
Abstract
miRNAs (or microRNAs) are small, endogenous, and noncoding RNAs construct of about 22 nucleotides. Cumulative evidence from biological experiments shows that miRNAs play a fundamental and important role in various biological processes. Therefore, the classification of miRNA is a critical problem in computational biology. Due to the short length of mature miRNAs, many researchers are working on precursor miRNAs (pre-miRNAs) with longer sequences and more structural features. Pre-miRNAs can be divided into two groups as mirtrons and canonical miRNAs in terms of biogenesis differences. Compared to mirtrons, canonical miRNAs are more conserved and easier to be identified. Many existing pre-miRNA classification methods rely on manual feature extraction. Moreover, these methods focus on either sequential structure or spatial structure of pre-miRNAs. To overcome the limitations of previous models, we propose a nucleotide-level hybrid deep learning method based on a CNN and LSTM network together. The prediction resulted in 0.943 (%95 CI ± 0.014) accuracy, 0.935 (%95 CI ± 0.016) sensitivity, 0.948 (%95 CI ± 0.029) specificity, 0.925 (%95 CI ± 0.016) F1 Score and 0.880 (%95 CI ± 0.028) Matthews Correlation Coefficient. When compared to the closest results, our proposed method revealed the best results for Acc., F1 Score, MCC. These were 2.51%, 1.00%, and 2.43% higher than the closest ones, respectively. The mean of sensitivity ranked first like Linear Discriminant Analysis. The results indicate that the hybrid CNN and LSTM networks can be employed to achieve better performance for pre-miRNA classification. In future work, we study on investigation of new classification models that deliver better performance in terms of all the evaluation criteria.
Similar content being viewed by others

Explore related subjects
Find the latest articles, discoveries, and news in related topics.Introduction
miRNAs (or microRNAs) are small, endogenous, and noncoding RNA constructs of about 22 nucleotides1. Cumulative evidence from biological experiments shows that miRNAs play a fundamental and important role in various biological processes such as regulation of gene expression by post-transcriptionally binding to 5'untranslated regions (UTR), coding sequences, or 3´UTR of target messenger RNAs (mRNAs)2,3. According to the latest release of an online miRNA database, miRBase (v22), there are 38,589 entries representing hairpin precursor miRNAs that express 48,860 mature miRNAs from 271 organisms such as humans, mice, rat, etc.4. The human genome, as a sub-category of the organism classification, contains 1917 annotated hairpin precursors, and 2654 mature sequences4. It is estimated that in mammals, approximately one-third of all protein-coding genes’ activities are controlled by miRNAs5. Several studies show that the deregulations of miRNAs are associated with many types of human diseases, e.g. cancer, cardiovascular diseases, or autoimmune diseases6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27. Due to these relationships between miRNAs and various diseases, studies to understand the functions, processes, and mechanisms of miRNAs are increasing dramatically28. Thus, how to classify miRNAs is a critical problem in computational biology.
The discovery of the first miRNA started in Caenorhabditis elegans in 1993 by Ambros and Ruvkun's studies. They found that the lin-4 was a small noncoding RNA but not a protein-coding RNA29,30,31. Seven years later, in 2000, the second miRNA, let-7, was reported. Experimental results show that let-7 consists of 21 nucleotide RNA and regulates timing in the transition from fourth level (L4) to adult C. elegans’ larval development32.
The biogenesis of miRNAs involves several steps and cellular mechanisms (Fig. 1), some in the nucleus and some in the cytoplasm. Since those processes have some different pathways, pre-miRNAs can be categorized into two categories: mirtrons and canonical miRNAs. Compared to mirtrons, canonical miRNAs are more conserved and easier to be identified33. The first step of the biogenesis of miRNAs begins with the transcription of miRNA genes that make up primary miRNA hairpins called pri-miRNA34,35,36. In the canonical pathway, pre-miRNAs with the hairpin structure are formed by the microprocessor complex consisting of Drosha and DGCR by dividing pri-miRNAs in the nucleus37,38. Then, the pre-miRNAs are produced in the nucleus and transported into the cytoplasm by exportin-5. Following this, pre-miRNAs are cleaved in the cytoplasm into small RNA duplexes by another RNase III enzyme Dicer and finally, mature miRNA is produced39,40. In the mirtron pathway, for bypassing the nuclear enzyme Drosha, it uses splicing to produce short pre-miRNA hairpin introns41,42. The next steps of those pre-miRNAs are in the same pathway as canonical miRNAs43. Mirtrons can also be divided into three categories: canonical, 3′ tailed, and 5′ tailed due to their sequence and structure42. Compared to canonical miRNAs, mirtron hairpins and small RNAs have numerous distinguishing features44,45.

Overview of miRNA biogenesis.
In previous studies, numerous computational methods, such as decision trees (DT), random forest (RF), and support vector machines (SVM), widely applied in miRNA identification and classification as applied in computational biology and healtcare46,47,48,49,50,51,52. Recently, Deep Learning (DL) methods are also frequently used to achieve better prediction accuracy compared with other traditional machine learning methods 53,54,55,56,57,58,59. The convolutional neural networks CNNs, a type of DL, have successfully employed for pre-miRNAs clasification33,56. For instance, Zheng et al.33 proposed a nucleotide-level CNN model. They encoded the sequences using “one-hot” encoding then padded each entry with the same shape. The model had convolutions and max-pooling layers. Their investigation showed that their CNN-based network feasible to apply to extract features from biological sequences. CNN-based methods outperform to identify the miRNAs and extract features automatically from the raw input data without detailed domain knowledge60,61,62,63. However, Park et al.64 show that the spatial information of these structures is as important as the structures that make up miRNAs. Therefore, they focused on only long-term dependencies and proposed an LSTM based framework to identify precursor miRNAs. Moreover, much research reveals that CNN-LSTM networks give a solution to use both structural characterization and spatial information together. A CNN-LSTM network is a combination of CNN layers for feature extraction on input data and LSTM layers to provide sequence prediction65. These networks are used in a variety of problems such as activity recognition, image description, video description, visual time series prediction, and generating textual annotations from image sequences65,66. Quang et al. proposed a hybrid CNN-LSTM framework67, DanQ, for predicting the function of DNA sequences. In this model, the convolution layer captures patterns, and the recurrent layer captures long-term dependencies. Similarly, Pan et al. proposed iDeepS68, to identify the binding sequence and structure patterns from RNA sequences. Their model extract features by using CNN and reveals possible long-term dependencies by using bi-directional LSTM (BLSTM). These successful studies show that utilizing both spatial and sequential features provides higher performance, especially in computational biology.
Many existing pre-miRNA classification methods focus on either sequential structure or spatial structure of pre-miRNAs. The main features that distinguish pre-miRNAs from each other are the types, number, and sequence order of amino acids that make up their fundamental structure. Hence, using a hybrid CNN-LSTM based network can give a solution to classify pre-miRNA facilitating with both spatial and sequential features of pre-miRNAs.
Materials and methods
In this study, we presented a hybrid deep learning method for pre-miRNA classification based on both sequential and spatial structure of pre-miRNA by integrating two different networks respectively: CNN and LSTM. We first described the problem of pre-miRNA classification. Then, we introduced the dataset, which is used to train and evaluate the proposed method. The dataset consisted of human mirtrons and canonical miRNAs sequences44. For consistency, the same sequence data were used as the previous models33,46. CNN extracted features from the input data automatically. Thus, it gave a solution to the problem of manual extraction of features. LSTM layer was used to perform temporal modeling following the CNN layer that convolved the input data. Next, we gave comprehensive details about CNN, LSTM, and CNN-LSTM networks. Finally, we described our proposed method and how to implement it in detail. The method was implemented in python using the Keras library (2.4.3) https://github.com/keras-team/keras, with the backend of TensorFlow (2.4.0).
Problem statement
Many existing pre-miRNA classification methods rely on manual future extraction. These methods focus on either spatial structure or sequential structure of pre-miRNAs. To overcome the limitations of previous models, we propose a nucleotide-level deep learning method based on a hybrid CNN and LSTM network together for pre-miRNAs classification. When we consider the structure and sequence of pre-miRNAs, it is clear that the problem is a binary sequence classification problem consisting of mirtrons and canonical miRNAs. In literature, several models including machine-learning methods have been developed to find a solution for the problematic classification. On the other hand, they have approximately 90% of accuracy. In this study, in the pre-miRNA classification, our goal was to show how to accurately predict classes with a hybrid CNN-LSTM network.
Convolutional neural networks
A CNN network is a type of deep learning that produces excellent performance and has been widely applied to many applications such as image classification69,70, object detection71,72, speech recognition73, computer vision74, video analysis75, and bioinformatics76,77. Apart from the traditional neural networks, CNN includes numerous layers that make it deeper. Moreover, CNN uses weights, biases, and outputs via a nonlinear activation. A typical CNN architecture fundamentally consists of convolutional layers, pooling layers, and fully connected layers63.
The convolution operation used in the convolutional layer is as follows:
where I for input matrix, K for a 2D filter of size m × n, and F for the output of a 2D feature map. And, the convolutional layer representation is with I*K.
Long short-term memory networks
An LSTM network is a class of recurrent neural network (RNN) that uses memory blocks that assist to run successfully and learn faster than traditional RNN78,79. LSTM networks find practical solutions for the vanishing and exploding gradient problems of RNNs80. Apart from the RNNs, a cell state is used in the LSTM network to save long-term states including input, forget, and output gates. Thus, the network can remember previous data and connect it with the present ones. Also, it solves complicated tasks difficult to find a solution by previous RNNs79,81.
CNN and LSTM networks
A CNN-LSTM model is a combination of CNN layers that extract the feature from input data and LSTMs layers to provide sequence prediction65. The CNN-LSTM is generally used for activity recognition, image labeling, and video labeling. Their common features are that they are developed for the application of visual time series prediction problems and generating textual annotations from image sequences65,66.
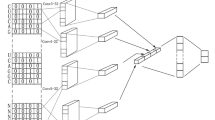
Figure 2 shows the basic architecture of the CNN-LSTM network with the input layer, visual feature extraction, sequence learning, and output layer, respectively65.

The basic architecture of the CNN-LSTM network.
Training and test datasets
The dataset consists of mirtrons and canonical miRNAs’ data. We combined two different datasets in our preprocessing data phase with 707 (63%) canonical miRNAs and 417 (37%) mirtrons. The first dataset (Dataset 1) consisted of mirtrons and canonical miRNAs derived from miRBase (v21) according to the annotation of Wen et.al.44. Moreover, the second dataset (Dataset 2) was derived also from the study of Wen et al.44, which included 201 entries, putative mirtrons data. In total, we used 1,124 entries in our proposed model. The same dataset used to be consistent with Zheng et al. and Rorbach et al.33,46.
Stratified k-folds cross-validation (CV) is a resampling procedure that splits of the dataset into folds according to the output categories and ensures that each fold has the same proportion. It is useful for imbalanced datasets82. Hence, we used stratified 5-folds CV for training and evaluating our model82. At each iteration, it divided the data into training and test sets with a 80–20% split. In the next iteration, it used the other percentile as the training and test set.
Table 1 shows the distribution of the training and test datasets at each iteration in stratified 5-folds CV.
The preprocessing of the data
The entry with the most sequences had 164 bases. Therefore, we prepared each sequence of entries with the maximum length (164) by padding. The word "N" was used for keeping the sequences in the same length. Like Zheng et al.33, “one-hot” encoding is used to encode each base of the sequences (Table 2). Next, we converted each sequence into a vector with a dimension of (164, 4) by the vectorization process.
The method architecture
We designed the architecture of our model with nine layers: an input layer, four CNN layers wrapped by the time-distributed layers, an LSTM layer, a dense layer, a dropout layer, and an output layer, respectively. Figure 3 shows the illustration of the architecture with visualization of our method. Before constructing the model, we ensured that each data has been transformed into an appropriate form to be used. In this case, we used the padding process to guarantee the length (which is 164) of each miRNA sequence similar by adding "N" for each blank. The next vectorization step was transforming the padded sequences to like a m × n matrix by using one-hot encoding.

Detailed architecture with visualization of the proposed methodology.
When all data are padded and vectorized, the network became ready for the feature extraction process. In this stage, three convolution layers were used to automatically extract features from input sequences using the relu activation function. In these convolutional layers, 128 filters were used. The kernel's height was selected as 6 and the kernel's width was selected 4 for convolution operation. This kernel size gives higher performance33. In these convolutional stages. We wrapped the convolution layers in a time-distributed wrapper to reshape input data by adding extra dimension at the end. For concatenation of all extracted features, we employed a flatten layer for passing to the LSTM layer. Then, one LSTM layer was designed with 100 units following a dropout layer (0.5) on the fully connected layer. Finally, for binary classification, the softmax activation function was used for specifying outputs. The model was optimized for 30 epochs, 6 for batch size, and 0.1 for validation split by training. The validation dataset monitors the convergence in the training process so that the training of the model can be canceled early according to the change in this convergence. Besides, adam optimizer with a 0,001 learning rate for optimization and categorical cross entropy for loss function was preferred during the optimization process. Adam is one of the gradient descent algorithms that calculate adaptive learning rates for each momentum-like parameter83 and categorical cross entropy is one of the loss functions preferred when there are two or more one-hot encoded label classes84. It optimizes multi-class classification models with a softmax activation function.
Table 3 shows the model summary including the input layer, convolution layers, flatten layer, LSTM layer, fully-connected layer, softmax layer and classification layer with the shape and the number of the parameters.
Method evaluation
In this study, the evaluation of our method was measured on the test dataset. We calculated five different measurements for performance in the analysis: accuracy (Acc.), sensitivity (Sen.), specificity (Spe.), F1 score, and Matthews Correlation Coefficient (MCC). They are calculated for evaluating predictive capability with the number of true positives (TP), true negatives (TN), false positives (FP), and false negatives (FN) by the following equations:
Accuracy indicates the overall correctness of prediction:
Sensitivity, true positive rate, indicates the ratio of correctly classified actual positives:
Specificity, true negative rate, indicates the ratio of correctly classified actual negatives:
F1-Score is a combination of the precision and recall of the model by harmonic mean:
Matthews Correlation Coefficient (MCC) is a binary classifier that measures the quality:
Results and discussion
Due to the automatic feature extraction without a comprehensive domain expert from pre-miRNAs sequences by using CNN and LSTM, we designed a hybrid method for the classification of pre-miRNAs. We started with preparing the dataset by converting the raw sequences to vectors using “one-hot” encoding. Next, all data were padded and vectorized. Then, we used three convolution layers to extract features automatically from input sequences using the relu activation function. For concatenation of all extracted features, we employed a flatten layer for passing to the LSTM layer. Then, one LSTM layer is designed with 100 units following a dropout layer (0.5) on the fully connected layer. Finally, for binary classification, the softmax activation function was used for the specifying outputs. Table 4 shows the performance results of our proposed network at each iteration. Additionally, we calculated mean, median, standard deviation, and confidence interval (CI) for each metric.
Table 5 shows the performance comparison of the average values of the proposed method with the previous methods. The prediction resulted in 0.943 (%95 CI ± 0.014) accuracy, 0.935 (%95 CI ± 0.016) sensitivity, 0.948 (%95 CI ± 0.029) specificity, 0.925 (%95 CI ± 0.016) F1 Score and 0.880 (%95 CI ± 0.028) MCC (Table 4) When compared to the closest results, our network revealed the best results for Acc., F1 Score, and MCC. These were 2.51%, 1.00%, and 2.43% higher than the closest result, respectively. The mean of sensitivity had the highest value like Linear Discriminant Analysis and ranked first. These ratios indicate that the hybrid CNN and LSTM networks can be employed to achieve better performance for pre-miRNA classification compared with previous methods. Even though the results show that our model has a higher ratio according to accuracy, sensitivity, F1 score, and MCC; we have a lower ratio (94.8%) of correctly classified true negatives. In imbalanced or skewed datasets, the number of examples of the minority class might not be sufficient for learning. As a result, the minority group is more often misclassified than the majority group85,86. The number of positive and negative samples in our training and test dataset is equally representative of the entire dataset. Thus, we solve the misclassification problem at the data preparation level.
This study is an investigation of the pre-miRNA classification problem through a convolutional neural network and long short-term memory network. In contrast to other methods, we took into account both the sequence structure and the spatial information of each entry. The preprocessing of data is the first but the most important stage of our study. Indeed, inappropriate preparation of the data will cause the network to be trained incorrectly and will make it difficult to obtain reliable results. Thus, we checked all outputs after the encoding, padding, and vectorization process. In addition, cascading the different neural networks is another issue in the model construction. Inappropriate network design may increase the bias and cause unexpected results. Therefore, we ensured that all the layers cascaded correctly.
Hyper-parameters determine the general characteristics of deep neural networks. The number of the hidden units, order of the layers, batch size, optimizer selection, and learning rate, etc. directly affect the performance of the methods. In this study, we utilized the previous researcher's experiments in addition to our experiments. For instance, Zheng et al.33 discovered that kernel size (6 × 4) and unit number (128) of the CNN network produced the best results according to other sizes and numbers in the pre-miRNA classification. When we tested hyperparameters like Zheng et al.33, we obtained similar performance results as they did. In our future work, we will take into account the experiences we have gained in these studies and we will do more extensive hyper-parameter optimization to ensure performance increase.
Despite the promising performance of our model, there are still some limitations. The first limitation comes from the total number of entries (1124) in the datasets. Even though the datasets have well-defined data, it is important to feed the method with more training and testing data to obtain more reliable results. The second limitation is the unbalanced ratio of classes. In this study, the number of positive samples (417) was less than the number of negative samples (707). The ratio of positive and negative samples was approximately 1:1.7. This imbalanced ratio may lead to limit accuracy and other metrices. Thus, we will focus on more comprehensive datasets in the future research.
We consider that the quality and size of the related dataset are important for training a model and achieving robust classification prediction. In future studies, enhanced datasets may lead to the construction of more successful models in terms of similar evaluation parameters.
Conclusion
In this paper, we proposed a nucleotide-level hybrid deep learning method based on a convolutional neural network and long-short term memory network together. In the data preprocessing phase, we used one-hot encoding to convert each base to a matrix of the same size by padding. Then, we employed three convolution layers wrapped by a time-distribution layer. For concatenation of all extracted features, we employed a flatten layer for passing to the LSTM layer. Then, we designed one LSTM layer following a dropout layer on the fully connected layer. Finally, for binary classification, the softmax activation function is used for specifying the outputs. Our results showed that the proposed method was successfully trained on the training dataset and had a better performance on the test dataset than the previous models.
The results indicated that the hybrid CNN and LSTM networks can be employed to achieve better performance for pre-miRNA classification. In future work, we will study on the investigation of new classification models that deliver better performance in terms of all the evaluation metrics.
References
Hammond, S. M. An overview of microRNAs. Adv. Drug Deliv. Rev. 87 (n.d.).
Bartel, D. P. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 116, 281–297 (n.d.).
Ambros, V. The functions of animal microRNAs. Nature. 431 (n.d.).
Kozomara, A., Birgaoanu, M. & Griffiths-Jones, S. MiRBase: from microRNA sequences to function. Nucleic Acids Res. https://doi.org/10.1093/nar/gky1141 (2019).
Filipowicz, W., Bhattacharyya, S. N. & Sonenberg, N. Mechanisms of post-transcriptional regulation by microRNAs: are the answers in sight?. Nat. Rev. Genet. https://doi.org/10.1038/nrg2290 (2008).
Meister, G. & Tuschl, T. Mechanisms of gene silencing by double-stranded RNA. Nature. 431 (n.d.).
Karp, X. & Ambros, V. Encountering MicroRNAs in cell fate signaling. Science (80-. ). 310, 1288–1289 (n.d.).
Garzon, R., Calin, G. A. & Croce, C. M. MicroRNAs in cancer. Annu. Rev. Med. https://doi.org/10.1146/annurev.med.59.053006.104707 (2009).
Zhao, Y., Samal, E. & Srivastava, D. Serum response factor regulates a muscle-specific microRNA that targets Hand2 during cardiogenesis. Nature https://doi.org/10.1038/nature03817 (2005).
Cheng, Y. & Zhang, C. MicroRNA-21 in cardiovascular disease. J. Cardiovasc. Transl. Res. https://doi.org/10.1007/s12265-010-9169-7 (2010).
Cheng, Y. et al. MicroRNAs are aberrantly expressed in hypertrophic heart: Do they play a pole in cardiac hypertrophy?. Am. J. Pathol. https://doi.org/10.2353/ajpath.2007.061170 (2007).
Sonkoly, E. et al. MicroRNAs: novel regulators involved in the pathogenesis of psoriasis?. PLoS ONE https://doi.org/10.1371/journal.pone.0000610 (2007).
Lee, Y. S. & Dutta, A. MicroRNAs in cancer. Annu. Rev. Pathol. Mech. Dis. https://doi.org/10.1146/annurev.pathol.4.110807.092222 (2009).
Peng, Y. & Croce, C. M. The role of micrornas in human cancer. Signal Trans. Target. Ther. 15004 (n.d.).
Qin, S. & Zhang, C. Micrornas in vascular disease. J. Cardiovasc. Pharmacol. 57 (n.d.).
Jamaluddin, M. S. Mirnas: roles and clinical applications in vascular disease. Expert. Rev. Mol. Diagn. 11 79–89 (n.d.).
Dalal, S. R., Kwon, J. H. The role of microrna in inflammatory bowel disease. Gastroenterol. Hepatol. 6 (n.d.).
Cheng, A. M., Byrom, M. W., Shelton, J., & Ford, L. P. Antisense inhibition of human miRNAs and indications for an involvement of miRNA in cell growth and apoptosis. Nucleic Acids Res. 33, 1290–1297 (n.d.).
Chapman, C. G. & Pekow, J. The emerging role of mirnas in inflammatory bowel disease: a review. Ther. Adv. Gastroenterol. 8, 4–22 (n.d.).
Hayes, J., Peruzzi, P. P., & Lawler, S. Micrornas in cancer: biomarkers, functions and therapy. Trends Mol. Med. 20, 460–469 (n.d.).
Kir, D., Schnettler, E., Modi, S., & Ramakrishnan, S. Regulation of angiogenesis by microRNAs in cardiovascular diseases. Angiogenesis (n.d.). https://doi.org/10.1007/s10456-018-9632-7.
Lu, J. et al. MicroRNA expression profiles classify human cancers. Nature https://doi.org/10.1038/nature03702 (2005).
Miska, E. A. How microRNAs control cell division, differentiation and death. Curr. Opin. Genet. Dev. 15 (n.d.).
Mandujano-Tinoco, E. A., Garcia-Venzor, A., Melendez-Zajgla, J., & Maldonado, V. New emerging roles of microRNAs in breast cancer. Breast Cancer Res. Treat. 10–1007 (n.d.).
Singh, R. P. The role of miRNA in inflammation and autoimmunity. Autoimmun. Rev. 12, 10–1016 (n.d.).
Calin, G. A. et al. Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers. Proc. Natl. Acad. Sci. USA. https://doi.org/10.1073/pnas.0307323101 (2004).
Li, C., Feng, Y., Coukos, G. & Zhang, L. Therapeutic microRNA strategies in human cancer. AAPS J. https://doi.org/10.1208/s12248-009-9145-9 (2009).
Alles, J. et al. An estimate of the total number of true human miRNAs. Nucleic Acids Res. https://doi.org/10.1093/nar/gkz097 (2019).
Lee, R. C., Feinbaum, R. L., & Ambros,V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. (1993). https://doi.org/10.1016/0092-8674(93)90529-Y.
Wightman, B., Ha, I., & Ruvkun, G. Posttranscriptional regulation of the heterochronic gene lin-14 by lin-4 mediates temporal pattern formation in C. Elegans. Cell. (1993). https://doi.org/10.1016/0092-8674(93)90530-4.
Lee, R., Feinbaum, R., & Ambros, V. A short history of a short RNA. Cell. (2004). https://doi.org/10.1016/s0092-8674(04)00035-2.
Reinhart, B. J. et al. The 21-nucleotide let-7 RNA regulates developmental timing in Caenorhabditis elegans. Nature https://doi.org/10.1038/35002607 (2000).
Zheng, X., Xu, S., Zhang, Y. & Huang, X. Nucleotide-level convolutional neural networks for pre-miRNA classification. Sci. Rep. https://doi.org/10.1038/s41598-018-36946-4 (2019).
Siomi, H. & Siomi, M. C. Posttranscriptional regulation of MicroRNA biogenesis in animals. Mol. Cell. https://doi.org/10.1016/j.molcel.2010.03.013 (2010).
Han, J. Molecular basis for the recognition of primary microRNAs by the Drosha-DGCR8 complex. Cell. 125, 10–1016 (n.d.).
O’Brien, J., Hayder, H., Zayed, Y. & Peng, C. Overview of microRNA biogenesis, mechanisms of actions, and circulation. Front. Endocrinol. (Lausanne). https://doi.org/10.3389/fendo.2018.00402 (2018).
Denli, A. M., Tops, B. B., Plasterk, R. H., Ketting, R. F., & Hannon, G. J. Processing of primary micrornas by the microprocessor complex. Nat. 432 (n.d.).
Gregory, R. I. et al. The microprocessor complex mediates the genesis of microRNAs. Nature https://doi.org/10.1038/nature03120 (2004).
Lee, Y. et al. The nuclear RNase III Drosha initiates microRNA processing. Nature https://doi.org/10.1038/nature01957 (2003).
Lund, E., Guttinger, S., Calado, A., Dahlberg, J. E., & Kutay, U. Nuclear export of microRNA precursors. Science (80-. ). 303, 10–1126 (n.d.).
Ruby, J. G., Jan, C. H. & Bartel, D. P. Intronic microRNA precursors that bypass Drosha processing. Nature https://doi.org/10.1038/nature05983 (2007).
Westholm, J. O. & Lai, E. C. Mirtrons: MicroRNA biogenesis via splicing. Biochimie https://doi.org/10.1016/j.biochi.2011.06.017 (2011).
Berezikov, E., Chung, W. J., Willis, J., Cuppen, E. & Lai, E. C. Mammalian mirtron genes. Mol. Cell. https://doi.org/10.1016/j.molcel.2007.09.028 (2007).
Wen, J., Ladewig, E., Shenker, S., Mohammed, J. & Lai, E. C. Analysis of Nearly One Thousand Mammalian Mirtrons Reveals Novel Features of Dicer Substrates. PLoS Comput. Biol. https://doi.org/10.1371/journal.pcbi.1004441 (2015).
Fromm, B. A uniform system for the annotation of vertebrate microRNA genes and the evolution of the human microRNAome. Annu. Rev. Genet. 49, 213–242 (n.d.).
Rorbach, G., Unold, O., & Konopka, B. M. Distinguishing mirtrons from canonical miRNAs with data exploration and machine learning methods. Sci. Rep. 8, 10–1038 (n.d.).
Gambhir, S., Malik, S. K. & Kumar, Y. Role of soft computing approaches in healthcare domain: a mini review. J. Med. Syst. https://doi.org/10.1007/s10916-016-0651-x (2016).
Peker, M., Şen, B. & Delen, D. Computer-aided diagnosis of Parkinson’s disease using complex-valued neural networks and mRMR feature selection algorithm. J. Healthc. Eng. https://doi.org/10.1260/2040-2295.6.3.281 (2015).
Şen, B. & Peker, M. Novel approaches for automated epileptic diagnosis using FCBF selection and classification algorithms. Turk. J. Electr. Eng. Comput. Sci. https://doi.org/10.3906/elk-1203-9 (2013).
Peker, M., Sen, B. & Delen, D. A novel method for automated diagnosis of epilepsy using complex-valued classifiers. IEEE J. Biomed. Heal. Inform. https://doi.org/10.1109/JBHI.2014.2387795 (2016).
Atasoy, F., Sen, B., Nar, F., & Bozkurt, I. Improvement of radial basis function ınterpolation performance on cranial ımplant design. Int. J. Adv. Comput. Sci. Appl. https://doi.org/10.14569/ijacsa.2017.080811 (2017).
Hofacker, I. L. Vienna RNA secondary structure server. Nucleic Acids Res. 31, 3429–3431 (n.d.).
Ng, K. L. S. & Mishra, S. K. De novo SVM classification of precursor microRNAs from genomic pseudo hairpins using global and intrinsic folding measures. Bioinformatics https://doi.org/10.1093/bioinformatics/btm026 (2007).
Jiang, P. et al. MiPred: Classification of real and pseudo microRNA precursors using random forest prediction model with combined features. Nucleic Acids Res. https://doi.org/10.1093/nar/gkm368 (2007).
Sacar Demirci, M. D., Baumbach, J., Allmer, J. On the performance of pre-microRNA detection algorithms. Nat. Commun. 8, 330. https://doi.org/10.1038/s41467-017-00403-z (n.d.).
Zheng, X., Fu, X., Wang, K. & Wang, M. Deep neural networks for human microRNA precursor detection. BMC Bioinform https://doi.org/10.1186/s12859-020-3339-7 (2020).
Do, B. T., Golkov, V., Gürel, G. E. & Cremers, D. Precursor microRNA identification using deep convolutional neural networks. BioRxiv. https://doi.org/10.1101/414656 (2018).
Cordero, J., Menkovski, V. & Allmer, J. Detection of pre-microRNA with convolutional neural networks. BioRxiv. https://doi.org/10.1101/840579 (2019).
Xue,C. Classification of real and pseudo microrna precursors using local structure-sequence features and support vector machine. BMC Bioinform. 6 (n.d.).
Huang, D. S. A constructive approach for finding arbitrary roots of polynomials by neural networks. IEEE Trans. Neural Netw. https://doi.org/10.1109/TNN.2004.824424 (2004).
Zhang, Y. et al. Using ensemble methods to deal with imbalanced data in predicting protein-protein interactions. Comput. Biol. Chem. https://doi.org/10.1016/j.compbiolchem.2011.12.003 (2012).
Albuquerque Vieira, J. P., & Moura, R. S. An Analysis of Convolutional Neural Networks for Sentence Classification (n.d.).
Krizhevsky, A., Sutskever, I., & Hinton, G. E. ImageNet classification with deep convolutional neural networks. Commun. ACM. 60, 10–1145 (n.d.).
Park, S., Min, S., Choi, H., & Yoon, S. deepMiRGene: deep neural network based precursor microRNA prediction (2016). http://arxiv.org/abs/1605.00017. Accessed June 6, 2021.
Donahue, J. et al. Long-term recurrent convolutional networks for visual recognition and description. IEEE Trans. Pattern Anal. Mach. Intell. https://doi.org/10.1109/TPAMI.2016.2599174 (2017).
Sainath, T. N., Vinyals, O., Senior, A., & Sak, H. Convolutional, long short-term memory, fully connected deep neural networks. İn ICASSP, IEEE International Conference on Acoustics, Speech, and Signal Processing—Proceedings. https://doi.org/10.1109/ICASSP.2015.7178838(2015).
Quang, D. & Xie, X. DanQ: A hybrid convolutional and recurrent deep neural network for quantifying the function of DNA sequences. Nucleic Acids Res. 44, e107–e107. https://doi.org/10.1093/nar/gkw226 (2016).
Pan, X., Rijnbeek, P., Yan, J. & BinShen, H. Prediction of RNA-protein sequence and structure binding preferences using deep convolutional and recurrent neural networks. BMC Genomics 19, 511. https://doi.org/10.1186/s12864-018-4889-1 (2018).
Jia, Y., Shelhamer, E., Donahue, J., Karayev, S., Long, J., Girshick, R., Guadarrama, S., & Darrell, T. Caffe: convolutional architecture for fast feature embedding. İn MM 2014, Proceedings of 2014 ACM Conference on Multimedia. https://doi.org/10.1145/2647868.2654889 (2014).
Zhao, Z. Q., Xie, B. J., Cheung, Y. M., & Wu, X. Plant leaf identification via a growing convolution neural network with progressive sample learning. İn Lecture Notes Computer Science (Including Subser. Lecture Notes Artificial Intellgent Lecture Notes Bioinformatics) (2015). https://doi.org/10.1007/978-3-319-16808-1_24.
Xiang, Y., Choi, W., Lin, Y., & Savarese, S. Subcategory-aware convolutional neural networks for object proposals and detection. İn Proceedings of 2017 IEEE Winter Conference on Application Computer Vision, WACV 2017 (2017). https://doi.org/10.1109/WACV.2017.108.
Galvez, R. L., Bandala, A. A., Dadios, E. P., Vicerra, R. R. P., & Maningo, J. M. Z. Object detection using convolutional neural networks. İn IEEE Reg. 10 Annual Interational Conference Proceedings/TENCON (2019). https://doi.org/10.1109/TENCON.2018.8650517.
Abdel-Hamid, O., Mohamed, A. R., Jiang, H., & Penn, G. Applying convolutional neural networks concepts to hybrid NN-HMM model for speech recognition. İn ICASSP, IEEE International Conference on Acoustics, Speech, and Signal Processing.: Proceedings (2012). https://doi.org/10.1109/ICASSP.2012.6288864.
Karpathy, A., Toderici, G., Shetty, S., Leung, T., Sukthankar, R., & Li, F. F. Large-scale video classification with convolutional neural networks. İn Proceedings of IEEE Computer Society Conference on Computer Vision and Pattern Recognition (2014). https://doi.org/10.1109/CVPR.2014.223.
Ngiam, J., Khosla, A., Kim, M., Nam, J., Lee, H., & Ng, A. Y. Multimodal deep learning. İn Proceedings 28th Internationl Conference on Machanical Learning ICML 2011 (2011).
Alipanahi, B., Delong, A., Weirauch, M. T. & Frey, B. J. Predicting the sequence specificities of DNA- and RNA-binding proteins by deep learning. Nat. Biotechnol. https://doi.org/10.1038/nbt.3300 (2015).
Zhou, J. & Troyanskaya, O. G. Predicting effects of noncoding variants with deep learning-based sequence model. Nat. Methods. https://doi.org/10.1038/nmeth.3547 (2015).
Gers, F. Long short-term memory in recurrent neural networks. Neural Comput. (2001).
Hochreiter, S. & Schmidhuber, J. Long short-term memory. Neural Comput. https://doi.org/10.1162/neco.1997.9.8.1735 (1997).
Hochreiter, S. The vanishing gradient problem during learning recurrent neural nets and problem solutions. Int. J. Uncertain. Fuzz. Knowl. Based Syst. https://doi.org/10.1142/S0218488598000094 (1998).
Greff, K., Srivastava, R. K., Koutnik, J., Steunebrink, B. R. & Schmidhuber, J. LSTM: a search space odyssey. IEEE Trans. Neural Netw. Learn. Syst. https://doi.org/10.1109/TNNLS.2016.2582924 (2017).
He, H., & Ma, Y. Imbalanced learning: foundations, algorithms, and applications (2013).
Kingma, P. E. & Ba, J. L. Adam: a method for stochastic optimization. İn 3rd The International Conference on Learning Representations ICLR 2015—Conference on Tracking Proceedings, International Conference on Learning Representations, ICLR (2015). https://arxiv.org/abs/1412.6980v9. Accessed June 7, 2021.
Probabilistic losses (n.d.). https://keras.io/api/losses/probabilistic_losses/#categoricalcrossentropy-class. Accessed June 7, 2021.
Johnson, J. M. & Khoshgoftaar, T. M. Survey on deep learning with class imbalance. J. Big Data. 6, 1–54. https://doi.org/10.1186/s40537-019-0192-5 (2019).
Kotsiantis, S., Kanellopoulos, D., Pintelas, P. E., Kanellopoulos, D., & Pintelas, P. Handling imbalanced datasets: a review data preprocessing view project machine learning and data mining view project handling imbalanced datasets: a review (n.d.). https://www.researchgate.net/publication/228084509. Accessed June 8, 2021.
Author information
Authors and Affiliations
Contributions
A.T. and B.S. conceived and developed the idea of the study and implemented mothods, contributed to interpretation and discussion of obtained results and commented on the manuscript and wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tasdelen, A., Sen, B. A hybrid CNN-LSTM model for pre-miRNA classification. Sci Rep 11, 14125 (2021). https://doi.org/10.1038/s41598-021-93656-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-93656-0
- Springer Nature Limited
This article is cited by
-
GeneAI 3.0: powerful, novel, generalized hybrid and ensemble deep learning frameworks for miRNA species classification of stationary patterns from nucleotides
Scientific Reports (2024)
-
Social media-based implosion of Silicon Valley Bank and its domino effect on bank stock indices: evidence from advanced machine and deep learning algorithms
Social Network Analysis and Mining (2024)
-
Predictive modelling of Parkinson’s disease progression based on RNA-Sequence with densely connected deep recurrent neural networks
Scientific Reports (2022)