
Abstract
We studied variation in adaptive traits and genetic association to understand the low P responses, including the symbiotic association of arbuscular mycorrhizal (AM) fungal colonization in Oryza species (O. sativa, O. nivara, and O. rufipogon). In the present experiment, we performed the phenotypic variability of the morphometric and geometric traits for P deficiency tolerance and conducted the association studies in GLM and MLM methods. A positive association between the geometric trait of the top-view area and root traits suggested the possibility of exploring a non-destructive approach in screening genotypes under low P. The AMOVA revealed a higher proportion of variation among the individuals as they belonged to different species of Oryza and the NM value was 2.0, indicating possible gene flow between populations. A sub-cluster with superior-performing accessions had a higher proportion of landraces (42.85%), and O. rufipogon (33.3%) was differentiated by four Pup1-specific markers. Association mapping identified seven notable markers (RM259, RM297, RM30, RM6966, RM242, RM184, and PAP1) and six potential genotypes (IC459373, Chakhao Aumbi, AC100219, AC100062, Sekri, and Kumbhi Phou), which will be helpful in the marker-assisted breeding to improve rice for P-deprived condition. In addition, total root surface area becomes a single major trait that helps in P uptake under deficit P up to 33% than mycorrhizal colonization. Further, the phenotypic analysis of the morphometric and geometric trait variations and their interactions provides excellent potential for selecting donors for improving P-use efficiency. The identified potential candidate genes and markers offered new insights into our understanding of the molecular and physiological mechanisms driving PUE and improving grain yield under low-P conditions.
Similar content being viewed by others


Introduction
Phosphorus (P) is significant in that it affects the growth and productivity of plants. A high amount of P is usually present in the soil, but availability is limited as P usually exists in the organic form, carbonate (alkaline) or oxide (acidic)1,2. Low P availability is the major constraint in agriculture3,4, with 20–30% of the applied P readily available to plants and the remaining converted to unavailable forms, thus creating water eutrophication5. As rice is the major P utilizer, enhancing P-use efficiency (PUE) is highly required for sustainable production. About 20 million hectares of world rice cultivation area are under P deficiency6, while 61.02% of Indian soil is low in P7. Global commercial phosphate reserves are estimated to become depleted in 300 to 400 years8. Around 90% of the P-based fertilizer and raw materials are imported, and the 10% indigenous rock deposits hardly satisfy India's domestic market9. After the introduction of high-yielding varieties (HYVs), the consumption of phosphate fertilizer gradually rose to 6.86 million metric tons in 2017–18 from less than 1 million tons (0.132 million MT) during 1965–66 (https://www.faidelhi.org/statistics/statistical-database). This suggests that there is a need to develop varieties that use P more efficiently and effectively to decrease the phosphate fertilizers needed for sustainable rice production10,11.
Phosphorus is involved in plant growth and is a critical component of nucleic acid, membrane phospholipids, ATP, and NADPH. Upon P starvation, remodeling of cellular processes and high turnover of phospholipids into galactolipids and sulfolipids have been observed12. In addition, environmental factors were also reported to have a substantial effect on P-starved plants. Further, arrest in primary root growth occurs by affecting the cell's proliferative capacity at the meristem12 and root tip due to abundance of iron in acidic soil and red-light-induced activation of P uptake mediated by phytochrome-B13. Supplementary to this, a reduction in tissue P concentration critically affects plant growth, which leads to plant death14. Therefore, screening genotypes in P-starving conditions, understanding the significance of traits and their genetic mechanism, and the nature of heritability are important for developing plants tolerant of soil with low P.
Phosphorus efficiency was divided into two separate components: P-acquisition efficiency (PAE) and internal P-use efficiency (PUE)15. PAE refers to the uptake of P from the soil, and PUE is the use of taken-up P for plant growth and development. PUE and PAE in modern rice cultivars can be improved by using the low-P tolerance naturally found in landraces or wild species genotypes. Wissuwa et al.16 mapped a major QTL, Pup1, on chromosome 12 for tolerance of P deficiency from aus-type rice variety Kasalath. In addition to this, Chin et al.17 developed several markers for P-uptake efficiency to assist in breeding programs, among them, marker OsPupK46-2 was particularly found associated with P-uptake efficiency. Gamuyao et al.18 termed OsPupK46-2 as phosphorus-starvation tolerance 1 (PSTOL1). PSTOL1 promotes high phosphorus uptake by enhancing early root growth18. Globally, plant breeders are introgressing only the PSTOL1 gene from Kasalath (aus) and African rice (O. glaberrima Steud.) to improve P uptake, which may narrow down the genetic variability of the PSTOL1 gene. In addition, reports have mentioned QTLs associated with P deficiency in root and shoot traits at the seedling stage in O. sativa L.19. However, research in response to P deficiency and trait variability contributes to tolerance against low P and PAE and research on QTLs between wild and cultivated rice is limited. Wild species are a secondary gene pool that is a potential reservoir for unique genes uncommon in improved genotypes. Mapping and the identification of additional QTLs responsible for low-P tolerance with a large effect and pyramiding of those QTLs may give an additional level of tolerance under soil deficient in P along with yield improvement. This can be achieved by exploring new alleles for PAE and PUE.
Wild species of rice and landraces of the upland ecosystem may possess the required genetic resources. In addition to PAE and traits involved in low-P tolerance, P's microbial contribution by symbiotic interaction also plays an important role in providing P to plants in exchange for carbon20. By exploring the soil beyond what is reachable to roots and acquiring immobile P, fungal hyphae are efficiently involved in catering to the need of up to 80% of the P required by the plant21,22,23. The impact of mycorrhizae on P uptake under upland soil has been proved and associated candidate genes were reported24. However, the genetic basis of the response of rice cultivars in a panel of the population for arbuscular mycorrhizal (AM) fungal colonization under low-P conditions is not well established. Earlier reports on QTL identification for traits related to PAE and PUE are from bi-parental mapping populations. Reports on association mapping for low P tolerance and exploration at different species levels are not available in rice. Association mapping can simultaneously map several QTLs and serve as an excellent tool for allele mining by exploring natural variability in the germplasm. This study aimed to understand genetic variation in the level of tolerance among a panel of a population (O. sativa, improved varieties and landraces, and wild species, O. nivara, and O. rufipogon) containing 120 genotypes shortlisted from 155 through phenotyping to explore the distribution of PSTOL1 across species and validated them under P-deficient conditions and identified QTLs for multiple traits (23) such as shoot and root system architecture, P accumulation, and colonization of AM fungi based on association mapping in response to low-P tolerance.
Results
The present experiment began in order to explore the genetic variation and identification of QTLs associated with low-P tolerance in a unique population panel consisting of cultivated and wild relatives of Oryza.
Phenotyping trait distribution pattern for adaptation to low P
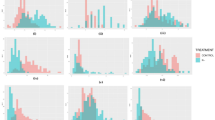
The frequency distribution of the population had a broad range of variation (Fig. 1). The mean and median were similar for most of the traits studied, and this indicates that the distribution frequency was normal. Among the various parameters studied, shoot length (−0.63), leaf length (–0.42), root length (−0.28), and shoot P content (−0.06) were negatively skewed, while the rest of the traits were positively skewed. The kurtosis values ranged from −0.63 to 8.71, the parameters were found to be > 3 TDW (total dry weight) (3.50), tiller number (3.69), shoot dry weight (3.87), root/shoot dry weight (6.89), root/shoot length (7.50), and root dry weight (8.71), indicating that the frequency distribution of the studied population for these traits was platykurtic. AM fungal root colonization ranged from 10% (improved genotypes) to 80% (landraces), while the average colonization percentage was significantly lower in improved genotypes (34%) than in landraces (41%) and wild genotypes (38%). The coefficient of variation (CV) of the entire population was reasonable and varied from 23% (SPAD) to 95% (root dry weight) (Table 1). The results obtained from the analysis of variance (ANOVA) revealed that significant (P < 0.0001) variation was observed among the genotypes for all 23 parameters studied (Table 1). The estimation of heritability (broad-sense) determines the insight into the extent of genetic control of the particular parameter and phenotypic accuracy to promote its breeding value. The estimated broad-sense heritability (h2) differed from 33.4% (maximum root length) to 74.8% (root P content) (Table 2). In addition, traits such as shoot dry weight, total dry weight, root-shoot length ratio, top-view plant area, total root length, root projected area, root surface area, root volume, mycorrhizal colonization, and root and shoot P content exhibited high h2 of > 60%.

Box plot showing the population distribution of traits measured under P deficiency for 155 rice genotypes. (a) Shoot length and leaf length; (b) leaf number (leaf no.) and maximum root length (max root lth); (c) leaf width (leaf wd) and root dry weight (RDW); (d) stem thickness and root P content (R_P); (e) shoot dry weight (SDW) and root-shoot dry weight ratio (R/S_DW); (f) tiller number (Tiller/pl) and root volume (RV); (g) total dry weight (TDW) and root-shoot length ratio (R/S_lth); (h) root tips and top-view area (TVA); (i) SPAD, total root projected area (TRPA), and mycorrhiza colonization (%) (M_col); (j) average root diameter (ARD) and shoot P content (S_P); and (k) total root length (T_root_lth) and (l) total root surface area (TRSA). The upper, median, and lower quartiles of the boxes represent the 75th, 50th, and 25th percentiles of the population, respectively. The square box inside the quartile box represents mean and the asterisk represents outliers.
Correlation analyses were executed to determine the correlation coefficient among the parameters associated with the traits responsible for low P in rice plants. Among the 23 traits studied, total dry weight had a positive correlation with 10 traits, followed by stem thickness, maximum root length, total root length, and total root surface area, which exhibited a positive association with eight traits (Fig. 2). In addition, a highly significant positive association was observed between shoot length and leaf length (r = 0.937), leaf number with tiller number (r = 0.8606), shoot dry weight (0.7066), and total dry weight (r = 0.7212). Similarly, shoot and root dry weight had a strong positive association between them. Also, the total root projected area exhibited a strong association with most of the root parameters, such as maximum root length (r = 0.8056), total root surface area (r = 0.9865), root volume (r = 0.8340), and number of root tips (r = 0.7378). The total root surface area also positively associated with root volume, root tips, and total root length. In contrast, root-shoot dry weight ratio exhibited a strong negative association with shoot length (r = −0.7486) and leaf length (r = −0.7114), wherein root P content also registered a negative association with shoot dry weight (r = −0.5524), root dry weight (r = −0.5198), and total dry weight (r = −0.5678).

Pearson correlation matrix among the 23 traits measured under P deficiency for 155 rice genotypes. The color denotes the correlation, where 1 represents a complete positive correlation (dark blue) and −1 represents a complete negative correlation (dark red) between two traits. A large circle denotes strong correlation and a small circle indicates a weaker correlation.
A multivariate principal component analysis was carried out to partition the traits associated with low P. A trait biplot analysis was composed and displayed for the first two principal components with a lay hold of 54.28% of the total variability depicted in Fig. 3. Principal component 1 (PC1) and PC2 accounted for 38.52% and 15.76%, respectively, toward the variability. Among the various traits studied, PC1 was strongly correlated with root- and shoot-related traits. PC1 increases with an increase in root volume, total root surface area, projected area, total root length, dry weight (shoot and root), stem thickness, and shoot and leaf length. It suggests that if root volume increases, the rest of the associated traits will increase, and genotypes would tend to have more root volume under P-starved conditions. PC2 had a positive association with SPAD and leaf width. However, root P content and root dry weight were negatively associated with PC1 and PC2, respectively. This suggests that the roots should translocate observed P and assimilate to expand the above-ground portion in genotypes under deficient P. Further, the biplot graph (Fig. 3) categorized the whole population into five major groups. Among these five groups, group 3 in quadrant 3 (bottom right) registered high biomass with low tissue P in shoot and root. On the contrary, group 2 in quadrant 2 exhibited more root growth with high root P. Other groups (1, 4, and 5) were presented on the opposite plane of the biplot and showed susceptibility under low P with high tissue P.

PCA biplot graph representing genotypes in two main principal components for traits measured under P-deprived conditions, and these two components explained 38.52% and 15.76% of the variance, respectively. The vector's direction and length indicate the traits' contribution to the first two components in the PCA. The transparency of the trait vectors represents the contribution to the variance in the dataset, ranging from 2% (lightest) to 6% (darkest). Genotypes were divided into five groups based on their level of tolerance. Groups 1 and 2 consisted of improved genotypes with high tissue P in root and more root growth, respectively. Group 3 consisted of the mixture of landraces, O. rufipogon, O.nivara, and positive checks. Group 4 consisted of a mixture of all species with more shoot P and Group 5 had O. nivara and O. rufipogon together with higher root-shoot dry weight ratio.
Marker segregation and diversity index
A total of 78 primer pairs consisting of microsatellites and gene-based markers covering all 12 chromosomes were used to assess the genetic diversity of 120 genotypes. Fifty-seven of 78 (73.07%) primers used were polymorphic among the 120 selected genotypes. A total of 154 alleles were amplified with 57 polymorphic markers, with an average of 2.70 alleles per locus and the number of alleles ranging from 2 to 5 (Table 2). The amplification size of the markers ranged from 70 bp (RM30) to 995 bp (K20-2). The major allele frequency of the primers ranged from 0.446 (RM410) to 0.991 (RM254). In addition, genetic diversity (He) ranged from 0.018 (RM254) to 0.861 (RM521), with an average of 0.407. Heterozygosity (Ho) ranged from 0 (RM254, K20-2, K20-1, K29-3, RM261, RM5485, RM47) to 1 (RM3307, RM5814), with an average of 0.175. The PIC values of all 57 polymorphic primer pairs ranged from 0.018 (RM254) to 0.585 (RM335), with an average of 0.326. Shannon index (I) ranged from 0.045 to 1.215, with an average of 0.641, and Nei's genetic diversity index (Nei) ranged from 0.015 to 0.655, with an average of 0.392. The average heterozygosity estimated by POPGENE ranged from 0.011 to 0.631, with an average of 0.364.
Analysis of molecular variance (AMOVA) and principal coordinate analysis (PCoA)
In this study, 120 genotypes were divided into five populations based on their ecology and species level (irrigated, lowland, upland, O. nivara, and O. rufipogon) to determine the genetic differentiation among them. Maximum variation (71%) was found among the individuals, followed by within individuals (23%), and minimum variation existed among the populations (6%) (Table 3). The deviation from Hardy–Weinberg's prediction was calculated using Wright's F statistics. The FIS and FIT values for all the loci were 0.757 and 0.771, respectively, while FST was 0.056 between the populations. The NM value of the assumed population was 2.003. The PCoA explained that the first two components accounted for 20.97% of the total genetic variation among the assumed population (Fig. 4). Most of the genotypes were plotted on the right side of the plot, including wild species. However, all wild accessions from populations 4 (O. nivara) and 5 (O. rufipogon) were plotted in the separate quadrant on the plot's right side. Twenty-four accessions of populations 1 (irrigated; 10 in number), 2 (shallow lowland; 6 in number), and 3 (upland; 8 in number) were plotted on the extreme left side of the plot. Nei genetic distance congregated the five populations into three major clusters. Populations 4 and 5 were grouped into separate clusters, while populations 1 and 2 stayed together. The highest pairwise Nei genetic distance was noticed between population 1 (irrigated) and population 4 (O. nivara) (0.1158), followed by population 5 (O. rufipogon) with population 1 (0.1046) and population 2 (lowland) with 4 (0.1023). Minimum genetic distance was observed between populations 1 and 2 (0.0248), followed by that between populations 2 and 3 (upland) (0.0269).

Principal coordinate analysis (PCoA) of the five sub-populations (Pop 1 (irrigated), Pop 2 (rainfed lowland), Pop 3 (upland), Pop 4 (O. nivara), and Pop 5 (O. rufipogon)) were plotted into three major clusters. (a) AMOVA showed maximum variation among the individuals, followed by within individuals and between populations. The genetic variability estimated by the fixation index revealed (Fst = 0.11) indicates the existence of moderate genetic differentiation within the population. (b) Nei genetic diversity among the assumed sub-population using principal coordinate analysis (PCoA). The assumed five sub-populations (Population 1 (irrigated), Population 2 (rainfed lowland), Population 3 (upland), Population 4 (O. nivara), and Population 5 (O. rufipogon)) were plotted into three major clusters.
Genetic diversity and population structure
Cluster and population structure analyses were carried out with the data generated with all 78 primers collectively, with and without 13 Pup1-specific markers. The cluster analysis based on unweighted neighbor-joining with 10,000 bootstraps with all the primers understudy grouped 120 genotypes into three major clusters (Fig. 5a). Cluster-I (blue) constitutes 45 genotypes divided into three major sub-clusters; all the wild cultivars fell into this cluster. Sub-cluster I-1 constitutes only two genotypes, Kamesh and ASD 16, followed by five genotypes (Kouni, AC100142, AC100175, Phalguni, and AC100010) grouped into Sub-cluster I-2. The rest of the 38 genotypes, including all the wild species and a few improved varieties (Meher, Sadabahar, Subhadra, Parijat) and landraces (Dular, IC459373, Sekri, Sukhapanki, Longmanabi A, Dular, and Harishankar), were grouped into Sub-cluster I-3. Cluster-II (green) was divided into six sub-clusters with 65 genotypes, most of which are improved varieties, including CR Dhan 801, whereas Cluster-III (red) was grouped as a separate cluster with nine genotypes (Akhiyaturfa, KumbhiPhou, ChakhaoAubi, KabukPhou, Rajeshwari, Longmanabi, Khitish, CR-Dhan103, LeimaPhou) of the northeastern states of India with two improved lines (Fig. 5a). Similarly, cluster analysis carried out with 65 low-P linked markers separated 120 genotypes into three groups (Fig. 5b). Cluster-I (green) represented only improved lines of 65 in number, whereas Cluster-II (red) grouped 47 genotypes comprising wild species and a few O. sativa (landraces and improved lines). Cluster-III (blue) had eight genotypes, with seven improved varieties and one wild accession.

Unrooted tree using unweighted neighbor-joining (UNJ) method depicting clustering pattern of a panel population of 120 accessions in response to all 78 primers collectively, with and without Pup1 markers. (a) 78 primers grouped genotypes into three major clusters. Cluster-I (blue) constitutes 45 genotypes that were divided into three major sub-clusters. Cluster-II (green) was divided into six sub-clusters with 65 genotypes, most of them being improved varieties, including CR Dhan 801 and Kasalath. Cluster-III (red) was grouped as a separate cluster with nine genotypes of northeastern states of India with two improved lines. (b) The cluster analysis with 65 low-P linked markers separated 120 genotypes into three groups. Cluster-I (green) represented only 65 improved lines Cluster-II (red) grouped 47 genotypes comprising wild species and a few O. sativa (landraces and improved lines) and Cluster-III (blue) had eight genotypes with seven improved varieties and one wild accession. (c) The cluster analysis with Pup1-specific markers grouped 120 genotypes into three major clusters. Cluster-I (red) consisted of 48 genotypes and was further divided into three sub-clusters. Cluster-II (green) separated 45 genotypes into three sub-clusters and Cluster-III (blue) represented 30 genotypes that were further divided into three sub-clusters with 14 (III-1), 13 (III-2), and 3 (III-3) genotypes. The positive checks Dular and Kasalath were grouped into sub-cluster III-2 with IC459373, multiple-stress-tolerant CR Dhan 801, Poongar, Sekri, Kouni, AC10062, AC100326, AC100284, AC 100281, AC 100135, and AC 100117.
Cluster analysis with 13 Pup1-specific markers grouped all 120 genotypes into three major clusters (Fig. 5c). Cluster-I (red) was further divided into three sub-clusters. A mono-genotypic cluster with genotype Hiranmayi was formed separately as Sub-cluster I-1, while Sub-cluster I-2 consisted of 15 genotypes with 12 O. sativa (landraces and improved varieties) and three wild accessions. In Sub-cluster I-3, 29 genotypes of improved varieties and landraces were grouped. On the other hand, Cluster-II (green) separated 45 genotypes into three sub-clusters: II-1 (10), II-2 (16), and II-3 (19). Sub-clusters II-2 and II-3 consisted of most of the wild accessions. Cluster-III (blue) represented 30 genotypes, which were further divided into three sub-clusters with 14 (III-1), 13 (III-2), and 3 (III-3) genotypes. Those with known low-P tolerance, such as Dular and Kasalath, were grouped into sub-cluster III-2 with IC459373, multiple-stress-tolerant CR Dhan 801, Poongar, Sekri, Kouni, AC10062, AC100326, AC100284, AC 100281, AC 100135, and AC 100117.
Similarly, the genetic relationship among the genotypes was also determined by a model-based simulation, STRUCTURE 2.3.4, separately for all 78 markers under study, 13 low-P gene-specific markers (Pup1), and 65 primers without Pup1 markers (Figs. 6a–c, 7a–c). The simulation, by using all 78 primers, identified the highest log-likelihood with the number of populations set at three (K = 3) (Pritchard et al. 2000). Structure harvester of Evano table (http://taylor0.biology.ucla.edu) analysis showed that, at K = 3, ΔK = 248.51, where the value was the highest in both independent burns (Fig. 6a). At K = 3, all 120 genotypes were divided into three sub-populations (SP). Among the 120 accessions studied, 25 accessions of wild species and landraces were placed in SP1, and 44 accessions fitted in SP2 comprising only improved genotypes. Similarly, 24 improved genotypes were grouped in SP3, and the rest of the wild species and landraces (27 genotypes) were grouped as an admixture (Fig. 7a). The fixation index (Fst) value ranged from 0.1880 in sub-population 1 of wild species to 0.5507 in sub-population 3 of improved fixed lines. The average distance (expected heterozygosity) was maximum (0.1261) between sub-population 1 of wild species and sub-population 3 of improved lines, whereas minimum distance was observed among sub-populations 1 to 2 (0.0797). The value of alpha (0.1270) reflects the relative admixture level between populations of less than 1, signifying origin mainly from one population (and each population is equally probable).

Graph of ∆K-value and ad hoc statistics related to the rate of change in the log probability of data between successive K-values. (a) 78 markers identified the highest log-likelihood with the number of populations set at three (K = 3) with ΔK = 248.51, (b) 65 markers linked to low P identified the highest log-likelihood with the number of populations set at three (K = 3) with ΔK of 179.01, and (c) Pup1-specific markers identified the highest log-likelihood with the number of populations set at two (K = 2) with ΔK = 106.29.

Distribution pattern of 120 rice accessions based on low-P linked markers and Pup1-specific markers determined by a model-based simulation, STRUCTURE 2.3.4. Grouping of accessions is based on (a) 78 markers, (b) 65 markers linked to low P, and (c) Pup1-specific markers. The number indicates the order of genotypes as mentioned in Table 1S.
On the other hand, without a Pup1-specific marker, the 65 low-P linked markers identified the highest log-likelihood with the number of populations set at three (K = 3), with ΔK of 179.01 (Fig. 6b). Among the 120 genotypes, 46 genotypes of improved varieties were placed in SP1, and 24 genotypes fitted in SP2 comprising wild species and upland (landrace and improved) varieties. Similarly, 24 improved genotypes were grouped in SP3, and the rest of the wild species, landraces, and improved (26) accessions were grouped as an admixture (Fig. 7b). Fst values of the sub-populations are 0.2111 for sub-population 2 (wild and upland), 0.2760 for sub-population 1 (improved), and 0.5471 for sub-population 3 (improved). Allele frequency divergence between sub-populations 1 and 3 was maximum (0.1283), while minimum divergence was observed between sub-populations 1 and 3 (0.0768). The average distance (expected heterozygosity) between genotypes is high in population 2 (0.2863) and minimum with sub-population 3 (0.1623). The alpha value was nearer to zero (0.1226), suggesting that no admixture and population might have originated from one population.
The simulation using 13 Pup1-specific markers identified the highest log-likelihood with the number of populations set at two (K = 2), with ΔK = 106.29 (Fig. 6c). At K = 2, all 120 genotypes were divided into two sub-populations. Twenty-five genotypes of only improved varieties were placed in SP1, 63 genotypes fit in SP2 comprising improved varieties (including CR Dhan 801), wild species, and a few landraces (Dular, Kasalath, and IC459373), and the rest of the 32 genotypes of wild species and improved varieties were grouped as an admixture (Fig. 7c). The Fst value of sub-population 1 was 0.6382, and that of sub-population 2 was 0.3718. The allele frequency divergence among the two sub-populations was 0.2847, and the average distance (expected heterozygosity) among genotypes in the same sub-population was high in population 2 (0.2252) and minimum in population 1 (0.1548). The alpha value of the Pup1-specific markers is 0.3285, which signifies origin mainly in one population (and each population is equally probable).
Association of markers with traits related to low P
In this study, both a generalized linear model (GLM) and mixed linear model (MLM) were used for association analysis at P-value < 0.005, FDR at the 5% level, and R2 > 7.5. In the GLM, 61 markers were associated with 110 QTLs (data not shown), and in MLM 16 markers were associated with 29 QTLs (Table 4). The R2 value of the associated markers for GLM ranged from 0.075 (RM283) to 0.190 (RM297), and for MLM it was from 0.076 (K29-3) to 0.149 (RM297). The marker F-value for GLM ranged from 7.95 (RM283) to 23.75 (RM297), and for MLM it ranged from 7.53 (K29-3) to 14.85 (RM297). A total of 19 QTLs for different traits were predicted on four chromosomes (1, 8, 11, and 12). A maximum of five QTLs (qARD8.1, qLL8.1, qSL8.1, qTRP8.1, and qTSA8.1) associated with average root diameter, leaf length, shoot length, total root projected area, and total surface area were identified on chromosome 8, followed by five QTLs (qNT11.1, qRDW11.3, qRV11.3, qSDW11.2, and qTDW11.2) found associated with tiller number, root dry weight, root volume, shoot dry weight, and total dry weight on chromosome 11 (Table 5). Five QTLs were harbored on chromosome 12, followed by four QTLs on chromosome 1, three on chromosome 3, and two (qRL6.1 and qTRL6.1) on chromosome 6. In the MLM, nine of 23 traits under study were associated with more than one marker. The traits tiller number, root biomass (RDW), and root length were associated with a maximum of three markers on different chromosomes. Similarly, AM fungal root colonization (C%) was associated with two markers on chromosomes 2 and 4, with phenotypic variability (PV) ranging from 8.0 to 10.0%.
Among 29 QTLs observed in the MLM, 12 were associated with root parameters such as root dry weight (RDW), root length (RL), total root length (TRL), total surface area (TSA), average root diameter (ARD), total root projected area (TRPA), root volume (RV), and root biomass (RDW), with recorded PV from 10.32% to 14.97% on six chromosomes (1, 3, 6, 8, 11, and 12). Out of 16 markers associated with 29 QTLs, marker RM6966 on chromosome 8 had an association with five traits, LL, ARD, TRPA, TSA, and SL (shoot length), and explained PV of 7.7% to 10.7%. Similarly, RM5926 on chromosome 11, RM297 on chromosome 1, RM242 on chromosome 9, and K41 on chromosome 12 are associated with four, three, two, and two different traits, respectively, linked with low-P tolerance. Among the Pup1-specific markers, K20-2, K29-3, and K41 on chromosome 12 were associated with root dry weight, stem thickness, root length, and top-view area, with PV of 7.6% to 11.6%. The marker PAP1 developed from NRRI on chromosome 11 was associated with tiller number with a PV of 7.7%.
Prediction of QTLs underlying candidate genes for traits associated with low-P tolerance
Twenty-nine QTLs related to traits measured under P starvation associated with 15 markers were considered to identify candidate genes (Table 6). Three markers, RM283 (chromosome 1), RM297 (chromosome 1), and RM242 (chromosome 9), associated with leaf number (qNL1.1), biomass (qRDW 1.1, qTDW 1.1, qSDW 1.1), leaf length (qLL9.), and tiller number (qNT 9.1) were found to have an association with growth promoter genes (auxin (Os09t0491740), brassinosteroid (Os01t0178500), and strigolactone (Os01t0746400)). Traits related to roots such as root length (qRL6.1), average root diameter (qARD8.1), root projected area (qTRPA8.1), and root surface area (qTSA8.1), associated with markers RM30 and RM6966, were found to be linked with candidate genes of phosphate starvation regulator (Os06t0664800) and phosphate transporter (OsPT6, Os08g0564000), respectively. On the other hand, tiller number (qNT3.1 and qNT11.1) was associated with markers RM2334 and PAP1 on chromosomes 3 and 11, respectively, linked with locus Os03t0672900 and Os11t0149100 responsible for the regulation of plant growth and purple acid phosphatase (EC:3.1.3.2) involved in the acquisition and use of organic P.
The network analysis of these candidate genes in the QTL interval region was analyzed using the riceFREND database, and it revealed the coexpression pattern of the genes (Fig. 1S). A total of seven candidate genes were explored to construct gene networks, and their interactions were disposed of in Supplementary Fig. S1. Besides, the loci information and functions of the associated genes were given in Table 2S. These reported genes are functionally associated with physiological and molecular pathways in biotic and abiotic stress tolerance mechanisms and also uptake the nutrient element regulated by the phytohormone biosynthesis pathways. Insilico expression analysis of these genes indicates that most genes are highly expressed in root tissues based on the RiceXPro database. The sum of nodes and edges of coexpressed genes of the seven-candidate genes were 72 and 134 respectively. Among the seven putative candidate genes, the locus Os08g0564000 located on chromosome 8 had functionally associated with eight known genes (OsRLCK266, OsWRKY32, OsRAM2, OsbHLH023, OsCP25, OsNPC4, OsERD6, and OsCP29). Similarly, other locus had functional associated with genes related to root growth and auxin.
Identification of rice germplasm with QTLs related to low-P tolerance traits such as root growth and shoot and root biomass
A total of seven rice accessions were found to tolerate P-deficient conditions, including tolerant checks Dular and Kasalath. Among them, the genotypes IC459373, Chakhao Aumbi, AC100219, AC100062, and Sekri exhibited a par or higher shoot and root dry weight with better root growth than the checks in P-starved conditions. Genotype IC459373 outperformed the checks, while Chakhao Aumbi and AC100219 performed significantly better under low P. Amid the favorable QTLs detected, six QTLs (Table 7) were found in Sekri, followed by five QTLs detected in Chakhao Aumbi having a high tiller number and biomass. The genotypes IC459373 and Kasalath had four QTLs each. Therefore, these accessions could be used as donors in a breeding program to improve PUE in rice.
Discussion
Researchers globally for long been trying to understand the critical role of P in plant growth and development mediated through signaling and metabolism to develop P-efficient cultivars in several crops. Considering the area occupied and the importance of rice, improving PUE is catching the attention of rice researchers. Because of finite P fertilizer and its availability only in specific regions around the globe4, the applied P fertilizer becomes unavailable to plants because of high reactivity with soil particles and microbial activity25,26. Exploring the large unavailable form of soil-bound P would be an alternate strategy to mitigate these factors. In addition, increasing the tolerance level through PUE, acquiring P from the soil by exploring the adjacent area and by modifying the root architecture, secretion of root exudates, and symbiotic association with AM fungi would be advantageous. Attempts were made to improve P efficiency in rice through classical plant breeding by transferring targeted traits with limited success27. The available germplasm in gene banks has sufficient genotypic variation to improve P efficiency, but the major obstruction in identifying and developing P-efficient genotypes is the lack of a screening facility with low-P soil. Predominantly, the identification of P-efficient genotypes is driven by biomass production of shoot or root under P-deficient soil. To overcome this impediment, QTLs/genes related to traits that improve P-use efficiency need to be identified to introgress them into an elite background. Wild species and landraces possess higher genetic diversity than modern/improved varieties, which serve as a reservoir of genetic diversity and help to use the beneficial alleles in breeding programs for crop development. This research work attempted to study the genetic diversity, structure, and association between markers and traits measured under low-P conditions among the selected subset of the population comprising wild species, landraces, and improved varieties with multiple traits.
Evaluation and identification of genotypes with tolerance to low P
This study included accessions of improved genotypes, landraces, O. nivara, and O. rufipogon from nine provinces of India. The frequency distribution and CV exhibited substantial variability, and adaptation to P starvation was observed among the 155 accessions of Oryza species. Similarly, ANOVA revealed the presence of significant variation among genotypes for all the parameters observed. In addition, traits such as biomass and tissue P measured under low P registered a high h2 of > 67%. Similarly, high heritability of 81% to 91% was reported by Wang et al.28 for total above-ground biomass, total above-ground P uptake, and P translocation efficiency in P-starved conditions. Therefore, the presence of moderate to high genetic advance with high h2 offers an opportunity to improve the selection of traits at the early generation based on shoot biomass, total root length, tissue P content, mycorrhizal colonization, and the geometric trait of top-view area for improving low-P tolerance in rice. The strong positive correlation between shoot and root biomass and inter-correlation among the root parameters show the possibility of improving P-efficient genotypes. The negative association between root P content and shoot dry weight (Fig. 8a) suggests that genotypes with desired root architecture QTLs such as Pup1 coupled with high-affinity P transporters (PHT1 family) would be supportive of using available P efficiently in P-deprived soil14. Besides, TRSA had become the higher contributor for improved P uptake under deficit P than mycorrhizal colonization. This was clear from Fig. 8b, and non-linear regression showed that 33% of P uptake would be through TRSA, while the role of mycorrhizae in P uptake was minimum (4%) in the present experiment. The multivariate principal component analysis also suggests the importance of roots under P-starved conditions to improve shoot growth. Genotypes such as upland landraces of northeast India, a few genotypes of improved upland varieties, O. nivara, O. rufipogon, and known tolerant genotypes Dular and Kasalath were categorized under group 3 with high biomass and low tissue P in both shoot and root, whereas genotypes of the improved varieties were put in groups 1 and 2 with high tissue P in the root and low P in the shoot. Conversely, O. nivara and O. rufipogon were grouped together (group 5) with high tissue P, root diameter, root-shoot weight ratio, and root-shoot length ratio, while group 4 had a mixture of all species with more shoot P. This suggests that genotypes of group 5 (O. nivara and O. rufipogon) can extract, upload, and translocate P from the rhizosphere to the shoot in low-P soil. The symbiotic association between AM fungi and plants (Fig. 9) was significantly higher in groups 3 and 2 (41%), in which they have a good root system (Fig. 3). The improved genotypes of group 1 had minimum colonization of 30% with a poor root system. It is well-known that rice plants with more root biomass positively correlate with AM fungal root colonization29.

Relationships between shoot biomass, total root surface area (TRSA) and mycorrhizal colonization with P under deficient condition. (a) The negative association between shoot biomass and root P concentration suggests that an increase in biomass (shoot) in deprived P was associated with the dilution effect of P. (b) The line indicates the fitted results representing the relationship between total P of plant tissue and possible parameters (TRSA and mycorrhizal colonization) involved in P uptake under P deficient condition. The contribution of improved P uptake of TRSA was high compared to mycorrhizal colonization.

Root and AM colonization in different rice genotypes found in low-P soil as shown by trypan blue staining. (a,e) Dular, (b,f) Kasalath, (c,g) Sekri, and (d,h) AC100219.
Genetic diversity and distribution of Pup1 among Oryza spp.
Elucidating molecular genetic diversity helps breeders estimate rice germplasm's genetic constitution and select donors to develop a systematic and effective breeding program. Fifty-six of 78 markers exhibited polymorphism (73%), which gave rise to 154 alleles, less than in our previous study30. We obtained 128 alleles from 39 polymorphic primers, and more than reported by Donde et al.31, 154 alleles from 65 polymorphic markers. The present average of 2.7 alleles per locus was lower than that of many other research reports; for example, Noyer et al.32 analyzed 419 rice accessions using 16 SSR markers and reported an average of 9.1 alleles per locus. The higher average number of alleles per locus than in the present study (2.24) may be due to the use of a larger number of genotypes. This study's average PIC value was 0.326, which was a bit higher than that of Xu et al.33 (0.31), who studied the association mapping of cold tolerance in improved japonica rice germplasm.
Cluster analysis based on unweighted pair group method with arithmetic mean (UPGMA) with all 78 markers and excluding gene-specific Pup1 grouped 120 genotypes into three clusters. Under both conditions (excluding and including the Pup1), the wild species were grouped into a single cluster and might have a similar type of P-efficiency mechanism under P deficiency. However, all explored wild species (30 in number) might have lower diversity because they share common geographical boundaries such as Odisha and neighboring province West Bengal of India. This coincides with several studies on assessing genetic diversity34,35,36. On the other hand, those wild species were separated from O. sativa L. (landraces and improved genotypes). Similarly, Xu et al.33 studied the genetic diversity in 103 rice germplasm accessions comprising O. rufipogon and O. sativa genotypes, and grouped both of them in separate clusters, which are in support of our study. The UPGMA cluster analysis with 13 gene-specific Pup1 markers grouped all 120 genotypes into three major clusters (Fig. 5c). Cluster-III (blue) grouped 25% (30 accessions) of the studied population, including the tolerant genotypes Dular and Kasalath having Pup1. However, this cluster again sub-divided into three sub-clusters (III-1 (14 in number), III-2 (13 in number), and III-3 (3 in number)) based on the presence of gene-specific Pup1 markers. Among the three sub-clusters, sub-cluster III-2 had grouped 13 genotypes that include six landraces (42.85% of the total population), five O. rufipogan (33.3%), one O. nivara (AC 100117) (6.66%), and one improved variety (CR Dhan 801) (1.31%). The performance of those 13 genotypes registered 28.42% shoot length, 83.41% root biomass, and 90.49% root volume, with 35.88% top-view area, higher than in the genotypes secured in the other two sub-clusters. On the other hand, out of 13 gene-specific Pup1 markers, K29-3, K41, K43, and K45 played an important role in differentiating genotypes between these sub-clusters. To corroborate this, Chin et al.17 reported K29-1, K29-3, K41, K43, K45, and K46-1 as core markers to differentiate genotypes that differ in Pup1 allele constitutions. Further, Neelam et al.37 substantiated that O. rufipogon collected from India has high PUE with a differential functional allele of PSTOL1.
By considering all 78 markers and excluding gene-specific Pup1 markers, the model-based simulation STRUCTURE 2.3.4 divided the population into three sub-populations, similar to the distance-based model. Conversely, the gene-specific Pup1 markers grouped the population into two sub-populations. The grouping of genotypes with distance and model-based analysis was similar at the genetic level with markers linked to low P, including exclusive markers or exclusive gene-specific Pup1 markers. It suggests that traits other than early root growth (Pup1-specific markers) in differentiating low-P tolerance must be documented. AMOVA revealed a higher proportion of variation among the individuals, while a lower proportion of variation was observed among the populations. A similar observation of a higher proportion of variation among the individuals was reported by Verma et al.38 and Islam et al.39. Genotypes of the present study belonged to different species of Oryza (sativa, rufipogon, and nivara), wherein the species sativa includes genotypes of landraces and from improved upland, lowland, and irrigated ecosystems, which resulted in a higher proportion of variation among the individuals than among the populations. Within-individuals that had registered higher variation indicated a high level of heterozygosity at each locus in wild genotypes and landraces. The Fst value was minimum in the case of sub-populations containing wild genotypes (0.20), while a high Fst index of 0.55 was observed in improved genotypes, suggesting that they are fixed and homozygous in nature. The alpha value determined by the model-based simulation STRUCTURE 2.3.4 under three circumstances was nearer to zero (0.12) in two instances, which suggests that the individuals are from one population, while the Pup1 gene marker represented with 0.3285 suggests that the variation arising in a specific region might be variation in the Pup 1 indel region. The value of NM (2.0) indicated possible gene flow between the populations and low genetic differentiation for the trait studied40,41. Therefore, a hypothesis has been proposed that the Pup1 region or gene with low-P tolerance might have been introgressed from O. rufipogon into O. sativa. This may be supported in this study by the frequency of tolerant accessions of O. rufipogon outnumbering those of O. nivara. Further, Neelam et al.37 substantiated that O. rufipogon (IRGC 106506) accessions performed significantly better under limited P, with 2.5 times higher root weight than the positive control.
In the present association study, both GLM and MLM were used to assess the association between traits and markers. However, the number and degree of association-related traits are based on the dataset and model used. MLM, unlike GLM, is more accurate and has a robust algorithm to improve the calibration. It integrates structure and kinship matrix, which rectifies the false-positive error expected due to population structure and relatedness42. Hence, MLM has been popularly used by several researchers for studying marker-trait association. In the GLM, 61 markers were associated with 110 QTLs; in the MLM, 16 markers were associated with 29 QTLs. The total number of QTLs and distribution of the markers are displayed in Fig. 10. The unique population exploited in this study was associated with 78 markers linked to low P with multiple traits screened under deficient P. Not much study was carried out on the traits selected in this study in reference to the association with tolerance of P deficiency. This resulted in the identification of novel and interesting QTLs for multiple traits. We reported 16 markers (RM6966, RM1272, RM200, RM2334, RM242, RM283, RM297, RM30, RM3343, RM521, RM574, RM5926, PAP1, K20-2, K29-3, and K41) associated with 29 QTLs for 17 traits (ARD, C%, LL, LW, NL, NT, RDW, RL, RV, SDW, SG, SL, TDW, TPA, TRL, TRPA, and TSA) under low-P conditions in rice. Overlapping of markers for tolerance of low P suggests a high correlation between the traits. For instance, marker RM6966 was found to be associated with several root-related traits necessary for low-P conditions. Similarly, RM242 and RM297 were linked to shoot and root biomass. Thus, the pleiotropic effect of markers unravels the genetic correlation among the traits30 measured under low P. Marker RM30 on chromosome 6 was observed to be significantly associated with root length (R2 = 9.9%), and was co-localized with a QTL reported for root elongation ratio under P deficiency with PV of 19.9%. The tolerance imparted by this QTL might be due to the positive regulation of the candidate gene Phosphate starvation regulator (Os06t0664800) at 0.15 Mb to the right of qRL6.1 on chromosome 6. The network analysis of locus Os06t0664800 by the riceFREND database revealed that it has 11 nodes and 19 edges. It was found that coexpression patterns of the genes were highly expressed in roots, specifically in protophloem sieve elements and promote root elongation (OsbHLH068)43. Similarly, marker RM6966 from this study suggested that three QTLs were controlling root traits of low-P tolerance in rice, which were co-localized with the QTLs qRS8b, qRDW8, and qRN8b reported by Li et al.19 for root traits located in the same genomic region under P deficiency with a PV of 11.04%. The R2 values of traits associated with marker RM6966 varied from 7.7% to 10.7%. The well-documented high-affinity phosphate transporter (OsPT6) gene located on chromosome 8 at 28.3 Mb within this QTL region proved to be involved in long-distance transport of P from root to shoot, and it aids in the accumulation of biomass44 under P-deficient conditions. Insilico coexpression pattern of the gene Os08g0564000 revealed that most of the genes involved in root development and signaling pathways during abiotic stress (phosphoesterase family protein; OsNPC4)45. The coexpression network and expression analysis suggest that the resulting gene network provides the molecular pathways and the biological function of each gene. This piece of information would be useful to explore the role of novel genes related to the target trait of interest in the breeding program.

Distribution of primers used for association mapping and detected QTLs on 11 chromosomes of rice. Distances on the map are in Mbp presented on the left-hand side of the chromosomes. Markers highlighted in red are found associated with adaptive traits under P-deprived conditions. [SL shoot length (cm), NT tillers plant−1, NL leaf number plant−1, LL leaf length (cm), LW leaf width (cm), SG stem thickness (mm), RL max. root length (cm), SPAD, SDW shoot dry weight (g), RDW root dry weight (g), TDW total dry weight (g), TRL total root length (cm), TRPA total root projected area (cm2), TSA total root surface area (cm2), ARD average root diameter (mm), RV root volume (cm3), RT root tips, TPA top-view area (mm2), MC mycorrhiza colonization (%), SP shoot P (mg g−1), RP root P (mg g−1)].
On the other hand, marker RM242 located on chromosome 9 was a hotspot for biomass accumulation under P-deficient conditions, for which two important QTLs, qLL 9.1 and qNT 9.1, were identified to be associated with low-P conditions, with R2 ranging from 8.50% to 9.97%. Marker RM242 was reported to be associated with increasing root length under irrigated conditions and drought stress and was used in a marker-assisted backcross46 to improve root length in rice genotype Kalinga III. Goncharova et al.42 also reported RM242 to be involved in increasing relative biomass and nitrogen-use efficiency. The locus Os09t0491740 is located 0.33 Mb away from marker RM242 involved in auxin efflux situated in the plasma membrane. Similarly, markers RM283 and RM297 were also linked with the growth promoter genes brassinosteroid (Os01t0178500) and strigolactone (Os01t0746400).
Four PAP markers (1 to 4) associated with purple acid phosphatase were developed at ICAR-NRRI to identify genotypes having candidate gene OsPAP21b. The gene OsPAP21b was reported to be up-regulated under phosphate deprivation47 and proved to be a major plant enzyme involved in releasing P from organophosphates that remain unavailable for plants before mineralization48. Among the four markers, PAP1 at 2.42 Mb on chromosome 11 exhibited polymorphism across genotypes, which co-localized with tiller number with PV of 7.6% under P deprivation. Among the Pup1-specific markers, the R2 value of K20-2 registered a maximum of 11.5% for stem thickness and 10.5% for geometric trait top-view area, followed by the Pup1 core marker K41, which exhibited association with stem thickness of 9%. Another core marker, K29-3, was associated with root dry weight. Therefore, it is believed that Pup1 is involved in the accumulation of biomass in addition to early rooting in P-deficient conditions. On the other hand, markers RM521 and RM1272 were associated with mycorrhizal colonization % on chromosomes 2 and 4, respectively, with an R2 value of 10.3% and 8.3%. The symbiotic association between the rice plant and soil fungi mycorrhiza is well known under P-deficient conditions and fulfills 80% of P21,22,23.
Here, several reliable associations between traits measured under P deprivation and molecular markers were established. Notably, six markers (RM297, RM30, RM6966, RM242, RM283, and PAP1) would be more useful in predicting the identification of genotypes under P-deprived conditions. Typically, an association panel is a diverse set of populations that harbors multiple alleles at any given locus (which requires identifying favorable alleles rather than using the whole genome for hybridization), which are identified phenotypically. Identification and breeding for favorable alleles of the trait of interest from wild genotypes and landraces into modern cultivars will help breeders arrive at a new improved variety with wider adaptability. Therefore, a set of five potential genotypes (IC459373, Chakhao Aumbi, AC100219, AC100062, and Sekri) having a set of favorable alleles would be used appropriately to improve the PUE of rice. Among the selective potential genotypes, IC459373 and AC100219 contain the QTLs qNT3.1 and qNT11.1 for tiller numbers, while Dular had only one QTL qNT11.1. However, the tiller number between the three genotypes were uniform even they had additional QTLs for the same trait. A more number of QTLs for same trait might have complimentary effect under stress condition for maximizing the root length, which is highly essential for low P condition. This may be observed in the correlation between tiller number and maximum root length (Fig. 2). Therefore, genotypes having multiple QTL for same trait may be explored to utilize them in the breeding program to improve the PUE in rice. As these genotypes (IC459373 and AC100219) having better performance with additional QTLs, they need to be studied further and we are in it. Besides , the identified alleles need to be evaluated to understand the advantage of using them and their effect on different genetic backgrounds.
Identifying suitable donors and improving PUE in rice, potential candidate genes, and markers associated with these traits under low P is a central research area in rice breeding. Therefore, in this study, we demonstrated a selected population panel having significant trait variations. The positive correlation between the traits explained the possibility of improving genotypes for P-deprived soil in a holistic manner. Notably, a negative association was observed between root P content and biomass. The trait total root surface area had become the major contributor for improved P uptake under deficit P than mycorrhizal colonization. The geometric trait of the top-view area exhibited a positive association with maximum root length and root volume, suggesting using a non-destructive approach in screening genotypes under low-P conditions, which is the first report to the authors knowledge as well as based on available scientific documentation. The genotypes were divided into three groups based on distance and model-based analysis, while gene-specific Pup1 markers classified the panel into two groups. This suggests that traits other than early root growth (Pup1-specific markers) in differentiating low-P tolerance must be documented. This unique panel serves to find the association between traits and primers and facilitates the identification of donors with a combination of adaptive traits necessary for low-P conditions. Further, the identified linked markers are highly valued when determining variation for a target trait. The markers RM259, RM297, RM30, RM6966, RM242, RM184, and PAP1 were highly associated with traits responsible for low-P conditions in rice. The multiple traits related to these genome regions are promising resources for improving and understanding PUE and are also more useful as a breeding tool in predicting genotypes under P-deprived conditions.
Methods
Plant materials
The population consisted of 155 rice accessions (Table 1S) comprising 41 wild species (21 Oryza rufipogon, 19 O. nivara, and 1 O. spontanea), 37 landraces, and 77 improved varieties originating from eight provinces of India. Seeds were obtained from ICAR-National Rice Research Institute (NRRI), Cuttack, Odisha; the Regional Research & Technology Transfer Station (RRTTS), Coastal zone, Bhubaneswar; and Orissa University of Agriculture and Technology (OUAT), Bhubaneswar. The seed materials were collected from the NRRI gene bank, and appropriate permission was obtained to collect and use them in the current study. Thus, the study complies with local & national regulations.
Growing conditions and experimental design
Phenotyping for phosphorus stress in a cement tank
All the accessions under study were screened in a cement tank for a low-P tolerance facility located at NRRI, Cuttack (20o27′09" N, 85o55′57" E, 26 masl). The genotypes were direct-seeded in tanks containing low-P soil (< 3 kg/ha; 0–15-cm layer, pH 4.9) with 20 cm X 15 cm between genotypes in three replicates during June 2019. Average day/night temperatures were 33.6/26.0 °C, and relative humidity was 85.9% in bright sunlight. Before sowing, seeds of all the accessions were heat-treated at 50 °C for 45 h in a hot-air oven to break seed dormancy. The soil was irrigated every other day and, 15 days after sowing, the seedlings were thinned, leaving only two seedlings per hill. Chlorophyll content was measured for three plants of each accession on the 44th day from the date of sowing by using a SPAD meter (SPAD-502, Konica Minolta). On the next day (45th day), the plants were uprooted and morphological traits, shoot length (cm), number of tillers, number of leaves, 3rd-leaf length (cm) and width (cm), stem thickness (mm), maximum root length (cm), and root-shoot length ratio were recorded for three plants of each genotype. Subsequently, root traits (total root length (cm), projected root area (cm2), root surface area (cm2), average root diameter (mm), root volume (cm3), and number of root tips) were recorded for each genotype per replication and analyzed by WinRHIZO Pro 2013e (LA 2400, Regent Instruments Inc.). The plant samples were dried in a hot-air oven at 60 °C for 5–6 days and shoot, root, and total dry weight and root-shoot dry weight ratio were recorded in grams. In addition, the plant's top-view area was measured using open-source Image J software49. Images of each accession were taken using a 12-megapixel Nikon camera at 1.5-m distance, and the pixels were changed into mm2 as the top-view area of the plant49,50. To quantify total P, the dried plants were powdered around 300 mg shoot and 90 mg root samples following the phospho molybdo vanadate colorimetric method. P concentration in the digest was determined using a Cystronicx UV Spectrophotometer at 420 nm, and total shoot and root P contents were determined on a mg/g dry weight basis. Additionally, to strengthen these data, mycorrhiza colonization was observed.
To study AM fungal root colonization, root samples were collected from all the genotypes, washed thoroughly, cut into small pieces (0.5 to 0.7 cm), and put in falcon tubes containing 15 to 20 ml of 10% KOH. Care was taken when the root samples became immersed inside the KOH solution. Further, the samples were autoclaved at 121 °C at 15 psi pressure for 15 min and washed under running tap water until the roots became clear in color. Each sample was treated with 10 to 15 ml of 2% HCL for 5 min and washed under running tap water. Finally, 10 to 15 ml of 0.05% trypan blue in lactoglycerol was added depending upon root volume and this was left overnight at room temperature. Root samples were rinsed with lactoglycerol (without stain) and slides were prepared with 10 root pieces on each slide for observation under a stereomicroscope to check the root colonization. Percentage of root colonization was determined as per the formula described by McGongigle et al.51 : Percentage of colonization = (number of root segments colonized/total number of root segments) × 100.
Statistical analysis
To study the effect of variation due to P in 155 accessions, descriptive statistics, box plot, ANOVA, and broad-sense heritability (H2) were calculated using Windostat 7.5 software with 23 traits measured under low-P conditions. Principal component analysis (PCA) was performed on a matrix of morphometric and geometric traits to assess the variability in traits for tolerance of P deficiency with 155 rice genotypes to differentiate the genotypes based on their performance under low-P conditions. The PCA analysis was executed using the PCA function from the FactoMine R package52 in R. The corrplot functions from the corrplot package53 in R (version 3.6.3) were used to find the effect of low P on the various genotypes.
Genotyping
DNA isolation, quantification, and PCR amplification
Based on the level of tolerance and phenotypic information, a panel of 120 accessions was shortlisted from 155 accessions for genotypic study based on their distribution pattern. The total genomic DNA was extracted from young leaves following the modified CTAB method54. Further, the DNA samples were quantified by a Nanodrop spectrophotometer (Thermo Scientific) and diluted to a working stock of 50 ng µl−1. PCR amplified the isolated DNA samples with a 10 μl reaction containing 20 ng of DNA, 10 mM Tris–HCl, 1.5 mM MgCl2, 0.2 unit of Taq DNA polymerase (New England Biolabs), 50 μM of dNTPs (New England Biolabs), and 0.1 μM each of forward and reverse primers using a T100 (Bio-Rad, USA) thermal cycler. Thermal cycler reaction was performed as mentioned: initial denaturation at 94 ºC for 10 min, the mixture was cycled 35 times at 94 ºC for 45 s for denaturation, followed by annealing at 55–60 ºC for 45 s (following the TM values of the primer) and 72 ºC of extension for 60 s, and followed by a final extension at 72 ºC for 10 min. The samples were run on a 3.5% agarose gel by using bromophenol blue as a dye and a 50 bp ladder (New England Biolabs) for 1 h in 0.5X tris–acetic acid–EDTA (TAE) buffer. The resolved PCR bands were documented in a gel documentation system (Gel Doc XR + , Bio-Rad, USA), and the images were stored for analysis. In this study, with the prior information, 78 low-P QTL-linked primers (Table 2), including 13 Pup1-specific markers55, were used to map the QTLs associated with traits related to low P and the genetic architecture of the population panel.
Genetic diversity, population structure, and association analysis
The molecular size of the amplified fragments was determined by image lab software using 50 bp DNA ladders as the standard. Amplification of DNA samples with the primers was scored as per the molecular size or '1' for amplified regions and '0' for unamplified regions. A data matrix with '0' and '1' or the molecular size was prepared depending on the amplification, and the data matrix underwent further analysis. Genetic diversity parameters, major allele frequency, gene diversity, heterozygosity, and the polymorphic information content (PIC) were calculated for each SSR locus using Power marker software version 3.2556. An un-rooted tree with a bootstrap value of 1000 was used to construct an unweighted neighbor-joining tree (Nei 1972) using Darwin 6.0 software57. Analysis of molecular variance (AMOVA) was generated using the GenAlex 6.502 program to describe the presence of molecular variance components within and between the population differentiation among the five assumed sub-populations58 and to estimate the F statistics comprising the deviations from Hardy–Weinberg expectation across the population (FIT), within-population (FIS), and for correlation of alleles between the sub-populations (FST). In addition, Shannon's Information index observed and expected homozygosity, observed and expected heterozygosity, Nei's genetic diversity index, and the number of migrants (NM) between the assumed sub-populations were calculated by POPGENE program version 1.3159.
The genotypic data of the genotypes under study were analyzed for possible population structure with the model-based program STRUCTURE 2.3.460 using a length of the burn-in period of 1,00,000, followed by 10,000 Markov chain Monte Carlo (MCMC) replications. At least ten runs of STRUCTURE were performed by setting the number of sub-populations (K) from K = 1 to K = 10. To find the true K-value, ad hoc statistics ∆K was followed61 using Structure Harvester version 0.6.9462. In this study, both model and distance-based cluster analysis were carried out with the data generated with all 78 primers collectively, excluding 13 Pup1-specific markers and only with Pup1-specific markers to realize the distribution of the PSTOL1 gene across species and the significance of other markers under low P. Further, to determine the genetic relatedness between traits measured under low P, the rice genotypes and primers were analyzed by the general linear model (GLM) and mixed linear model (MLM) in TASSEL version 5.2.6363. False discovery rate (FDR) was used to obtain q-values (adjusted P-values) as described by earlier studies64. A significant association between markers and traits was identified based on their R2 and P-value.
Candidate gene analysis underlying a QTL region and coexpression of gene assay
The associated traits and SSR markers aligned to the IRGSP 1.0 genome in The Rice Annotation Project (https://rapdb.dna.affrc.go.jp/viewer/gbrowse/irgsp1/) to retrieve the genes associated with linked markers. The sequence on either side of each linked marker extended from 500 kb left and 500 kb right and was marked as a QTL region and meticulously searched to find the genes associated with low P tolerance. The interval on either side of the associated marker can be called an interval of the QTL genome65. Furthermore, we have performed the coexpression of gene assay for each putative candidate gene based on the guide gene approach using the riceFREND database (https://ricefrend.dna.affrc.go.jp/) to identify the coexpressed genes. This approach provided the details of coexpressed genes, functional information associated with reported genes, and transcription factors. The direct link of this database with RiceXPro (https://ricexpro.dna.affrc.go.jp/) helps to understand the insilico expression of these candidate genes. This integrative process in two databases allows the number of nodes of the gene interactions, and the ranking of nodes helps identify the groups of functionally related genes66.
Ethics approval and consent to participate
The authors declare that this research review was conducted in the absence of any commercial or economic associations that could be construed as potential conflicts of interest.
Data availability
The comprehensive collected data and information supporting the conclusions of this research article are provided as figures, tables, and supplementary tables.
References
Kochian, L. V. Plant nutrition: Rooting for more phosphorus. Nature 488, 466–467 (2012).
Zeng, H. et al. Genome-wide identification of phosphate-deficiency-responsive genes in soybean roots by high-throughput sequencing. Plant Soil 398, 207–227 (2016).
Tiessen, H. Phosphorus in the global environment BT. in The Ecophysiology of Plant-Phosphorus Interactions. (eds. White, P. J. & Hammond, J. P.). 1–7. https://doi.org/10.1007/978-1-4020-8435-5_1. (Springer, 2008).
Cordell, D., Drangert, J.-O. & White, S. The story of phosphorus: Global food security and food for thought. Glob. Environ. Chang. 19, 292–305 (2009).
Schindler, D. W., Carpenter, S. R., Chapra, S. C., Hecky, R. E. & Orihel, D. M. Reducing phosphorus to Curb Lake eutrophication is a success. Environ. Sci. Technol. 50, 8923–8929 (2016).
Neue, H. U., Lantin, R. S., Cayton, M. T. C. & Autor, N. U. Screening of rices for adverse soil tolerance BT. in Genetic Aspects of Plant Mineral Nutrition. (eds. El Bassam, N., Dambroth, M. & Loughman, B. C.). 523–531. https://doi.org/10.1007/978-94-009-2053-8_72. (Springer Netherlands, 1990).
Muralidharudu, Y. et al. GIS Based Soil Fertility Maps of Different States of India. (2011).
Van Kauwenbergh, S. J. World Phosphate Rock Reserves and Resources. (2010).
Sharma, V. P. & Thaker, H. Demand for fertilizer in India: Determinants and outlook 2020. Ind. J. Agric. Econ 66, 638–661 (2011).
White, P. J., Broadley, M. R. & Gregory, P. J. Managing the nutrition of plants and people. Appl. Environ. Soil Sci. 2012, 104826 (2012).
Veneklaas, E. J. et al. Opportunities for improving phosphorus-use efficiency in crop plants. New Phytol. 195, 306–320 (2012).
Essigmann, B., Güler, S., Narang, R. A., Linke, D. & Benning, C. Phosphate availability affects the thylakoid lipid composition and the expression of SQD1 a gene required for sulfolipid biosynthesis in Arabidopsis thaliana. Proc. Natl. Acad. Sci. U. S. A. 95, 1950–1955 (1998).
Sakuraba, Y. et al. A phytochrome-B-mediated regulatory mechanism of phosphorus acquisition. Nat. Plants 4, 1089–1101 (2018).
Anandan, A. et al. Trait variations and expression profiling of OsPHT1 gene family at the early growth-stages under phosphorus-limited conditions. Sci. Rep. 11, 13563 (2021).
Wang, X., Shen, J. & Liao, H. Acquisition or utilization, which is more critical for enhancing phosphorus efficiency in modern crops?. Plant Sci. 179, 302–306 (2010).
Wissuwa, M., Wegner, J., Ae, N. & Yano, M. Substitution mapping of Pup1: A major QTL increasing phosphorus uptake of rice from a phosphorus-deficient soil. Theor. Appl. Genet. 105, 890–897 (2002).
Chin, J. H. et al. Development and application of gene-based markers for the major rice QTL phosphorus uptake 1. Theor. Appl. Genet. 120, 1073–1086 (2010).
Gamuyao, R. et al. The protein kinase Pstol1 from traditional rice confers tolerance of phosphorus deficiency. Nature 488, 535–539 (2012).
Li, J., Xie, Y., Dai, A., Liu, L. & Li, Z. Root and shoot traits responses to phosphorus deficiency and QTL analysis at seedling stage using introgression lines of rice. J. Genet. Genomics 36, 173–183 (2009).
Smith, S. E. & Read, D. 17-Mycorrhizas in Agriculture, Horticulture and Forestry. (eds. Smith, S. E. & Read, D. B. T.-M. S.). Vol. 611-XVIII. https://doi.org/10.1016/B978-012370526-6.50019-2. (Academic Press, 2008).
MacLean, A. M., Bravo, A. & Harrison, M. J. Plant signaling and metabolic pathways enabling arbuscular mycorrhizal symbiosis. Plant Cell 29, 2319–2335 (2017).
Choi, J., Summers, W. & Paszkowski, U. Mechanisms underlying establishment of arbuscular mycorrhizal symbioses. Annu. Rev. Phytopathol. 56, 135–160 (2018).
Lanfranco, L., Fiorilli, V. & Gutjahr, C. Partner communication and role of nutrients in the arbuscular mycorrhizal symbiosis. New Phytol. 220, 1031–1046 (2018).
Yang, S. Y. et al. Nonredundant regulation of rice arbuscular mycorrhizal symbiosis by two members of the phosphate transporter1 gene family. Plant Cell 24, 4236–4251 (2012).
Richardson, A. E. & Simpson, R. J. Soil microorganisms mediating phosphorus availability update on microbial phosphorus. Plant Physiol. 156, 989–996 (2011).
Panda, S. et al. Proofing direct-seeded rice with better root plasticity and architecture. Int. J. Mol. Sci. 22, 6058 (2021).
Wissuwa, M., Mazzola, M. & Picard, C. Novel approaches in plant breeding for rhizosphere-related traits. Plant Soil 321, 409 (2008).
Wang, K. et al. Identification of quantitative trait loci for phosphorus use efficiency traits in rice using a high density SNP map. BMC Genet. 15, 155 (2014).
Panneerselvam, P. et al. Understanding interaction effect of arbuscular mycorrhizal fungi in rice under elevated carbon dioxide conditions. J. Basic Microbiol. 59, 1217–1228 (2019).
Anandan, A., Anumalla, M., Pradhan, S. K. & Ali, J. Population structure, diversity and trait association analysis in rice (Oryza sativa L.) germplasm for early seedling vigor (ESV) using trait linked SSR markers. PLoS ONE 11, 152406 (2016).
Donde, R. et al. Identification of QTLs for high grain yield and component traits in new plant types of rice. PLoS ONE 15, e0227785–e0227785 (2020).
Noyer, J. L., Tharreau, D., Ahmadi, N., Feyt, H. & Luce, C. The use of microsatellite markers to examine the diversity of the genetic resources of rice (Oryza sativa) adapted to European conditions. in International Symposium on Molecular Markers for Characterizing Genotypes and Identifying Cultivars in Horticulture. 221–235. (2000).
Xu, J. H., Cheng, C., Tsuchimoto, S., Ohtsubo, H. & Ohtsubo, E. Phylogenetic analysis of Oryza rufipogon strains and their relations to Oryza sativa strains by insertion polymorphism of rice SINEs. Genes Genet. Syst. 82, 217–229 (2007).
Joshi, S. P., Gupta, V. S., Aggarwal, R. K., Ranjekar, P. K. & Brar, D. S. Genetic diversity and phylogenetic relationship as revealed by inter simple sequence repeat (ISSR) polymorphism in the genus Oryza. Theor. Appl. Genet. 100, 1311–1320 (2000).
Rakshit, S. et al. Large-scale DNA polymorphism study of Oryza sativa and O. rufipogon reveals the origin and divergence of Asian rice. Theor. Appl. Genet. 114, 731–743 (2007).
Huang, P. et al. Phylogeography of Asian wild rice, Oryza rufipogon: A genome-wide view. Mol. Ecol. 21, 4593–4604 (2012).
Neelam, K. et al. Novel alleles of phosphorus-starvation tolerance 1 gene (PSTOL1) from Oryza rufipogon confers high phosphorus uptake efficiency. Front. Plant Sci. 8, 1–12 (2017).
Verma, H., Borah, J. L. & Sarma, R. N. Variability assessment for root and drought tolerance traits and genetic diversity analysis of rice germplasm using SSR markers. Sci. Rep. 9, 16513 (2019).
Islam, M. Z. et al. Diversity and population structure of red rice germplasm in Bangladesh. PLoS ONE 13, e0196096–e0196096 (2018).
Slatkin, M. Estimating levels of gene flow in natural populations. Genetics 99, 323–335 (1981).
Barbhuiya, A. R., Khan, M. L. & Dayanandan, S. Genetic structure and diversity of natural and domesticated populations of Citrus medica L. in the Eastern Himalayan region of Northeast India. Ecol. Evol. 6, 3898–3911 (2016).
Yu, K. et al. Features of rice (Oryza sativa L.) varieties for organic farming in connection with marker assisted breeding (review). Agric. Biol. 55, 847–860 (2020).
Chen, H.-C. et al. The function of OsbHLH068 is partially redundant with its homolog, AtbHLH112, in the regulation of the salt stress response but has opposite functions to control flowering in Arabidopsis. Plant Mol. Biol. 94, 531–548 (2017).
Zhang, F. et al. Overexpression of rice phosphate transporter gene OsPT6 enhances phosphate uptake and accumulation in transgenic rice plants. Plant Soil 384, 259–270 (2014).
Singh, A. et al. Comprehensive genomic analysis and expression profiling of phospholipase C gene family during abiotic stresses and development in rice. PLoS ONE 8, e62494 (2013).
Steele, K. A., Price, A. H., Shashidhar, H. E. & Witcombe, J. R. Marker-assisted selection to introgress rice QTLs controlling root traits into an Indian upland rice variety. Theor. Appl. Genet. 112, 208–221 (2006).
Zhang, Q., Wang, C., Tian, J., Li, K. & Shou, H. Identification of rice purple acid phosphatases related to posphate starvation signalling. Plant Biol. 13, 7–15 (2011).
Mehra, P., Pandey, B. K. & Giri, J. Improvement in phosphate acquisition and utilization by a secretory purple acid phosphatase (OsPAP21b) in rice. Plant Biotechnol. J. 15, 1054–1067 (2017).
Anandan, A. et al. Non-destructive phenotyping for early seedling vigor in direct-seeded rice. Plant Methods 16, 1–18 (2020).
Bhatta, B. B. et al. Improvement of phosphorus use efficiency in rice by adopting image-based phenotyping and tolerant indices. Front. Plant Sci. 12, 717107. https://doi.org/10.3389/fpls.2021.717107 (2021).
McGonigle, T. P., Miller, M. H., Evans, D. G., Fairchild, G. L. & Swan, J. A. A new method which gives an objective measure of colonization of roots by vesicular—arbuscular mycorrhizal fungi. New Phytol. 115, 495–501 (1990).
Lê, S., Josse, J. & Husson, F. FactoMineR: An R package for multivariate analysis. J. Stat. Softw. 25, 1–18 (2008).
Wei, T. et al. R package “corrplot”: Visualization of a correlation matrix. Statistician 56, 316–324 (2017).
Murray, M. G. & Thompson, W. F. Rapid isolation of high molecular weight plant DNA. Nucleic Acids Res. 8(19), 4321–5. https://doi.org/10.1093/nar/8.19.4321 (1980).
Mahender, A., Anandan, A. & Pradhan, S. K. Traits-related QTLs and genes and their potential applications in rice improvement under low phosphorus condition. Arch. Agron. Soil Sci. 64, 449–464. https://doi.org/10.1080/03650340.2017.1373764 (2018).
Liu, K. & Muse, S. V. PowerMarker: An integrated analysis environment for genetic marker analysis. Bioinformatics 21, 2128–2129 (2005).
Perrier, X. & Jacquemoud-Collet, J. P. DARwin Software. (2006).
Peakall, R. & Smouse, P. E. GenAlEx 6.5: Genetic analysis in Excel. Population genetic software for teaching and research—An update. Bioinformatics 28, 2537–2539 (2012).
Yeh, F. C., Yang, R. C. & Boyle, T. POPGENE Version 1.32: Microsoft Window-Based Freeware for Population Genetics Analysis. (1999).
Pritchard, J. K., Stephens, M. & Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 155, 945–959 (2000).
Evanno, G., Regnaut, S. & Goudet, J. Detecting the number of clusters of individuals using the software structure: A simulation study. Mol. Ecol. 14, 2611–2620 (2005).
Earl, D. A. & von Holdt, B. M. STRUCTURE HARVESTER: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 4, 359–361 (2012).
Bradbury, P. J. et al. TASSEL: Software for association mapping of complex traits in diverse samples. Bioinformatics 23, 2633–2635 (2007).
Benjamini, Y. & Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B 57, 289–300 (1995).
Kumar, S. et al. Identification of drought tolerant Rice genotypes by analysing drought tolerance indices and morpho-physiological traits. SABRAO J. Breed Genet. 46, 217–230 (2014).
Serin, E. A. R., Nijveen, H., Hilhorst, H. W. M. & Ligterink, W. Learning from co-expression networks: Possibilities and challenges. Front. Plant Sci. 7, 444. https://doi.org/10.3389/fpls.2016.00444 (2016).
Acknowledgements
The authors are profoundly grateful to the Director, ICAR-National Rice Research Institute, Cuttack, for providing all the necessary facilities and support for this work. Also, we thank and acknowledge IRRI Communications for the English-language editing.
Funding
The authors would like to thank and acknowledge the Bill & Melinda Gates Foundation (BMGF) for providing a research grant to JA for the Green Super Rice Project under ID OPP1130530. We would also like to thank the ICAR-National Rice Research Institute, Cuttack, Odisha, for providing funds to AA for the institutional project.
Author information
Authors and Affiliations
Contributions
A.A., R.N., S.S., and B.B.B. conducted the literature review and carried out the phenotypic experiment; A.A. and R.N. collected the phenotypic traits and performed the statistical analysis; A.A., H.S., and J.M. collected the rice germplasm; A.A., R.N., C.P., P.P., and A.M. interpreted the results and prepared the manuscript; and A.A., A.M., and J.A. revised the manuscript with inputs from other authors and reviewers. All authors have read and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Anandan, A., Nagireddy, R., Sabarinathan, S. et al. Multi-trait association study identifies loci associated with tolerance of low phosphorus in Oryza sativa and its wild relatives. Sci Rep 12, 4089 (2022). https://doi.org/10.1038/s41598-022-07781-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-07781-5
- Springer Nature Limited
This article is cited by
-
Exploring genetic diversity, population structure and stability for yield related traits in rice germplasm of northeastern India
Genetic Resources and Crop Evolution (2024)
-
Identification of novel marker-trait associations and candidate genes for combined low phosphorus and nitrogen-deficient conditions in rice at seedling stage
Tropical Plant Biology (2024)