
Abstract
Heart rate (HR) and sinoatrial node (SAN) function are modulated by the autonomic nervous system. HR regulation by the parasympathetic nervous system (PNS) is impaired in diabetes mellitus (DM), which is denoted cardiovascular autonomic neuropathy. Whether blunted PNS effects on HR in type 2 DM are related to impaired responsiveness of the SAN to PNS agonists is unknown. This was investigated in type 2 diabetic db/db mice in vivo and in isolated SAN myocytes. The PNS agonist carbachol (CCh) had a smaller inhibitory effect on HR, while HR recovery time after CCh removal was accelerated in db/db mice. In isolated SAN myocytes CCh reduced spontaneous action potential firing frequency but this effect was reduced in db/db mice due to blunted effects on diastolic depolarization slope and maximum diastolic potential. Impaired effects of CCh occurred due to enhanced desensitization of the acetylcholine-activated K+ current (IKACh) and faster IKACh deactivation. IKACh alterations were reversed by inhibition of regulator of G-protein signaling 4 (RGS4) and by the phospholipid PIP3. SAN expression of RGS4 was increased in db/db mice. Impaired PNS regulation of HR in db/db mice occurs due to reduced responsiveness of SAN myocytes to PNS agonists in association with enhanced RGS4 activity.
Similar content being viewed by others


Introduction
Heart rate (HR), which is a critical determinant of cardiac performance, is determined by the intrinsic properties of the sinoatrial node (SAN) and is modulated by the autonomic nervous system (ANS)1,2. The sympathetic nervous system (SNS) increases HR by enhancing SAN activity while the parasympathetic nervous system (PNS) reduces HR via the actions of acetylcholine on muscarinic receptors (M2R) in the SAN2.
In diabetes mellitus (DM), cardiovascular complications are highly prevalent, leading to death and morbidity in DM patients. HR regulation by the ANS is known to be impaired in DM patients, which has been attributed to a condition denoted cardiovascular autonomic neuropathy (CAN)3,4. CAN, which can be associated with damage to the nerves that innervate the heart, can affect up to 90% of DM patients and increases mortality by two- to five-fold compared to DM patients without CAN3,5. Although nerve damage may be involved in the impairments in HR regulation by the PNS, it is also possible that these impairments may occur due to alterations in PNS signaling within the SAN.
SAN myocytes generate spontaneous action potentials (APs), which set the intrinsic HR, in association with a diastolic depolarization (DD) that occurs between successive APs2. The DD is generated by a number of ionic currents including the hyperpolarization-activated current (If), which is generated by hyperpolarization-activated cyclic nucleotide gated (HCN) channels, and a rapidly-activating delayed rectifier K+ currents (IKr), which is generated by ether-a-go-go (ERG) channels1,6. The PNS reduces HR via the activation of inhibitory G proteins (Gi) associated with M2Rs. Key mediators of this reduction in HR are the acetylcholine-activated K+ current (IKACh; generated by Kir3.1 and Kir3.4 channels) and If1,7. Specifically, activation of IKACh by the βγ subunit of the Gi protein results in hyperpolarization of the maximum diastolic potential (MDP) in SAN myocytes while inhibition of If downstream of Gαi and a reduction in cyclic AMP (cAMP) reduces the slope of the DD in SAN myocytes. Both effects contribute importantly to a slowing of spontaneous AP firing in SAN myocytes.
Previous studies have shown that regulation of HR by the PNS is impaired in type 1 DM (T1DM) and that this occurs in association with altered responsiveness to PNS agonists in the SAN8,9. Type 2 DM (T2DM) accounts for up to 90% of all DM patients and is also associated with impaired PNS activity4,10. Importantly, blunted PNS activity and CAN present earlier in T2DM patients compared to T1DM4; however, the basis for this is unknown making studies of dysregulation of HR by the PNS specifically in T2DM essential. Accordingly, the purpose of this study was to investigate the regulation of HR and SAN function by the PNS using db/db mice, a model of obesity and T2DM11.
Methods
An expanded methods section is available in the Supplementary Information.
Animals
This study used male and female littermate wildtype and db/db (strain: C57BL/gj-Leprdb) mice between 16 and 20 weeks of age. The db/db mice contain a mutation in the leptin receptor (Lepr) gene leading to hyperphagia11. These db/db mice exhibit the expected features of T2DM including obesity and hyperglycemia, as we12 and others13,14 have shown. All experimental procedures were approved by the University of Calgary Animal Care and Use Committee or the Dalhousie University Committee for Laboratory Animals and were in accordance with the guidelines of the Canadian Council on Animal Care and the ARRIVE guidelines.
Intracardiac electrophysiology and electrocardiogram recording
HR was measured from lead II surface electrocardiograms (ECGs) and corrected SAN recovery time (cSNRT) was measured using an octapolar electrophysiology catheter in anesthetized mice as described previously8,15,16 and in the Supplementary Information.
Patch-clamping of isolated sinoatrial node myocytes
Isolated SAN myocytes were used to record spontaneous APs and ionic currents, including If and IKr, using the whole cell patch-clamp technique in current clamp or voltage clamp mode, respectively. The protocols and solutions for these experiments are described in the Supplementary Information.
Quantitative polymerase chain reaction and Western blotting
Quantitative gene expression was measured in isolated SAN tissue as previously described16,17. Western blotting was performed using isolated SAN tissue as described previously16. The experimental protocols for these techniques are described in the Supplementary Information.
Statistical analysis
All data are presented as means ± SEM. Data were analyzed using two-way ANOVA with the Holm–Sidak posthoc test or Student’s t-test as indicated in each figure legend. P < 0.05 was considered significant.
Results
Effects of carbachol on heart rate and sinoatrial node function in db/db diabetic mice
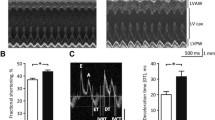
ECG recordings in anesthetized mice (Fig. 1A) demonstrate that HR was lower in db/db mice at baseline and that the ability of the PNS agonist carbachol (CCh; 0.1 mg/kg intraperitoneal injection) to reduce HR was reduced compared to wildtype mice (Fig. 1B,C). The kinetics of the effects of CCh on HR, and the return of HR to baseline after application of the muscarinic (M2) receptor blocker atropine (10 mg/kg, intraperitoneal injection), are presented in Fig. 1D–F. These data further demonstrate that the ability of CCh to reduce HR is impaired in db/db mice and that the return to baseline HR after application of atropine occurs faster in db/db mice (Fig. 1D–F). Impaired HR regulation by CCh in db/db mice was also studied by directly assessing SAN function, as determined by corrected SAN recovery time (cSNRT), in vivo (Fig. 2A). cSNRT was longer at baseline in db/db mice. Application of CCh (0.1 mg/kg) prolonged cSNRT; however, this response was reduced in db/db mice (Fig. 2B,C). These data indicate that the effects of CCh on HR are reduced in db/db mice and that this is associated with impaired responsiveness of the SAN to PNS agonists.

Effects of carbachol on heart rate in db/db mice in vivo. (A) Representative surface ECGs in baseline conditions and after application of CCh (0.1 mg/kg) in anesthetized wildtype and db/db mice. (B) Summary of heart rate at baseline and after CCh in wildtype (n = 9) and db/db (n = 13) mice. *P < 0.05 vs baseline; +P < 0.05 vs. wildtype by two-way ANOVA with Holm–Sidak posthoc test. (C) Change in heart rate after CCh application in wildtype and db/db mice. *P < 0.05 vs. wildtype by Student’s t-test. (D) Time course of changes in heart rate after application of CCh and atropine (10 mg/kg) in wildtype (n = 5) and db/db (n = 6) mice. (E) Magnified region of data from panel D illustrating the changes in heart rate upon application of atropine in wildtype and db/db mice. For panels (D) and (E) *P < 0.05 vs. wildtype by two-way repeated measures ANOVA with Holm–Sidak posthoc test. (F) Recovery time for heart rate after application of atropine. *P < 0.05 vs. wildtype by Student’s t-test.

Effects of carbachol on sinoatrial node recovery time in db/db mice in vivo. (A) Representative recordings illustrating assessment of corrected sinoatrial node recovery time (cSNRT) at baseline and after CCh (0.1 mg/kg) in wildtype and db/db mice. (B) Summary of cSNRT at baseline and after CCh in wildtype (n = 7) and db/db (n = 11) mice. *P < 0.05 vs baseline; +P < 0.05 vs. wildtype by two-way ANOVA with Holm–Sidak posthoc test. (C) Change in cSNRT after CCh application in wildtype and db/db mice. *P < 0.05 vs. wildtype by Student’s t-test.
Effects of CCh on SAN myocyte electrophysiology in db/db mice
To determine the basis for the impaired effects of CCh on HR, spontaneous APs were recorded in isolated SAN myocytes from db/db and wildtype mice (Fig. 3A). SAN AP frequency was lower at baseline in db/db mice. CCh (50 nM) reduced SAN AP frequency, but this effect was smaller in db/db SAN myocytes compared to wildtype (Fig. 3B). Consistent with this, CCh elicited a smaller reduction in DD slope (Fig. 3C) and failed to produce a statistically significant hyperpolarization of the MDP (Fig. 3D) in db/db SAN myocytes. These responses were similar in SAN myocytes isolated from male and female db/db and wildtype mice (Supplementary Fig. S1).

Effects of carbachol on spontaneous action potential firing in isolated sinoatrial node myocytes from db/db mice. (A) Representative spontaneous APs at baseline and after CCh (50 nM) in wildtype and db/db SAN myocytes. Dashed lines are at 0 mV. (B) Summary of spontaneous AP frequency at baseline and after CCh in wildtype and db/db SAN myocytes. (C) Summary of DD slope at baseline and after CCh in wildtype and db/db SAN myocytes. (D) Summary of maximum diastolic potential (MDP) at baseline and after CCh in wildtype and db/db SAN myocytes. For panels (B–D) n = 14 wildtype and 13 db/db SAN myocytes; *P < 0.05 vs baseline; +P < 0.05 vs wildtype by two-way ANOVA with Holm–Sidak posthoc test.
The absence of a normal hyperpolarization of the MDP suggests a key role for IKACh in the blunted response to CCh in db/db mice. Thus, IKACh was investigated next. Figure 4A illustrates representative recordings at baseline, at the peak of the CCh (10 µM) response and 2 min after the peak CCh response when IKACh has undergone desensitization. Peak IKACh (i.e. peak of the CCh activated IK) was not different between wildtype and db/db SAN myocytes (Fig. 4B). In contrast, plotting the time course of the effects of CCh on IKACh (measured at − 100 mV; Fig. 4C) revealed that IKACh desensitization (i.e. the reduction in IKACh amplitude that occurs in the presence of CCh following peak response) was increased (Fig. 4D) and IKACh recovery time (i.e. deactivation) during CCh washout was faster (Fig. 4E) in db/db SAN myocytes compared to wildtype. There were no differences in expression of Chrm2 mRNA or M2R protein in the SAN in db/db mice (Supplementary Fig. S2). While the mRNA expression of Kcnj3 was reduced, there were no differences in the expression of Kcnj5, or the corresponding proteins, Kir3.1 and Kir3.4, in the SAN in db/db mice (Supplementary Fig. S3). These data demonstrate that IKACh is impaired in db/db SAN myocytes due to enhanced desensitization and faster deactivation kinetics.

IKACh properties in isolated SAN myocytes from db/db mice. (A) Representative IK recordings during a voltage ramp from + 50 mV to − 120 mV (holding potential = − 80 mV) at baseline, at the peak of the CCh (10 µM) response and 2 min after the peak CCh response in wildtype and db/db SAN myocytes. (B) Peak IKACh I–V relationship in wildtype (n = 28) and db/db (n = 25) SAN myocytes. P = 0.998 for wildtype vs. db/db by two-way repeated measures ANOVA with Holm–Sidak posthoc test. (C) Time course of CCh stimulated IKACh, measured at − 100 mV, in wildtype and db/db SAN myocytes. (D) Summary of IKACh desensitization in wildtype and db/db SAN myocytes. (E) Summary of IKACh recovery time during CCh washout in wildtype and db/db SAN myocytes. For panels (D,E) *P < 0.05 vs. wildtype by Student’s t-test.
CCh can also reduce SAN AP firing by inhibiting If. Accordingly, the effects of CCh on If in db/db SAN myocytes were investigated. CCh (10 µm) reduced If density in wildtype and db/db SAN myocytes (Fig. 5A–C). Comparison of these effects illustrates that If tended to be smaller at baseline in db/db SAN myocytes and that If was reduced similarly in both genotypes following application of CCh (Fig. 5D). CCh reduced If in association with a hyperpolarizing shift in the If activation curve (Fig. 5E,F). There were no differences in the voltage dependence of activation (V1/2(act)) at baseline or after CCh in wildtype and db/db SAN myocytes (Fig. 5G) indicating that the effects of CCh on If were similar between both groups of mice. There were also no differences in the mRNA expression or protein levels of HCN1, HCN2 or HNC4 in the SAN of db/db mice (Supplementary Fig. S4). These data demonstrate that impaired SAN responsiveness to PNS agonists is not related to altered effects of CCh on If.

Effects of carbachol on the hyperpolarization-activated current (If) in db/db SAN myocytes. (A) Representative If recordings at baseline and after CCh (10 µM) application in wildtype and db/db SAN myocytes. Voltage clamp protocol shown at bottom of recordings. (B) If IV curves at baseline and after CCh application in wildtype SAN myocytes (n = 15). (C) If IV curves at baseline and after CCh application in db/db SAN myocytes (n = 16). For panels (B) and (C) *P < 0.05 vs. baseline by two-way repeated measures ANOVA with Holm–Sidak posthoc test. (D) If density at − 120 mV at baseline and after CCh application in wildtype and db/db SAN myocytes. *P < 0.05 vs baseline by two-way ANOVA with Holm–Sidak posthoc test. (E) If steady-state activation curves at baseline and after CCh application in wildtype SAN myocytes (n = 15). (F) If steady-state activation curves at baseline and after CCh application in db/db SAN myocytes (n = 16). (G) If V1/2(act) at baseline and after CCh application in wildtype and db/db SAN myocytes. *P < 0.05 vs. baseline by two-way ANOVA with Holm–Sidak posthoc test. Refer to Supplementary Table 1 for additional analysis of If steady-state activation.
Since baseline spontaneous AP firing and DD slope were reduced in db/db SAN myocytes, and this wasn’t accounted for by differences in baseline If, other mechanisms were explored. Repolarizing IK was reduced in db/db SAN myocytes (Supplementary Fig. S5). Furthermore, voltage clamp protocols designed to detect IKr tail currents revealed that IKr is reduced in db/db SAN myocytes (Supplementary Fig. S5). This reduction occurred without changes in V1/2(act) or slope factor (k) for IKr tail currents (Supplementary Fig. S5). IKr has been shown to be a critical determinant of SAN spontaneous AP firing and DD slope18 suggesting that the reduction in IKr could account for baseline differences in spontaneous AP firing in db/db mice.
Altered RGS4 and PIP3 signaling underlie impaired IKACh in db/db SAN myocytes
Previous studies have shown that IKACh desensitization and deactivation kinetics are critically affected by regulator of G protein signaling 4 (RGS4) in the SAN19. Furthermore, RGS4 is inhibited by phosphatidylinositol (3,4,5) P3 (PIP3)20 which is activated by insulin-mediated PI3K signaling21,22. Therefore, impaired insulin and PI3K signaling in T2DM could result in enhanced RGS4 activity due to a loss of PIP3-mediated inhibition of RGS4. To test this hypothesis, wildtype and db/db SAN myocytes were dialyzed with the RGS4 inhibitor CCG-4986 (10 µM) for 10 min prior to studying the effects of CCh (10 µM) on IKACh (Fig. 6A). Time course plots demonstrate that IKACh desensitization and deactivation kinetics were normalized in db/db SAN myocytes after CCG-4986 application (Fig. 6B). Summary data demonstrate that there were no differences in peak IKACh density (Fig. 6C), IKACh desensitization (Fig. 6D) or IKACh recovery time (Fig. 6E) between wildtype and db/db SAN myocytes following inhibition of RGS4 with CCG-4986. RGS4 mRNA expression and protein levels were increased in the SAN in db/db mice (Fig. 7).

Effects of RGS4 inhibition on IKACh kinetics in db/db SAN myocytes. (A) Representative IK recordings illustrating peak and desensitized responses to CCh (10 µM) in wildtype and db/db SAN myocytes pre-treated with the RGS4 inhibitor CCG-4986 (10 µM). (B) Time course of CCh stimulated IKACh, measured at − 100 mV, in wildtype and db/db SAN myocytes in the presence of CCG-4986. (C) Summary of peak IKACh at − 100 mV in wildtype and db/db SAN myocytes in the presence of CCG-4986. (D) Summary of IKACh desensitization in wildtype and db/db SAN myocytes in the presence of CCG-4986. (E) Summary of IKACh recovery time during CCh washout in wildtype and db/db SAN myocytes in the presence of CCG-4986. For panels (C–E) n = 16 wildtype and 18 db/db SAN myocytes; data analyzed by Student’s t-test.

RGS4 gene and protein expression in the SAN in db/db mice. (A) mRNA expression of rgs4 in wildtype (n = 5) and db/db (n = 5) SAN. *P < 0.05 vs wildtype by Student’s t-test. (B) Representative Western blot and summary protein expression for RGS4 in the SAN of wildtype (n = 6) and db/db (n = 6) mice. *P < 0.05 vs. wildtype by Student’s t-test. Uncropped Western blot provided in Supplementary Fig. S6.
Next, wildtype and db/db SAN myocytes were dialyzed with PIP3 (1 µM) for 10 min prior to application of CCh (10 µM) to investigate IKACh properties (Fig. 8A). Similar to RGS4 inhibition, time course plots demonstrate that IKACh desensitization and deactivation were normalized in db/db SAN myocytes treated with PIP3 (Fig. 8B). On average, peak IKACh density (Fig. 8C), IKACh desensitization (Fig. 8D) and IKACh recovery time (Fig. 8E) were not different between wildtype and db/db SAN myocytes when treated with PIP3.

Effects of PIP3 on IKACh kinetics in db/db SAN myocytes. (A) Representative IK recordings illustrating peak and desensitized responses to CCh in wildtype and db/db SAN myocytes dialyzed with PIP3 (1 µM). (B) Time course of CCh stimulated IKACh, measured at − 100 mV, in wildtype and SAN myocytes dialyzed with PIP3. (C-E ) Summary of peak IKACh at − 100 mV (C), IKACh desensitization (D) and IKACh recovery time during CCh washoff (E) in wildtype (n = 16) and db/db (n = 19) SAN myocytes dialyzed with PIP3. Data in panels (C–E) analyzed by Student’s t-test. (F) Representative IK recordings illustrating peak and desensitized responses to CCh in wildtype and db/db SAN myocytes dialyzed with PIP2 (1 µM). (G) Time course of CCh stimulated IKACh, measured at − 100 mV, in wildtype and SAN myocytes dialyzed with PIP2. (H–J) Summary of peak IKACh at − 100 mV (H), IKACh desensitization (I) and IKACh recovery time during CCh washoff (J) in wildtype (n = 18) and db/db (n = 19) SAN myocytes dialyzed with PIP2. For panels (H–J) *P < 0.05 by Student’s t-test.
In control experiments, wildtype and db/db SAN myocytes were dialyzed with phosphatidylinositol 4,5-bisphosphate (PIP2), a phospholipid that does not directly inhibit RGS4 or mediate the actions of insulin-dependent PI3K signaling. PIP2 (1 µM) was dialyzed into SAN myocytes for 10 min prior to application of CCh to investigate IKACh (Fig. 8F). Time course plots demonstrate that in the presence of PIP2, IKACh desensitization remained enhanced and IKACh deactivation was faster in db/db SAN myocytes (Fig. 8G). Summary data show that in the presence of PIP2, peak IKACh density was not different (Fig. 8H), but that IKACh desensitization tended to be increased (Fig. 8I) and recovery time was faster (Fig. 8J) in db/db SAN myocytes compared to wildtype.
Discussion
The present study demonstrates that db/db mice display impaired responsiveness to the PNS agonist CCh in the SAN leading to impaired HR regulation, indicating that this mouse model of T2DM reproduces the phenotype of impaired ANS regulation of the heart seen in T2DM patients. Furthermore, the present study provides novel insight into the cellular and molecular mechanisms leading to blunted PNS regulation of HR in T2DM. In isolated SAN myocytes, CCh displayed a reduced ability to slow spontaneous AP firing in association with a smaller reduction in DD slope and less hyperpolarization of the MDP in db/db mice. In fact, CCh failed to produce a statistically significant hyperpolarization in db/db SAN myocytes. These responses were similar between male and female db/db mice.
Consistent with these effects, IKACh was centrally involved in the impaired effects of CCh on AP firing in SAN myocytes. Analysis of IKACh properties revealed that the peak IKACh density was not altered in db/db SAN myocytes. Similarly, there were no major changes in gene or protein expression of M2R or Kir3 channels in the SAN in db/db mice. Rather, the major alterations in IKACh in db/db SAN myocytes were an increase in desensitization and faster deactivation kinetics. These findings are consistent with our studies in vivo, which demonstrated that the effects of CCh were reduced in db/db mice (likely in association with enhanced desensitization of IKACh) and that reversal of the effects of CCh upon application of atropine was much more rapid in db/db mice (likely in association with faster IKACh deactivation). Both of these effects resulted in impaired PNS effects on HR (and SAN function) over the full time-course of CCh application and removal. Our findings are also consistent with previous studies demonstrating that IKACh plays a critical role in mediating SAN and HR responses to CCh23,24.
IKACh is well known to undergo a process of desensitization whereby current amplitude decreases in the presence of M2R agonists such as CCh25,26,27. IKACh desensitization, as well as deactivation are critically affected by regulator of G protein signaling (RGS) proteins, including RGS4, which is highly expressed in the SAN19,28. Genetic ablation of RGS4 leads to enhanced PNS signaling due to reductions in IKACh desensitization and slower IKACh deactivation19. Thus, increased IKACh desensitization and faster deactivation in db/db SAN myocytes (i.e. the opposite of RGS4 ablation) suggested enhanced RGS4 activity in db/db mice. This was confirmed using CCG-4986, which is a selective RGS4 inhibitor at the doses used in this study29,30. These findings are consistent with previous studies demonstrating an essential role for RGS proteins in regulated PNS signaling in the heart via effects on IKACh19,28,31,32,33.
RGS4 is importantly regulated by PIP3, which exerts an inhibitory effect on RGS4 activity20,34,35. In the normal heart, PIP3 is generated from PIP2 through the actions of PI3Kα (p100α isoform)21. In T2DM, impaired insulin signaling results in reduced PI3K activity, which would result in less PIP3 generation22,36. This loss of PIP3 would lead to less inhibition of RGS4 and could underlie increased RGS4 activity in the SAN of db/db mice. In support of this, bypassing insulin signaling and directly dialyzing db/db SAN myocytes with PIP3 normalized IKACh desensitization and deactivation. Thus, the studies conducted here demonstrate that the kinetic properties of IKACh are altered in T2DM due to the loss of PIP3 mediated inhibition of RGS4 and that this is a major determinant of impaired PNS signaling to the SAN and HR regulation in T2DM.
CCh can also reduce spontaneous AP firing by inhibiting If, which was also investigated in the present study. As expected, CCh reduced If density in association with a hyperpolarizing shift in the V1/2(act); however, this effect was similar in wildtype and db/db SAN myocytes. This indicates that HCN channel function was not altered by a change in RGS4 activity in T2DM. CCh can also modulate SAN activity via other mechanisms such as L-type Ca2+ currents and SR Ca2+ handling1,6. These targets were not investigated in this study; therefore, whether the effects of CCh on these targets are altered in db/db mice and whether these targets are regulated by RGS4 is presently unknown. These could be areas for future study. Nevertheless, the absence of altered CCh effects on If (which is also an important mediator of PNS signaling in the SAN) in db/db mice suggests that the effects of RGS4 are selective for Kir3 channels. In addition to RGS4, RGS6 has also been shown to regulate IKACh kinetics in the SAN33,37. Whether RGS6 is regulated by PIP3 in similar ways to RGS4 and whether RGS6 contributes to impaired PNS signaling in DM is not currently known.
PNS signaling in the SAN has also been previously investigated in T1DM using Akita mice8. The present study demonstrates that while there are similarities in how PNS signaling is altered in Akita and db/db mice, there are also important differences. Similar to the present study in db/db mice, HR regulation by CCh was impaired in Akita mice in association with impaired responsiveness of SAN myocytes to CCh. IKACh desensitization and deactivation also showed similar changes in Akita SAN myocytes to those identified here in db/db mice and these changes were reversible by RGS4 inhibition or application PIP3 in both models of DM. Conversely, Akita mice showed no differences in expression of RGS4 in the SAN8, whereas RGS4 gene and protein expression were increased in the SAN in db/db mice. This indicates that RGS4 plays a central role in impaired PNS signaling in the SAN in T1DM and T2DM, but that there are some differences in how RGS4 is altered in the two forms of DM. The increase in RGS4 expression, in combination with increased RGS4 activity due to loss of PIP3 signaling, demonstrates that alterations in RGS4 are more complex and multifaceted in db/db mice compared to Akita mice. This could explain, at least in part, the finding that CAN often develops earlier in T2DM compared to T1DM patients4. Collectively, these data demonstrate that RGS4 signaling is particularly important in the SAN in type 2 diabetic db/db mice due to multiple alterations including increases in its gene and protein expression as well as its functional regulation by PI3K-PIP3 signaling.
The present study also shows that baseline HR and spontaneous AP firing in SAN myocytes are reduced in db/db mice. This is consistent with previous studies showing lower HR and impaired SAN function in animal models of T1DM and T2DM12,38,39,40,41,42. In the present study, baseline If density tended to be lower in db/db SAN myocytes; however, there were no differences in baseline V1/2(act) or in the gene and protein expression of HCN1, HCN2 or HCN4 in db/db mice suggesting If does not play a major role in baseline HR differences. IKr plays an integral role in the SAN where it controls AP repolarization, contributes to the DD slope and helps determine AP firing frequency18. We measured IKr from tail currents, which showed V1/2(act) values very similar to those reported in previous studies for this current18, and found that IKr is reduced in db/db SAN myocytes, suggesting that this could contribute to baseline differences in HR and SAN activity in db/db mice. Additional studies will be required to investigate the roles of If, IKr and other ionic mechanisms not assessed in this study in baseline differences in SAN AP firing in db/db mice. Reduced basal HR in vivo in the presence of impaired PNS signaling suggests additional alterations in db/db mice. Baseline HR is determined by intrinsic SAN function, the balance between sympathetic and parasympathetic nervous system regulation of the SAN and other factors such as circulating factors that affect the SAN. Our study suggests that even though PNS signaling is impaired, baseline HR is reduced in part due to impaired intrinsic SAN function. Furthermore, sympathetic nervous system activity was not investigated in this study, but may also be altered in db/db mice. As such, future studies investigating sympathetic regulation of HR and baseline HR differences in db/db mice are warranted.
In summary, the present study provides new insight into the basis for impaired PNS regulation of HR in T2DM. The effects of CCh on HR were reduced in association with impaired responsiveness of SAN myocytes to CCh. IKACh, which is a key mediator of the effects of PNS activation in the SAN, displayed enhanced desensitization and faster deactivation kinetics in db/db SAN myocytes resulting in blunted effects of CCh on HR throughout the duration of CCh exposure and removal. These IKACh alterations occurred due to enhanced RGS4 activity and the loss of PIP3 signaling in db/db SAN myocytes. These findings, which identify potential new targets for intervention in diabetic patients, should be taken into consideration when interpreting CAN and blunted ANS signaling to the heart in T2DM.
Data availability
Any data or experimental reagents are available from the corresponding author upon reasonable request.
References
Mangoni, M. E. & Nargeot, J. Genesis and regulation of the heart automaticity. Physiol. Rev. 88, 919–982 (2008).
MacDonald, E. A., Rose, R. A. & Quinn, T. A. Neurohumoral control of sinoatrial node activity and heart rate: Insight from experimental models and findings from humans. Front. Physiol. 11, 170 (2020).
Vinik, A. I. & Ziegler, D. Diabetic cardiovascular autonomic neuropathy. Circulation 115, 387–397 (2007).
Bakkar, N. Z. et al. Cardiac autonomic neuropathy: A progressive consequence of chronic low-grade inflammation in type 2 diabetes and related metabolic disorders. Int. J. Mol. Sci. 21, 9005 (2020).
Maser, R. E., Mitchell, B. D., Vinik, A. I. & Freeman, R. The association between cardiovascular autonomic neuropathy and mortality in individuals with diabetes: A meta-analysis. Diabetes Care 26, 1895–1901 (2003).
Lakatta, E. G., Maltsev, V. A. & Vinogradova, T. M. A coupled SYSTEM of intracellular Ca2+ clocks and surface membrane voltage clocks controls the timekeeping mechanism of the heart’s pacemaker. Circ. Res. 106, 659–673 (2010).
DiFrancesco, D. Pacemaker mechanisms in cardiac tissue. Annu. Rev. Physiol. 55, 455–472 (1993).
Krishnaswamy, P. S. et al. Altered parasympathetic nervous system regulation of the sinoatrial node in Akita diabetic mice. J. Mol. Cell. Cardiol. 82, 125–135 (2015).
Park, H. J. et al. Role of SREBP-1 in the development of parasympathetic dysfunction in the hearts of type 1 diabetic Akita mice. Circ. Res. 105, 287–294 (2009).
Kahn, S. E., Cooper, M. E. & Del Prato, S. Pathophysiology and treatment of type 2 diabetes: Perspectives on the past, present, and future. Lancet 383, 1068–1083 (2014).
Hsueh, W. et al. Recipes for creating animal models of diabetic cardiovascular disease. Circ. Res. 100, 1415–1427 (2007).
Bohne, L. J. et al. Electrical and structural remodeling contribute to atrial fibrillation in type 2 diabetic db/db mice. Heart Rhythm 18, 118–129 (2021).
Carley, A. N. et al. Treatment of type 2 diabetic db/db mice with a novel PPARgamma agonist improves cardiac metabolism but not contractile function. Am. J. Physiol. Endocrinol. Metab. 286, E449-455 (2004).
Hafstad, A. D., Solevag, G. H., Severson, D. L., Larsen, T. S. & Aasum, E. Perfused hearts from Type 2 diabetic (db/db) mice show metabolic responsiveness to insulin. Am. J. Physiol. Heart Circ. Physiol. 290, H1763-1769 (2006).
Egom, E. E. et al. Impaired sinoatrial node function and increased susceptibility to atrial fibrillation in mice lacking natriuretic peptide receptor C. J. Physiol. 593, 1127–1146 (2015).
Mackasey, M. et al. Natriuretic peptide receptor-C protects against angiotensin II-mediated sinoatrial node disease in mice. JACC Basic Transl. Sci. 3, 824–843 (2018).
Moghtadaei, M. et al. The impacts of age and frailty on heart rate and sinoatrial node function. J. Physiol. 594, 7105–7126 (2016).
Clark, R. B. et al. A rapidly activating delayed rectifier K+ current regulates pacemaker activity in adult mouse sinoatrial node cells. Am. J. Physiol. Heart Circ. Physiol. 286, H1757-1766 (2004).
Cifelli, C. et al. RGS4 regulates parasympathetic signaling and heart rate control in the sinoatrial node. Circ. Res. 103, 527–535 (2008).
Ishii, M., Inanobe, A. & Kurachi, Y. PIP3 inhibition of RGS protein and its reversal by Ca2+/calmodulin mediate voltage-dependent control of the G protein cycle in a cardiac K+ channel. Proc. Natl. Acad. Sci. U. S. A. 99, 4325–4330 (2002).
Oudit, G. Y. et al. The role of phosphoinositide-3 kinase and PTEN in cardiovascular physiology and disease. J. Mol. Cell. Cardiol. 37, 449–471 (2004).
Bertrand, L., Horman, S., Beauloye, C. & Vanoverschelde, J. L. Insulin signalling in the heart. Cardiovasc. Res. 79, 238–248 (2008).
Wickman, K., Nemec, J., Gendler, S. J. & Clapham, D. E. Abnormal heart rate regulation in GIRK4 knockout mice. Neuron 20, 103–114 (1998).
Mesirca, P. et al. The G-protein-gated K+ channel, IKACh, is required for regulation of pacemaker activity and recovery of resting heart rate after sympathetic stimulation. J. Gen. Physiol. 142, 113–126 (2013).
Lomax, A. E., Rose, R. A. & Giles, W. R. Electrophysiological evidence for a gradient of G protein-gated K+ current in adult mouse atria. Br. J. Pharmacol. 140, 576–584 (2003).
Bender, K. et al. Acute desensitization of GIRK current in rat atrial myocytes is related to K+ current flow. J. Physiol. 561, 471–483 (2004).
Shui, Z., Boyett, M. R., Zang, W. J., Haga, T. & Kameyama, K. Receptor kinase-dependent desensitization of the muscarinic K+ current in rat atrial cells. J. Physiol. 487(Pt 2), 359–366 (1995).
Doupnik, C. A., Davidson, N., Lester, H. A. & Kofuji, P. RGS proteins reconstitute the rapid gating kinetics of gbetagamma-activated inwardly rectifying K+ channels. Proc. Natl. Acad. Sci. U. S. A. 94, 10461–10466 (1997).
Roman, D. L., Blazer, L. L., Monroy, C. A. & Neubig, R. R. Allosteric inhibition of the regulator of G protein signaling-Galpha protein–protein interaction by CCG-4986. Mol. Pharmacol. 78, 360–365 (2010).
Roman, D. L. et al. Identification of small-molecule inhibitors of RGS4 using a high-throughput flow cytometry protein interaction assay. Mol. Pharmacol. 71, 169–175 (2007).
Fu, Y., Huang, X., Piao, L., Lopatin, A. N. & Neubig, R. R. Endogenous RGS proteins modulate SA and AV nodal functions in isolated heart: Implications for sick sinus syndrome and AV block. Am. J. Physiol. Heart Circ. Physiol. 292, H2532-2539 (2007).
Fu, Y. et al. Endogenous RGS proteins and Galpha subtypes differentially control muscarinic and adenosine-mediated chronotropic effects. Circ. Res. 98, 659–666 (2006).
Yang, J. et al. RGS6, a modulator of parasympathetic activation in heart. Circ. Res. 107, 1345–1349 (2010).
Ishii, M., Fujita, S., Yamada, M., Hosaka, Y. & Kurachi, Y. Phosphatidylinositol 3,4,5-trisphosphate and Ca2+/calmodulin competitively bind to the regulators of G-protein-signalling (RGS) domain of RGS4 and reciprocally regulate its action. Biochem. J. 385, 65–73 (2005).
Popov, S. G., Krishna, U. M., Falck, J. R. & Wilkie, T. M. Ca2+/Calmodulin reverses phosphatidylinositol 3,4, 5-trisphosphate-dependent inhibition of regulators of G protein-signaling GTPase-activating protein activity. J. Biol. Chem. 275, 18962–18968 (2000).
Abel, E. D. Insulin signaling in heart muscle: Lessons from genetically engineered mouse models. Curr. Hypertens. Rep. 6, 416–423 (2004).
Posokhova, E., Wydeven, N., Allen, K. L., Wickman, K. & Martemyanov, K. A. RGS6/Gss5 complex accelerates IKACh gating kinetics in atrial myocytes and modulates parasympathetic regulation of heart rate. Circ. Res. 107, 1350–1354 (2010).
Zhang, Y. et al. Electrical conduction system remodeling in streptozotocin-induced diabetes mellitus rat heart. Front. Physiol. 10, 826 (2019).
Luo, M. et al. Diabetes increases mortality after myocardial infarction by oxidizing CaMKII. J. Clin. Investig. 123, 1262–1274 (2013).
Soltysinska, E., Speerschneider, T., Winther, S. V. & Thomsen, M. B. Sinoatrial node dysfunction induces cardiac arrhythmias in diabetic mice. Cardiovasc. Diabetol. 13, 122 (2014).
Howarth, F. C. et al. The pattern of mRNA expression is changed in sinoatrial node from Goto-Kakizaki Type 2 diabetic rat heart. J. Diabetes. Res. 2018, 8454078 (2018).
Polina, I. et al. Loss of insulin signaling may contribute to atrial fibrillation and atrial electrical remodeling in type 1 diabetes. Proc. Natl. Acad. Sci. U. S. A. 117, 7990–8000 (2020).
Acknowledgements
We acknowledge Megan McRae, Erin Seto, Adam Kirkby, and Sara Rafferty for technical support and experimental assistance. This work was supported by the Canadian Institutes of Health Research to R.A.R. (MOP 142486 and PJT 166105). Y.L. holds a Canadian Institutes of Health Research Fellowship. H.J.J. was supported by a Killam Postdoctoral Fellowship and a Libin Cardiovascular Institute Postdoctoral Fellowship.
Author information
Authors and Affiliations
Contributions
The conception of the experiments was by Y.L. and R.A.R. The design of the experiments was by Y.L., H.J.J., P.S.K., O.B. and R.A.R. The collection, analysis and interpretation of the data was by Y.L., H.J.J., P.S.K., O.B. and R.A.R. Drafting the article or revising it critically for important intellectual content was done by Y.L., H.J.J. and R.A.R. All authors approved the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Liu, Y., Jansen, H.J., Krishnaswamy, P.S. et al. Impaired regulation of heart rate and sinoatrial node function by the parasympathetic nervous system in type 2 diabetic mice. Sci Rep 11, 12465 (2021). https://doi.org/10.1038/s41598-021-91937-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-91937-2
- Springer Nature Limited