness of these facts, smoking still causes 480,000 deaths per year in the United States alone2. Cancers of the lung, in particular non-small cell cancer, and of the mouth, larynx, esophagus, stomach, kidney, pancreas, liver, bladder, cervix, colon and rectum, and acute myeloid leukemia are associated with tobacco smoking2,4. An increased prevalence of emphysema, chronic bronchitis, asthma and other pulmonary specific diseases is also seen in long-term smokers5,6,7. Tobacco smoking is also linked to non-pulmonary conditions such as cardiovascular disease8,9, cerebrovascular disease10, and diabetes11, as well as reproductive issues12. Smoking also exerts a detrimental effect on the gastrointestinal tract, impacting the mucosal immune response and increasing the risk for Crohn’s disease (CD)13.
Given the relationship between smoking and the respiratory system, it is not surprising that smoking has been shown to affect the respiratory tract microbiota. Although human data are sparse, smoking has been suggested to increase the relative abundance of the genera Veillonella and Megasphaera in the upper respiratory tract microbiome14. Clinical studies of respiratory tract microbiota in smoking-related conditions such as lung cancer have shown variable results, likely due to significant differences in the microbiota of the upper and lower respiratory tract15, as well as differences in sampling and sequencing techniques among studies. However, a recent meta-analysis suggests increased abundances of the phyla Actinobacteria and Firmicutes in the respiratory microbiota of lung cancer patients, including lower relative abundance of the genus Prevotella, and higher Streptococcus, when compared to healthy controls16. Studies in mice have also shown that exposure to cigarette smoke significantly alters murine respiratory microbiota, with decreases in the genera Lactobacillus, Kluyvera, and Nesterenkonia and increases in Trichococcus and Escherichia-Shigella17,18.
Human studies using stool samples have shown that smoking also affects the fecal microbiome, although the results of these studies are variable. For example, Lee et al. found decreased relative abundance of the phyla Firmicutes and Proteobacteria, and increased relative abundance of Bacteroidetes, in the fecal microbiome of smokers19. Further, these changes may be significantly mitigated following smoking cessation20.
Within the gastrointestinal tract, the small intestine is primarily responsible for digestion and nutrient absorption, and plays important roles in immune regulation and maintaining gut barrier integrity21,22,23. We have previously shown that stool is a poor surrogate for the small bowel microbiome24. However, the small intestine can only be accessed through invasive and expensive techniques such as endoscopy. As a result, few studies of the small intestinal microbiome have been performed to date, and only one explored the impact of smoking on the small intestine25. Shanahan et al. examined the effects of smoking on the duodenal mucosa-associated microbiome using duodenal biopsies 25, and found that current smokers exhibited significantly decreased bacterial diversity and increased relative abundance of phylum Firmicutes, including species from the genera Streptococcus and Veillonella, as well as increased relative abundance of the genus Rothia, and decreased relative abundance of the genera Prevotella and Neisseria25.
It is known that gut mucosa-associated and luminal microbial populations differ from each other, and fulfill different roles26,27,28. For example, the mucosal microbiota are in close contact with gut epithelial cells, lending to roles in nutrient uptake and immune function, whereas luminal microbes are more involved in digestion29,30. In addition, the mucous in the luminal microbiome harbours Gram negative bacteria, including Gram negative species from phylum Proteobacteria which we have consistently found to be disruptors of the small intestinal luminal microbiome31,32,33, but the viscosity of this mucus has been a barrier to fully characterizing luminal microbial populations. We recently developed and validated techniques to optimize DNA yields and 16S rRNA gene sequencing for small intestinal luminal aspirates that break down this mucus, rendering its microbial content more accessible34, and have used these in a series of studies to characterize changes in the small intestinal microbiome in small intestinal bacterial overgrowth (SIBO)31, with age and the aging process32, and with proton pump inhibitor (PPI) use35, amongst others.
In this study, we used these validated techniques to characterize and compare the composition of the duodenal luminal microbiome in never-smokers, current smokers, and ex-smokers who had not smoked in 10 or more years.
Results
Demographics
Duodenal aspirates from a total of 78 REIMAGINE subjects were included in this study. These included current smokers (CS, N = 24), ex-smokers who had not smoked in 10 or more years (XS, N = 27), and never-smokers (NS, N = 27). Groups were matched by sex (number of males/females), age (± 5 years), and body mass index (BMI, ± 3) (Table 1). Current smokers who provided data reported an average of 17.59 ± 17.44 pack-years of smoking, and ex-smokers who provided data reported an average of 11.16 ± 21.43 pack-years of smoking (see Table 1). The average number of years since quitting for ex-smokers was 26 ± 11.48 years. No subjects were taking antibiotics at the time of endoscopy. Proton pump inhibitor (PPI) use was significantly higher in the CS and XS groups when compared to NS (P = 0.03, Table 1). When the primary reasons for endoscopy were compared between groups, biliary disease, a category which includes pancreatitis, was listed for 7 subjects in the CS group, and further exploration of their charts revealed that these subjects had acute pancreatitis, recurrent pancreatitis, or a history of pancreatitis. These findings indicated an increased prevalence of pancreatitis when compared to never-smokers (P = 0.02, OR: 10.94, 95% Cl: 1.56–127.20). In contrast, the prevalence of pancreatitis was not significantly increased in ex-smokers who had quit smoking for 10 or more years when compared to never-smokers (P = 0.35, OR: 3.41, 95%Cl: 0.47–45.79) (Table 1). None of the other reasons for endoscopy were significantly different among groups.
Differences in the duodenal luminal microbiome in current smokers compared to never-smokers
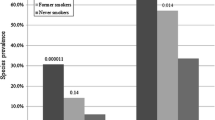
The average retained and identified reads with 97% similarity considering the SILVA database (after quality filtering and removal of chimeric reads) did not differ between groups (NS = 199,134; CS = 169,834 and XS = 180,206; P = 0.58). While there were no significant differences in overall microbial alpha and beta diversity in the duodenal luminal microbiome of the CS group when compared to the NS and XS groups (Supplemental Fig. 1A, B), specific taxonomic differences were identified between the groups. At the phylum level, the duodenal luminal microbiome in aspirate samples from all three groups (NS, CS, and XS) was dominated by Firmicutes (~ 63%), followed by the phyla Proteobacteria (~ 13%), Actinobacteria (10%), Bacteroidetes (~ 6%), Fusobacteria (~ 5%), and Patescibacteria (~ 1%) (Fig. 1A). The relative abundance of phylum Bacteroidetes was lower in the CS group (~ 3%) than in the NS group (~ 9%) (fold change (FC) = −3.78, P = 2.2E-3, false discovery rate-adjusted (FDR-adj.) P = 0.01, Fig. 1B). Although the same trend was observed when the XS group was compared to the NS group (FC = −1.68, P = 0.22, FDR-adj. P = 0.52), the relative abundance of Bacteroidetes in the duodenal luminal microbiome of the XS group (~ 5%) was closer to that in the NS group (~ 9%, Fig. 1B).

(A) Relative abundance of different phyla in the duodenal luminal microbiome of never-smokers (NS), current smokers (CS), and ex-smokers (XS). (B) Log-transformed abundance of phylum Bacteroidetes in the duodenal luminal microbiome of NS (red), CS (green), and XS (blue).
The common duodenal luminal microbiome was defined as the most widespread components of the microbiome found across at least 40% of subjects. At the family level, this common duodenal luminal microbiome was significantly different in the CS group when compared to the NS group (Fig. 2A,B). The prevalences of certain duodenal families were found to be positively associated with microbial diversity, including Prevotellaceae (phylum Bacteroidetes), Neisseriaceae (phylum Proteobacteria) and Porphyromonadaceae (phylum Bacteroidetes) (Fig. 3A), and the relative abundances of these families were lower in the CS group than in the NS group (Prevotellaceae FC = −2.80, P = 0.03, FDR-adj. P = 0.05, Neisseriaceae FC = −3.41, P = 9.45E-3, FDR-adj. P = 0.05, and Porphyromonadaceae FC = −6.53, P = 5.36E-4, FDR-adj. P = 4.37E-3) (Fig. 4, Supplemental Table 1). As a result, Porphyromonadaceae was no longer part of the common duodenal luminal microbiome in the CS group (Fig. 2B). The prevalences of other duodenal microbial families were found to be associated with decreased microbial diversity, including Enterobacteriaceae (phylum Proteobacteria) and Lactobacillaceae (phylum Firmicutes) (Fig. 3B). The relative abundance of family Enterobacteriaceae was higher in the duodenal microbiome of the CS group than in the NS group (Enterobacteriaceae FC = 14.77, P = 1.32E-4, FDR-adj. P = 1.37E-3, Supplemental Table 1). The relative abundance of family Lactobacillaceae also trended towards being higher in the CS group vs. the NS group, but the P-value reached significance only when a non-parametric statistical test was used (FC = 2.08, Mann–Whitney P = 0.036) (Fig. 4, Supplemental Table 1).

The common duodenal luminal microbiome at the family level in never-smokers (A), current smokers (B) and ex-smokers (C).

Spearman associations between duodenal microbial families in never-smokers (A), current smokers (B), and ex-smokers (C).

Relative abundance of different families in the duodenal luminal microbiome of never-smokers (NS), current smokers (CS), and ex-smokers (XS). Families Enterobacteriaceae and Lactobacillaceae are highlighted.
Analysis of Spearman correlations suggested that the families Enterobacteriaceae and Lactobacillaceae had negative associations with most families in the common duodenal luminal microbiome of the CS group, including Porphyromonadaceae, Prevotellaceae and Neisseriaceae (Fig. 3B, Supplemental Fig. 2), and that there were significantly more negative associations among families overall in the CS group than in the NS group (Supplemental Fig. 2).
Differences in the duodenal luminal microbiome are reduced in ex-smokers
Many of the differences seen in the duodenal luminal microbiome of the CS group when compared to the NS group were not seen or were less pronounced in the XS group. As a result, the duodenal luminal microbiome profile in the XS group was more similar to that of the NS group (Fig. 2C, Supplemental Table 1). This included similar relative abundances of the disruptors Enterobacteriaceae and Lactobacillaceae (Supplemental Fig. 3) and of families associated with duodenal microbial diversity (Porphyromonadaceae, Prevotellaceae and Neisseriaceae). Analysis of Spearman correlations suggested a higher number of positive associations between families in the XS group (Fig. 3C, Supplemental Fig. 3). Of note, the XS group in this study was limited to subjects who had quit smoking 10 or more years ago.
Differences are seen in duodenal luminal genera and species in current smokers, never-smokers, and ex-smokers
Analyses at the genus and species levels revealed that the higher relative abundance of family Enterobacteriaceae in the duodenal luminal microbiome of the CS group appeared to be primarily driven by the Gram-negative genera Escherichia-Shigella and Klebsiella. The relative abundance of an unknown species from genus Escherichia-Shigella was 20.51-fold higher in the duodenal microbiome of the CS group compared to the NS group (FDR-adj. P = 1.20E-3), but was lower in the XS group compared to CS (FC = −7.68, FDR-adj. P = 1.06E-9) (Supplemental Table 1). As a result, the relative abundance of this unknown Escherichia-Shigella species was even lower in the XS group than in the NS group (FC = −3.33, FDR-adj. P = 0.02). The relative abundance of the unknown species from genus Klebsiella was significantly higher in the CS group when compared to the NS group (FC = 9.9, FDR-adj. P = 0.02, Supplemental Table 1), and was lower in the XS group compared to the CS group (FC = −3.40, FDR-adj. P = 0.02). There was no significant difference in the relative abundance of this species in the XS group when compared to the NS group (P = 0.1, Supplemental Table 1).
The higher relative abundance of family Lactobacillaceae (phylum Firmicutes) in the CS group was driven by several species from the genus Lactobacillus, which was 4.81-fold higher in the CS group when compared to the NS group (Fig. 5A, FDR-adj. P = 0.01, Supplemental Table 1). The top 3 species identified as most likely to be Lactobacillus that had higher relative abundance in the CS group when compared to NS group (of a total of 11) were L. panis (FC = 6.4, FDR-adj. P = 2.65E-7), L. oris (FC = 6.88, FDR-adj. P = 4.62E-7) and L. murinus (FC = 7.28, FDR-adj. P = 2.38E-6). In addition, the relative abundance of genus Lactobacillus tended to be higher in the CS group when compared to the XS group, but this did not reach statistical significance (FC = 2.17, P = 0.06, FDR-adj. P = 0.28, Fig. 5A, Supplemental Table 1). There was no significant difference in the relative abundance of genus Lactobacillus in the XS group when compared to the NS group (Fig. 5B, P = 0.1, FDR-adj. P = 0.36).

Log-transformed abundances of family Lactobacillaceae (A) and genus Lactobacillus (B) in the duodenal luminal microbiome of never-smokers (NS) (red), current smokers (CS) (green), and ex-smokers (XS) (blue).
Changes in family Prevotellaceae were partially due to an unknown species from genus Prevotella, the relative abundance of which was significantly lower in the CS group relative to the NS group (FC = −19.6, FDR-adj. P = 9.84E-7), but interestingly, was higher in the XS group when compared to the CS group (FC = 10.81, FDR-adj. P = 7.15E-3, Supplemental Fig. 4, Supplemental Table 1). The relative abundance of this unknown Prevotella species in the XS group was closer to that in the NS group (FC = −1.81, FDR-adj. P = 0.02, Supplemental Fig. 4, Supplemental Table 1).
The lower relative abundance of family Neisseriaceae in the duodenal luminal microbiome in the CS group vs. the NS group was partially due to two commensal species, Neisseria subflava (FC = −4.17, FDR-adj. P = 8.36E-4) and N. cinerea (FC = −5.46, FDR-adj. P = 8.50E-4) (Supplemental Table 1). The relative abundances of these species were not significantly different in the XS group when compared to the NS group (N. cinerea FC = −1.33, P = 0.11, FDR-adj. P = 0.44; N. subflava FC = −1.66, P = 0.07, FDR-adj. P = 0.34) (Supplemental Fig. 5, Supplemental Table 1). Of note, Neisseria species were classified using Greengenes Database 2013 because these species could not be identified using the SILVA Database v132.
The relative abundance of family Porphyromonadaceae, which was part of the core duodenal luminal microbiome in the NS group, fell out of the core duodenal microbiome in the CS group (Fig. 2B) and in the XS group (Fig. 2C). This was partially due to lower relative abundance of an unknown species from genus Porphyromonas. The relative abundance of this unknown Porphyromonas species was lower in the CS group relative to the NS group (FC = −3.86, FDR-adj. P = 9.15E-5), and was also lower in the XS group when compared to the NS group (FC = −2.03, P = 0.01, FDR-adj. P = 0.08).
In addition to Porphyromonas species, the relative abundances of other taxa from phylum Bacteroidetes were lower in the CS group relative to the NS group, and were also lower in the XS group, 10 or more years after quitting smoking. These included genus Chryseobacterium (family Weeksellaceae), which was lower in the CS group than in the NS group (FC = −4.06, P = 3.26E-4, FDR-adj. P = 2.64E-3, Supplemental Table 1), but was not significantly different in the XS group when compared to the NS group (P = 0.69, FDR-adj. P = 1, Supplemental Table 1).
Although no statistical differences were observed in Kyoto Encyclopedia of genes and Genomes (KEGG) predicted microbial metabolic pathways when analyses were performed across all three groups and between group pairs (Supplemental Tables 2 to 5), discovery analysis based on microbial pathways enriched in the CS group as compared to the NS group revealed a pattern associated with toluene and xylene degradation (linear discriminant analysis (LDA) scores > 2) (Supplemental Fig. 6).
The duodenal luminal microbiome of current smokers has a microbial signature similar to the oral and respiratory microbiota reported in smoking-related conditions
As the duodenal luminal microbiome can be colonized by a variety of oral and respiratory microbes, and given the direct relationship between tobacco smoking, the oral and respiratory microbiomes17,18,36, and the development of certain human diseases5,6,7, microbial taxa previously found to be associated with smoking-related diseases were also quantitated in the duodenal aspirates from our subjects.
The relative abundance of species Bulleidia extructa (phylum Firmicutes), which is five times more prevalent in the subgingival microbiome of subjects with smoking-associated periodontitis than in healthy controls37, was significantly higher in the duodenal luminal microbiome of the CS group when compared to the NS group (FC = 4.55, FDR-adj. P = 1.00E-4). While the relative abundance of B. extructa was lower in the XS group than in the CS group (FC = −2.47, P = 0.01, FDR-adj. P = 0.09), it was still significantly higher in the XS group than in the NS group (FC = 3.25, FDR-adj. P = 7.74E-3, Supplemental Table 1).
In addition, the relative abundance of an unknown species from genus Filifactor (phylum Firmicutes), which is more abundant in the subgingival microbiome of smokers vs. never-smokers in subjects with chronic moderate periodontitis38, was also higher in the duodenal luminal microbiome of the CS group compared to the NS group (FC = 2.21, P = 0.04, FDR-adj. P = 0.2), and lower in the XS group when compared to the CS group (FC = −5.88, FDR-adj. P = 2.03E-6, Supplemental Table 1). The relative abundance of this species was also lower in the XS group when compared to the NS group (FC = −3.67, FDR-adj. P = 4.87E-3, Supplemental Table 1).
Discussion
In this study, we demonstrate that the duodenal luminal microbiome in current smokers is significantly different from that in never-smokers. Given the critical roles played by small intestinal microbes in digestion, nutrient absorption, immune regulation, and maintenance of gut barrier integrity23,39, coupled with the fact that an estimated 942 million men and 175 million women ages 15 or older are current smokers40, these findings suggest that smoking may have implications for metabolic and gastrointestinal health beyond the conditions currently recognized. Interestingly, our findings also indicate that many, but not all, of the alterations in the duodenal luminal microbiome in current smokers were less pronounced or absent in ex-smokers who had stopped smoking 10 or more years earlier, which may suggest that the effects of smoking are lessened after 10 or more years of stopping smoking.
Although there were no phylum-level changes that reached significance between the three groups (current smokers (CS), never-smokers (NS), and ex-smokers who had not smoked in 10 or more years (XS)), there was a trend towards lower relative abundance of Bacteroidetes in the CS group when compared to the NS group, and to a lesser extent in the XS group when compared to the NS group. Analysis at the family level led to a number of interesting and statistically significant findings. We found that the core duodenal luminal microbiome, i.e. the most widespread components of the microbiome found in all subjects, was significantly altered in the CS group when compared to the NS group. Specifically, we found that certain bacterial families were positively correlated with microbial diversity, including Porphyromonadaceae (phylum Bacteroidetes), Prevotellaceae (phylum Bacteroidetes), and Neisseriaceae (phylum Proteobacteria), and that the relative abundances of all three of these families were significantly lower in current smokers than in never-smokers. The lower relative abundances of these three families appeared to be driven primarily by higher relative abundances of taxa from two other bacterial families, Lactobacillaceae (phylum Firmicutes) and Enterobacteriaceae (phylum Proteobacteria), and were associated with a greater number of negative associations between microbial families in current smokers. Our studies of the small intestinal luminal microbiota have consistently shown that increased relative abundance of both Enterobacteriaceae and Lactobacillaceae can disrupt microbial diversity and significantly impact microbial profiles 31,32,33, and our current findings further support this.
An encouraging finding was that many of the negative changes in the duodenal luminal microbiome identified in current smokers appear to be significantly less pronounced or absent in ex-smokers who had not smoked in 10 or more years. As a result, the duodenal microbial profile in the XS group was more similar to the NS group than the CS group, with fewer negative associations between families. At the family level, there were no significant differences in the overall relative abundances of Prevotellaceae, Neisseriaceae or Porphyromonadaceae in the XS group when compared to the NS group. The relative abundances of the disruptive genera Klebsiella and Lactobacillus (most likely L. panis, L. oris, and L. murinus) were higher in the CS group, but were not significantly different in the XS group when compared to the NS group. The relationship between these findings and the documented mitigation of smoking-associated disease risks in ex-smokers1,2,3 remains to be determined.
The relative abundances of unknown species from the genera Prevotella and Porphyromonas were lower in both the CS and the XS groups than in the NS group, and the relative abundance of Bulledia extructa was higher in both the CS and the XS groups than in the NS group, suggesting that these might represent permanent changes in the duodenal microbiome of former smokers. The relative abundance of the disruptive genus Escherichia-Shigella was even lower in the XS group than in the NS. We hypothesized that this may represent a reconfiguration of the small intestinal luminal microbiome in ex-smokers who have quit for 10 or more years. As subjects taking antibiotics were excluded from the study, this is not an antibiotic effect, but further explorations are required to determine the reasons for this difference.
It is increasingly understood that there is considerable bi-directional cross-talk between the gut and the lungs, referred to as the ‘gut-lung axis’, and that gut microbiota can influence the lungs and lung immune responses through the effects of microbial components (such as lipopolysaccharides (LPS)) and metabolites (such as short chain fatty acids (SCFA))41,42. Supporting this, individuals with gut conditions such as inflammatory bowel disease (IBD) have increased prevalence of pulmonary diseases43, and individuals with chronic obstructive pulmonary disease (COPD), cystic fibrosis (CF), and asthma have increased prevalence of gastrointestinal conditions including irritable bowel syndrome (IBS)44,45. Interestingly, some of the microbes we found to be altered in the duodenal luminal microbiome of the CS group have previously been shown to be altered in the respiratory microbiota of smokers and individuals with pulmonary conditions46. For example, the relative abundance of the genera Neisseria and Porphyromonas is lower both in the duodenal luminal microbiome of our CS group and in the respiratory microbiome of smokers46. Genus Prevotella, which is highly abundant in the respiratory microbiome46, was lower in the CS group and is also noted to be decreased in the respiratory microbiome of lung cancer patients16 and the fecal microbiome of tuberculosis patients47. Lastly, we identified higher relative abundance of Enterobacteriaceae and an Escherichia-Shigella species (likely E. coli) in the duodenal luminal microbiome of our CS group, both of which are also higher in the gut microbiota of mice with respiratory influenza infections48. Taken together, these results demonstrate that some of the microbial changes we identified in the duodenal luminal microbiome of smokers parallel differences seen in respiratory and fecal microbiota of smokers and individuals with lung cancer and other pulmonary conditions, as well as in animal models, and may warrant further investigation.
There are also consistencies between our findings and changes in the oral microbiota in smokers36. We found that the relative abundance of B. extructa (phylum Firmicutes) and an unknown species from genus Filifactor (phylum Firmicutes) was higher in the CS group, both of which are associated with periodontitis and further increased in the subgingival microbiome of smokers with periodontitis37,38. The relative abundance of Klebsiella was also higher in the CS group, which is consistent with an early study which suggested that cigarette smoking promoted the growth of Gram-negative bacteria, including Klebsiella species, in the oral cavity49, and another study which found that K. pneumoniae was the second most commonly identified bacterial species in sputum of smokers with lung cancer that develop pneumonia50. Lastly, the relative abundance of Lactobacillus was higher in the CS group, and is also higher in the oral microbiome of current smokers36. Interestingly, changes in the small bowel microbiome in the CS group were linked to changes in predicted microbial pathways associated with toluene and xylene degradation, two volatile organic compounds (VOCs) present in tobacco smoke51. These findings may suggest that smoking has effects on the microbiome along an extended length of the gastrointestinal tract.
Like the lungs, the duodenum was thought until recently to have relatively low microbial biomass52. In fact, the true biomass is hard to determine as the viscosity of small intestinal samples creates challenges for microbial recovery, especially when using kits designed for stool samples. To combat this, we recently developed and validated novel techniques that facilitate increased recovery of microbes from small intestinal luminal aspirates, as well as improved DNA isolation and library preparation for 16S rRNA sequencing34. Through the use of these techniques, we have shown that there are greater numbers of microbes in duodenal luminal fluid than previously thought, and that the microbial populations in duodenal luminal fluid are significantly different from those in stool24,33. Moreover, luminal microbial populations also differ in the different segments of the small intestine24.
Shanahan et al.25 previously examined the effects of smoking on the duodenal mucosa-associated microbiome, and there are similarities and differences between those findings and our findings regarding the duodenal luminal microbiome. Shanahan et al. found significantly lower bacterial diversity in the duodenal mucosa-associated microbiome, as well as higher relative abundance of phylum Firmicutes, and lower relative abundance of Bacteroidetes and Actinobacteria, in current smokers vs. never-smokers25, whereas we did not find any significant differences in overall microbial alpha and beta diversity in the duodenal luminal microbiome of current smokers when compared to never-smokers and ex-smokers. We also did not find any significant differences at the phylum level, although there was a trend towards lower relative abundance of phylum Bacteroidetes in the CS group. At the genus level, Shanahan et al.25 found significantly higher relative abundance of the genera Streptococcus, Veillonella, and Rothia in duodenal mucosa-associated microbiome of current smokers vs. never-smokers, but we did not find significant differences in these genera in the duodenal luminal microbiome. We found higher relative abundances of Lactobacillus, and the Gram-negative genera Escherichia-Shigella and Klebsiella, in the duodenal luminal microbiome of current smokers, but Shanahan et al. did not report alterations in these genera in the duodenal mucosa-associated microbiome25. These differences are not unexpected, given the differences in luminal vs. mucosal microbial populations previously noted26,27. Importantly, there were also a number of consistent findings, including the lower relative abundance of Prevotella and Neisseria in current smokers vs. never-smokers, although the similar relative abundances of Lactobacillus and Klebsiella species we found in ex-smokers and never-smokers in our cohort were not reported by Shanahan et al.25. All of the ex-smokers in our cohort had quit smoking 10 or more years ago, which may explain why these and other taxa we found to be altered in current smokers were more similar in ex-smokers and never-smokers.
Our study has limitations. The subjects were undergoing upper endoscopy for a variety of reasons, including assessment of intestinal complaints as well as for screening purposes due to familial and other risk factors, and may not be fully representative of normal healthy individuals. Sample sizes were also limited, which may have affected some of the post correction tests applied to P-values during metagenomic and pathway prediction analyses, as a large sample size is usually required to achieve good power while controlling for false positives at a low level (i.e. low false discovery rate (FDR))53. In addition, this was a cross-sectional study with a single sample per subject. Current smokers were matched to ex-smokers and never-smokers by age, BMI, and gender, antibiotic users were excluded, and other potential influences on the gut microbiome, including PPI use and reasons for endoscopy, were also compared between groups. However, we cannot rule out other potential confounders that could affect the microbiome over time in each of the groups, and larger studies would be required to address this fully. PPI use was significantly higher in the CS and XS groups when compared to the NS group, which is consistent with previous findings of increased prevalence of PPI use amongst smokers54,55. However, we did not find any significant differences in those small intestinal luminal taxa we have previously identified as affected by PPI use35, suggesting that our current findings are not driven by PPI use. Apart from the higher prevalence of pancreatitis in current smokers, which is consistent with the fact that smoking is a known risk factor for pancreatitis56, there were no other significant differences in primary diagnosis/reason for endoscopy between the groups.
In conclusion, this study demonstrates that smoking affects the duodenal luminal microbiome, and many of the differences in the duodenal luminal microbiome in current smokers and never-smokers are not seen, or are significantly less pronounced, in ex-smokers. We found that smoking is associated with significantly higher relative abundances of the disruptive genera Escherichia-Shigella, Klebsiella and Lactobacillus, as well as lower relative abundance of genera associated with microbial diversity such as Prevotella and Neisseria, both of which are also lower in the duodenal mucosa-associated microbiome25. In addition, some of the taxa we identified as having higher relative abundance in the duodenal luminal microbiome of smokers are similar to alterations in the oral microbiota of smokers, particularly smokers with periodontitis, as well as alterations in the respiratory and fecal microbiota of smokers, lung cancer patients, and patients with various respiratory conditions, raising the possibility that they may represent perturbations of the gut-lung axis in smokers. While many of the duodenal luminal microbiome differences seen in current smokers were not seen in ex-smokers who had stopped smoking 10 or more years ago, some appeared to be permanent. While these findings may have implications for small intestinal functions in metabolic and gastrointestinal health, the clinical implications of these changes remain to be determined.
Methods
Study subjects
Subjects for this study were identified from the REIMAGINE (Revealing the Entire Intestinal Microbiota and its Associations with the Genetic, Immunologic, and Neuroendocrine Ecosystem) study. REIMAGINE is an ongoing study to explore the impact of alterations in the small bowel microbiota on different conditions and diseases. Subjects between the ages of 18–85 years old undergoing a standard of care upper endoscopy (esophagogastroduodenoscopy – EGD) without colon preparation are eligible for inclusion31,34. The study protocol is approved by the Institutional Review Board at Cedars-Sinai Medical Center (Pro00035192), and all subjects provide informed written consent prior to participation.
REIMAGINE subjects complete a detailed questionnaire following the EGD. This questionnaire includes self-documented family and medical history, and includes smoking history24,31,34. All medical information provided by subjects is verified by previous medical records. Subjects who completed the smoking section of the questionnaire were eligible for inclusion in this study. Based on their responses, current smokers (CS), never-smokers (NS), and ex-smokers (XS) who had not smoked in 10 or more years were identified. The cut-off of 10 or more years for ex-smokers was chosen as the U.S. Department of Health and Human Services suggests that the risks for cancers of the bladder, esophagus, or kidney decrease within 10 years of quitting smoking, and the risk for lung cancer decreases by 50% within 10–15 years of quitting smoking1. Groups were matched by age (± 5 years), body mass index (BMI) (± 3 kg/m2), and sex. All data were de-identified prior to analysis. Subjects were excluded if current antibiotic use was reported or if subjects had used antibiotics within 6 months prior the procedure.
Duodenal luminal samples
Luminal fluid was aspirated from the duodenum during the EGD procedure, using a sterile aspiration double-lumen catheter (Hobbs Medical, Inc.) specifically designed for small bowel aspiration34. To minimize the chance of contamination from the mouth, esophagus, and stomach, the endoscopist collected duodenal aspirates using a sterile inner catheter that was manipulated through a sterile bone wax cap only after reaching the second portion of the duodenum34.
Aspirate processing
Duodenal aspirates were mixed with an equal volume of sterile 1X dithiothreitol (DTT) and vortexed for about 30 s until liquified as described previously34. The samples were then centrifuged at maximum speed (> 13,000 RPM) for 5 min, followed by the removal of the supernatant. 1 mL of sterile All Protect reagent (Qiagen, Hilden, Germany) was added to the microbial pellet. The pellets under All Protect were then stored at −80 °C until the DNA extraction was performed34.
DNA extraction and isolation
The duodenal aspirate pellets were thawed on ice and processed as previously described34. Briefly, microbial DNA was isolated from each sample using the MagAttract PowerSoil DNA KF Kit (Qiagen) on a KingFisher Duo (Thermo Fisher Scientific, Waltham, MA, USA)34. DNA quantification was carried out using the Qubit™ ds high sensitivity DNA BR Assay kit (Invitrogen by Thermo Fisher Scientific) on a Qubit™ 4 Fluorometer (Invitrogen, Carlsbad, CA, USA).
Library preparation and 16S rRNA sequencing and analysis
The 16S V3 and V4 regions were amplified with specific primers (S-D-Bact-0341-b-S-17 and S-D-Bact-0785-a-A-21)57 modified to include Illumina sequencing adapters34. Library quality was determined on an Agilent 2100 Bioanalyzer System. The MiSeq System (Illumina, San Diego, California) was used to perform paired-end sequencing of amplicons (2 × 301 cycles) with the MiSeq reagent v3 Kit with 600 cycles and 5% to 10% PhiX (Illumina). 16S V3 and V4 libraries from blank samples were also sequenced, as previously described34.
Sequencing and statistical analysis
CLC Genomic Workbench v. 10.1.1 and CLC Microbial Genomics Module v. 2.5 (Qiagen) software was used for Operational Taxonomic Unit (OTU) clustering and taxonomic analysis against the SILVA database v132 with 97% similarity. Greengene Database 2013 v.13_8 was used only where species level classifications were not available in the SILVA database. Default parameters were used for minimum occurrences (2) and chimera crossover cost (3), and the creation of new OTUs was not allowed. Low depth samples (< 5,000 sequences per sample) were removed from the analysis. Microbial alpha diversity indices including Shannon’s, Simpson’s, and Observed were calculated after low depth samples were removed from the analysis. Inter-sample variability (beta diversity) was calculated using the Bray–Curtis metric using MicrobiomeAnalyst58,59 tools.
Predictions for significant differentially abundant OTUs between different groups were performed using a rarefied OTU table. Multiple comparisons and statistical analyses were performed using CLC Microbial Genomics Module v.2.5 (Qiagen) and MicrobiomeAnalyst58,59. The OTU table was rarefied to the minimal number of reads assigned to a sample, and a Negative Binomial GLM model was used to obtain maximum likelihood estimates for the fold change (FC) of a log-transformed OTU between different groups. The Wald test was used for determination of significance, and P-values were corrected using False Discovery Rate (FDR). PERMANOVA analysis with Bonferroni-corrected P-values (permutations: 99,999) was used to compare beta diversity differences between groups. FC were log-transformed. A P-value < 0.05 after controlling for FDR or Bonferroni was considered to be statistically significant. Two-tailed Spearman r correlations, Mann–Whitney tests, and graph construction were carried out with rarefied OTU tables using GraphPad Prism 7.02 (GraphPad Software, La Jolla, CA, USA) and IBM SPSS Statistics Version. Fisher’s Exact test was used to determine associations between categorical variables between different groups using GraphPad Prism 7.02 (GraphPad Software).
Core microbiome (common microbiome) analysis and correlation constructions were performed using the Core Microbiome tool and correlation network tool available at MicrobiomeAnalyst58,59. Relative abundances were calculated based of the percentage of a given taxon in the total taxa identified in the duodenal luminal microbiome. The core microbiome at phylum level and family level was calculated based on the most common phyla in at least 40% of samples. Correlations and associations between microbial taxa were generated based on Spearman rank correlation, using a correlation threshold of at least 0.3 and P-values lower than 0.05. Microbial metabolic pathways were analyzed based on Kyoto Encyclopedia of genes and Genomes (KEGG) collection databases, using PICRUSt260. Microbial predicted pathways were compared across all three groups using the Kruskal–Wallis test and between two groups using the Mann–Whitney test. P-values were adjusted using FDR. Association analyses were also performed using the globaltest algorithm61 available at MicrobiomeAnalyst. Linear Discriminant Analysis (LDA) Effective Size (LEfSe)62 (Galaxy Version 1.0) was used to discover metagenomic biomarkers for predicted pathways.
Ethics approval
This study was performed in accordance with the principles of the Declaration of Helsinki. The study was approved by the Institutional Review Board at Cedars-Sinai Medical Center (Pro00035192).
Consent to participate
Informed written consent was obtained from all individual participants included in the study.
Data availability
The datasets generated during this study are available at the National Center for Biotechnology Information (NCBI) BioProject Repository (https://www.ncbi.nlm.nih.gov/bioproject) under BioProject ID PRJNA732897.
References
U.S. Department of Health and Human Services. Smoking Cessation. A Report of the Surgeon General. (Atlanta, GA, 2020).
Office on Smoking and Health, National Center for Chronic Disease Prevention and Health Promotion. Tobacco-Related Mortality (2020).
Schabath, M. B. & Cote, M. L. Cancer progress and priorities: Lung cancer. Cancer Epidemiol. Biomark. Prev. 28, 1563–1579. https://doi.org/10.1158/1055-9965.Epi-19-0221 (2019).
Office on Smoking and Health, National Center for Chronic Disease Prevention and Health Promotion. 2014 Surgeon General's Report: The Health Consequences of Smoking—50 Years of Progress (2014).
Tubío-Pérez, R. A., Torres-Durán, M., Pérez-Ríos, M., Fernández-Villar, A. & Ruano-Raviña, A. Lung emphysema and lung cancer: What do we know about it?. Ann. Transl. Med. 8, 1471. https://doi.org/10.21037/atm-20-1180 (2020).
Forey, B. A., Thornton, A. J. & Lee, P. N. Systematic review with meta-analysis of the epidemiological evidence relating smoking to COPD, chronic bronchitis and emphysema. BMC Pulm. Med. 11, 36. https://doi.org/10.1186/1471-2466-11-36 (2011).
Stapleton, M., Howard-Thompson, A., George, C., Hoover, R. M. & Self, T. H. Smoking and asthma. J. Am. Board Fam. Med. 24, 313–322. https://doi.org/10.3122/jabfm.2011.03.100180 (2011).
Burns, D. M. Epidemiology of smoking-induced cardiovascular disease. Prog. Cardiovasc. Dis. 46, 11–29. https://doi.org/10.1016/s0033-0620(03)00079-3 (2003).
Gallucci, G., Tartarone, A., Lerose, R., Lalinga, A. V. & Capobianco, A. M. Cardiovascular risk of smoking and benefits of smoking cessation. J. Thorac. Dis. 12, 3866–3876. https://doi.org/10.21037/jtd.2020.02.47 (2020).
Roy, A., Rawal, I., Jabbour, S. & Prabhakaran, D. in Cardiovascular, Respiratory, and Related Disorders (2017).
Chang, S. A. Smoking and type 2 diabetes mellitus. Diabetes Metab. J. 36, 399–403. https://doi.org/10.4093/dmj.2012.36.6.399 (2012).
Soares, S. R. & Melo, M. A. Cigarette smoking and reproductive function. Curr. Opin. Obstet. Gynecol. 20, 281–291. https://doi.org/10.1097/GCO.0b013e3282fc9c1e (2008).
Berkowitz, L. et al. Impact of cigarette smoking on the gastrointestinal tract inflammation: Opposing effects in Crohn’s disease and ulcerative colitis. Front. Immunol. 9, 74. https://doi.org/10.3389/fimmu.2018.00074 (2018).
Lim, M. Y. et al. Analysis of the association between host genetics, smoking, and sputum microbiota in healthy humans. Sci. Rep. 6, 23745. https://doi.org/10.1038/srep23745 (2016).
Goddard, A. F. et al. Direct sampling of cystic fibrosis lungs indicates that DNA-based analyses of upper-airway specimens can misrepresent lung microbiota. Proc. Natl. Acad. Sci. U.S.A. 109, 13769–13774. https://doi.org/10.1073/pnas.1107435109 (2012).
Zheng, X., Sun, X., Liu, Q., Huang, Y. & Yuan, Y. The composition alteration of respiratory microbiota in lung cancer. Cancer Invest. 38, 158–168. https://doi.org/10.1080/07357907.2020.1732405 (2020).
Li, K. J. et al. Dysbiosis of lower respiratory tract microbiome are associated with inflammation and microbial function variety. Respir. Res. 20, 272. https://doi.org/10.1186/s12931-019-1246-0 (2019).
Zhang, R. et al. Effects of smoking on the lower respiratory tract microbiome in mice. Respir. Res. 19, 253. https://doi.org/10.1186/s12931-018-0959-9 (2018).
Lee, S. H. et al. Association between Cigarette smoking status and composition of gut microbiota: Population-based cross-sectional study. J. Clin. Med. 7, 282. https://doi.org/10.3390/jcm7090282 (2018).
Biedermann, L. et al. Smoking cessation induces profound changes in the composition of the intestinal microbiota in humans. PLoS ONE 8, e59260. https://doi.org/10.1371/journal.pone.0059260 (2013).
Ko, H.-J. & Chang, S.-Y. Regulation of intestinal immune system by dendritic cells. Immune Netw. 15, 1–8. https://doi.org/10.4110/in.2015.15.1.1 (2015).
Rios, D. et al. Antigen sampling by intestinal M cells is the principal pathway initiating mucosal IgA production to commensal enteric bacteria. Mucosal Immunol. 9, 907–916. https://doi.org/10.1038/mi.2015.121 (2016).
Burgueno, J. F. & Abreu, M. T. Epithelial Toll-like receptors and their role in gut homeostasis and disease. Nat. Rev. Gastroenterol. Hepatol. 17, 263–278. https://doi.org/10.1038/s41575-019-0261-4 (2020).
Leite, G. G. S. et al. Mapping the segmental microbiomes in the human small bowel in comparison with stool: A REIMAGINE study. Dig. Dis. Sci. 65, 2595–2604. https://doi.org/10.1007/s10620-020-06173-x (2020).
Shanahan, E. R. et al. Influence of cigarette smoking on the human duodenal mucosa-associated microbiota. Microbiome 6, 150. https://doi.org/10.1186/s40168-018-0531-3 (2018).
Ringel, Y. et al. High throughput sequencing reveals distinct microbial populations within the mucosal and luminal niches in healthy individuals. Gut Microbes 6, 173–181. https://doi.org/10.1080/19490976.2015.1044711 (2015).
Wu, M. et al. The differences between luminal microbiota and mucosal microbiota in mice. J. Microbiol. Biotechnol. 30, 287–295. https://doi.org/10.4014/jmb.1908.08037 (2020).
Yasuda, K. et al. Biogeography of the intestinal mucosal and lumenal microbiome in the rhesus macaque. Cell Host Microbe 17, 385–391. https://doi.org/10.1016/j.chom.2015.01.015 (2015).
Eckburg, P. B. et al. Diversity of the human intestinal microbial flora. Science 308, 1635–1638. https://doi.org/10.1126/science.1110591 (2005).
Ko, H. J. & Chang, S. Y. Regulation of intestinal immune system by dendritic cells. Immune Netw 15, 1–8. https://doi.org/10.4110/in.2015.15.1.1 (2015).
Leite, G. et al. The duodenal microbiome is altered in small intestinal bacterial overgrowth. PLoS ONE 15, e0234906. https://doi.org/10.1371/journal.pone.0234906 (2020).
Leite, G. et al. Age and the aging process significantly alter the small bowel microbiome. Cell Rep. 36, 109765. https://doi.org/10.1016/j.celrep.2021.109765 (2021).
Barlow, J. T. et al. Quantitative sequencing clarifies the role of disruptor taxa, oral microbiota, and strict anaerobes in the human small-intestine microbiome. Microbiome 9, 214. https://doi.org/10.1186/s40168-021-01162-2 (2021).
Leite, G. G. S. et al. Optimizing microbiome sequencing for small intestinal aspirates: Validation of novel techniques through the REIMAGINE study. BMC Microbiol. 19, 239. https://doi.org/10.1186/s12866-019-1617-1 (2019).
Weitsman, S. et al. Effects of proton pump inhibitors on the small bowel and stool microbiomes. Dig. Dis. Sci. https://doi.org/10.1007/s10620-021-06857-y (2021).
Yang, Y. et al. Cigarette smoking and oral microbiota in low-income and African-American populations. J. Epidemiol. Community Health 73, 1108–1115. https://doi.org/10.1136/jech-2019-212474 (2019).
Camelo-Castillo, A. J. et al. Subgingival microbiota in health compared to periodontitis and the influence of smoking. Front. Microbiol. 6, 119. https://doi.org/10.3389/fmicb.2015.00119 (2015).
Moon, J. H., Lee, J. H. & Lee, J. Y. Subgingival microbiome in smokers and non-smokers in Korean chronic periodontitis patients. Mol. Oral Microbiol. 30, 227–241. https://doi.org/10.1111/omi.12086 (2015).
Kiela, P. R. & Ghishan, F. K. Physiology of intestinal absorption and secretion. Best Pract. Res. Clin. Gastroenterol. 30, 145–159. https://doi.org/10.1016/j.bpg.2016.02.007 (2016).
Office on Smoking and Health, National Center for Chronic Disease Prevention and Health Promotion. Current Cigarette Smoking Among Adults in the United States. (2020).
Dang, A. T. & Marsland, B. J. Microbes, metabolites, and the gut-lung axis. Mucosal Immunol. 12, 843–850. https://doi.org/10.1038/s41385-019-0160-6 (2019).
Budden, K. F. et al. Emerging pathogenic links between microbiota and the gut-lung axis. Nat. Rev. Microbiol. 15, 55–63. https://doi.org/10.1038/nrmicro.2016.142 (2017).
Wang, H. et al. Gut-lung crosstalk in pulmonary involvement with inflammatory bowel diseases. World J. Gastroenterol. 19, 6794–6804. https://doi.org/10.3748/wjg.v19.i40.6794 (2013).
Rutten, E. P. A., Lenaerts, K., Buurman, W. A. & Wouters, E. F. M. Disturbed intestinal integrity in patients with COPD: Effects of activities of daily living. Chest 145, 245–252. https://doi.org/10.1378/chest.13-0584 (2014).
Roussos, A., Koursarakos, P., Patsopoulos, D., Gerogianni, I. & Philippou, N. Increased prevalence of irritable bowel syndrome in patients with bronchial asthma. Respir. Med. 97, 75–79. https://doi.org/10.1053/rmed.2001.1409 (2003).
Morris, A. et al. Comparison of the respiratory microbiome in healthy nonsmokers and smokers. Am. J. Respir. Crit. Care Med. 187, 1067–1075. https://doi.org/10.1164/rccm.201210-1913OC (2013).
Luo, M. et al. Alternation of gut microbiota in patients with pulmonary tuberculosis. Front. Physiol. 8, 822. https://doi.org/10.3389/fphys.2017.00822 (2017).
Wang, J. et al. Respiratory influenza virus infection induces intestinal immune injury via microbiota-mediated Th17 cell-dependent inflammation. J. Exp. Med. 211, 2397–2410. https://doi.org/10.1084/jem.20140625 (2014).
Ertel, A., Eng, R. & Smith, S. M. The differential effect of cigarette smoke on the growth of bacteria found in humans. Chest 100, 628–630. https://doi.org/10.1378/chest.100.3.628 (1991).
Heo, J. W. et al. Smoking is associated with pneumonia development in lung cancer patients. BMC Pulm. Med. 20, 117. https://doi.org/10.1186/s12890-020-1160-8 (2020).
Chambers, D. M., Ocariz, J. M., McGuirk, M. F. & Blount, B. C. Impact of cigarette smoking on volatile organic compound (VOC) blood levels in the U.S. population: NHANES 2003–2004. Environ. Int. 37, 1321–1328. https://doi.org/10.1016/j.envint.2011.05.016 (2011).
Sender, R., Fuchs, S. & Milo, R. Revised estimates for the number of human and bacteria cells in the body. PLoS Biol. 14, e1002533. https://doi.org/10.1371/journal.pbio.1002533 (2016).
Tong, T. & Zhao, H. Practical guidelines for assessing power and false discovery rate for a fixed sample size in microarray experiments. Stat Med 27, 1960–1972. https://doi.org/10.1002/sim.3237 (2008).
Kim, J. J., Jang, E. J., Park, J. & Sohn, H. S. Association between proton pump inhibitor use and risk of fracture: A population-based case-control study. PLoS ONE 15, e0235163. https://doi.org/10.1371/journal.pone.0235163 (2020).
Hvid-Jensen, F. et al. Lifestyle factors among proton pump inhibitor users and nonusers: A cross-sectional study in a population-based setting. Clin. Epidemiol. 5, 493–499. https://doi.org/10.2147/CLEP.S49354 (2013).
Aune, D., Mahamat-Saleh, Y., Norat, T. & Riboli, E. Tobacco smoking and the risk of pancreatitis: A systematic review and meta-analysis of prospective studies. Pancreatology 19, 1009–1022. https://doi.org/10.1016/j.pan.2019.09.004 (2019).
Klindworth, A. et al. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 41, e1. https://doi.org/10.1093/nar/gks808 (2013).
Chong, J., Liu, P., Zhou, G. & Xia, J. Using MicrobiomeAnalyst for comprehensive statistical, functional, and meta-analysis of microbiome data. Nat Protoc 15, 799–821. https://doi.org/10.1038/s41596-019-0264-1 (2020).
Dhariwal, A. et al. MicrobiomeAnalyst: A web-based tool for comprehensive statistical, visual and meta-analysis of microbiome data. Nucleic Acids Res 45, W180–W188. https://doi.org/10.1093/nar/gkx295 (2017).
Douglas, G. M. et al. PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 38, 685–688. https://doi.org/10.1038/s41587-020-0548-6 (2020).
Goeman, J. J., van de Geer, S. A., de Kort, F. & van Houwelingen, H. C. A global test for groups of genes: Testing association with a clinical outcome. Bioinformatics 20, 93–99. https://doi.org/10.1093/bioinformatics/btg382 (2004).
Segata, N. et al. Metagenomic biomarker discovery and explanation. Genome Biol. 12, R60. https://doi.org/10.1186/gb-2011-12-6-r60 (2011).
Acknowledgements
The authors thank Dr Nipaporn Pichetshote and Dr Bianca Chang for their assistance in obtaining samples, and Dr Maria Jesus Villanueva-Milan, Shreya Celly, Maritza Sanchez, Stacy Weitsman MS, and Walter Morales for their assistance with sample processing and analysis. We would also like to thank the Monica Lester Charitable Trust and the Elias, Genevieve, and Georgianna Atol Charitable Trust for their generous support of our research program.
Funding
This study was supported in part by funds from the Monica Lester Charitable Trust and the Elias, Genevieve, and Georgianna Atol Charitable Trust.
Author information
Authors and Affiliations
Contributions
G.L., M.P. and R.M. conceived and designed the study. A.F. and A.H. recruited subjects and performed clinical procedures, and RM, AR and MP supervised the clinical studies and obtained informed consent. G.L., G.P., G.B., and M.L.P. performed laboratory experiments. G.L., M.P. and J.W. analysed the data. G.L., A.H., A.F., G.P., M.L.P., J.W., and G.B. prepared the original draft, and G.L., G.B., M.P., and R.M. reviewed and edited the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Leite, G., Barlow, G.M., Hosseini, A. et al. Smoking has disruptive effects on the small bowel luminal microbiome. Sci Rep 12, 6231 (2022). https://doi.org/10.1038/s41598-022-10132-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-10132-z
- Springer Nature Limited
This article is cited by
-
When smoke meets gut: deciphering the interactions between tobacco smoking and gut microbiota in disease development
Science China Life Sciences (2024)
-
Dysbiosis of gut microbiota due to diet, alcohol intake, body mass index, and gastrointestinal diseases in India
Applied Microbiology and Biotechnology (2023)
-
Coronavirus Disease 2019 (COVID-19) Pandemic Lifestyle Changes May Have Influenced Small Bowel Microbial Composition and Microbial Resistance
Digestive Diseases and Sciences (2023)