
Abstract
Honeybees are one of the most environmentally important insects, as their pollination of various plant species contributes to the balance among different ecosystems. It has been studied extensively for their unique attribute of forming a caste society. Unlike other insects, honeybees communicate socially by secreting pheromones or by exhibiting specific patterns of motion. In the honeybee industry, the Asian honeybees (Apis cerana) and the Western honeybees (Apis mellifera) are dominant species. However, molecular research on the transcriptomes of A. cerana has not been studied as extensively as those of A. mellifera. Therefore, in this study, caste-specific transcriptional differences were analyzed, which provides a comprehensive analysis of A. cerana. In our dataset, we analyzed gene expression profiles using organs from worker, drone, and queen bees. This gene-expression profile helped us obtain more detailed information related to organ-specific genes, immune response, detoxification mechanisms, venom-specific genes, and ovary development. From our result, we found 4096 transcripts representing different gene-expression pattern in each organ. Our results suggest that caste-specific transcripts of each organ were expressed differently even under natural conditions. These transcriptome-wide analyses provide new insights into A. cerana and that promote honeybee research and conservation.
Similar content being viewed by others


Introduction
Considering the vital role of honeybees in supporting ecosystems and thus sustaining lifeforms, including human life, honeybees are irreplaceable by other insects1,2, because they are the largest group crop pollinators3,4 and provide honey5,6, beeswax, propolis7, royal jelly8, and many other benefits9,10,11. A honeybee colony is composed of a single queen and thousands of workers and a couple of hundred drones, which provides a classic model for understanding the sociality12,13,14,15,16, communication17,18, and sex-determination19,20,21 of insects. Genetically, females, queen and worker bees, develop from fertilized eggs, whereas drones develop from unfertilized eggs22. Previous studies of honeybees described a simple genetic system of haplodiploid development and showed that different diets and cell sizes determine the physiological characteristics, such as body size, behavior, physiology, and lifespan23, of the three castes of worker, drone, and queen bee. The relationship between honeybees behavior and the gene expressions24 of odorant receptors25,26,27 and hormones28,29 in the brains of workers and queen bees were investigated in numerous studies that investigated the social aspect of bee behavior. While developing into three castes, royal jelly was revealed to trigger larvae to develop into a queen and control the gene expression of the juvenile hormone, which plays a key role in sex-determination30,31.
Honeybees live in a densely populated environment, have close connections, and share food with nest-mates32. This causes pathogens to spread easily and very quickly within the colony. Therefore, honeybees are highly susceptible to pathogens. Researchers have focused to understand how honeybees resist disease and protect their health. Honeybees have evolved two strategies for protecting their colonies from pathogens; (1) an innate immune system33,34 and (2) social immunity35,36. First, honeybees protect their colony by protecting themselves, which is achieved with an innate immune system. As one example, workers are major pollinators that forage on wildflowers and crops. While foraging, honeybees are exposed to harmful agricultural pesticides and pathogen, such as viral and bacterial infections37, which may be transmitted to the rest of the bee colony. Studies on the innate immune system of bees have elucidated the stronger innate immune response of younger forger bees compared to older forager bees38. Second, the social immunity of bees has been studied as a function of health maintenance. Worker bees detect smells and remove diseased or infected brood/adults, get rid of dead adults, foreign objects, and pathogens from their hive, and clean the surface of their body35,36.
Honeybees are considered interesting social insect models and have been extensively studied on the subjects such as insect-communication system, flight behavior, and developmental biology. Despite these diverse studies on honeybees, there is still insignificant understanding of the sex determination of haplodiploid system at the molecular level. There are two genetically same types of female honeybees: worker bees and the queen bee. Worker bees can lay eggs usually in the absence of a queen. Unlike the queen that can mate and store sperms in spermatheca to lay fertilized eggs39, worker bees cannot mate. Therefore, only the queens are able to lay fertilized eggs whereas workers’ eggs remain unfertilized40,41. Egg-viability studies have shown that sex determination is associated with a difference in cell size and dietary habits. Most research into sex determination has focused on phenotypes during honeybee development and is limited to studying the early stages or the organ levels22,42. Therefore, it would be highly informative to investigate the dynamics of expressions from the two kinds of honeybee ovaries, i.e., queen ovary and worker ovary, which could provide insights into the haplodiploid development of honeybees.
In this study, organ-specific RNA-seq was performed to analyze transcriptome data of several organs (one from the queen, five from a worker, and five from a drone) of the Asian honeybees. The gene expressions of worker-drone organs, worker-queen organs, and drone-queen organs were compared and a dataset of differentially expressed genes was validated using quantitative reverse transcription polymerase chain reaction (qRT-PCR). Specifically, our analyses focused on the expressions of genes related to the mechanisms of odorants, vision, hormones, growth factors, the immune-response, detoxification, venom, and sex determination.
Results
Transcriptome profiling of organs from worker, drone, and queen of Apis cerana
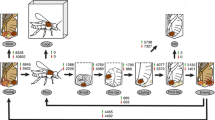
High-throughput sequencing was performed on several organs of A. cerana: worker brain (WB), worker ventriculus (WV), worker rectum (WR), worker ovary (WO), worker venom gland (WVG), drone brain (DB), drone ventriculus (DV), drone rectum (DR), drone testis (DT), drone mucus gland (DMG), and queen ovary (QO). High-throughput sequencing was performed using the Illumina HiSeq 2500 platform (Fig. 1). The sequencing results showed that the two biological replicates of each sample have an average of 14 million reads. After removing the adaptor and low-quality tags, approximately 92% clean reads were obtained from each library (Supplementary Table S1).

Schematic overview of the experimental design and analysis pipeline. Organs collected directly from adult honeybees for mRNA-seq: brain, ventriculus, rectum from workers and drones, venom gland from worker, and reproductive organs (testis, mucus gland, and ovaries) from three castes. The collected organs shown in gray.
A comparison of the genomes of A. cerana with two other species of insects, i.e., Drosophila melanogaster and A. mellifera in the NCBI v2.0 database (ACSNU-2.0), provided the coding domain sequence and the read count for gene expression of A. cerana using the Kallisto software. The result showed a total of 10,651 genes. High-throughput sequencing analysis indicated that worker, drone, and queen bees had differentially expressed genes. After removing the hypothetical protein, ribosomal protein family, and genes having less than 10 transcripts per million (TPM) reads, a total of 4,096 differentially expressed genes were found among all three castes. Raw and processed data are publicly available in the NCBI/GEO database: (http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE164333.
Common transcript patterns between worker-drone, worker-queen, and drone-queen
One of the major goals of our study was to obtain high-quality transcriptome data that could be used to predict global changes in the gene expressions of honeybees under natural conditions. Transcriptome profiling showed 269, 196, and 324 genes differentially expressed in the brain, ventriculus, and rectum, respectively, of workers and drones. Venn diagram data showed a total of 269 brain-associated genes in WB and DB, among which 255 genes were only expressed in WB, 11 genes were only expressed in DB, and only three genes were expressed commonly in both WB and DB (Fig. 2a,b). Among the 269 genes in brain, 14 genes were correlated with caste differentiation such as hormone, visual sense, and neuronal signal peptides. Among these genes, ten genes, including odorant receptor (OR) genes were up-regulated in WB. The differential expression of transcripts in the brain indicates that they affect honeybee behaviors29,43,44. These results also suggest that workers can sense a wider range of odors types.

Venn diagram of gene numbers among organs. (a) Venn diagram of brain genes between workers and drones. (b) Number of down-regulated and up-regulated brain genes between workers and drones. (c) Venn diagram of gut genes between worker and drone organs. (d) Number of down-regulated and up-regulated gut genes between workers and drones. (e) Venn diagram of genes between female and male reproductive organs. (f) Number of down-regulated and up-regulated genes between female and male reproductive organs. WB worker brain, WV worker ventriculus, WR worker rectum, WO worker ovary, WVG worker venom gland, DB drone brain, DV drone ventriculus (midgut), DR drone rectum, DMG drone mucus gland, DT drone testis, QO queen ovary.
Differences in the diets of each caste reportedly affect gut enzymes45. We found 39, 78, 238, and 32 genes specifically expressed in WV, DV, WR, and DR, respectively. Among these genes, 30 genes were exclusively expressed in ventriculus, whereas, only five genes were expressed in the rectums of both worker and drone (Fig. 2c). In ventriculus, more transcripts were overexpressed in DV compared to WV (Fig. 2d). In the rectum, however, more transcripts were overexpressed in WR compared to DR (Fig. 2d). The ventriculus and rectum, which are typical digestive organs of honeybees, showed substantial differences in the gene expression profile transcripts, despite being anatomically connected.
The reproductive organs of drones (haploid), workers (diploid), and queen (diploid) were compared. Three commonly expressed genes were found in both DT and DMG, and 18 commonly expressed genes in both WO and QO. The numbers of specific transcripts expressed in DT, DMG, WO, and QO were found in 49, 21, 27, and 418 genes, respectively (Fig. 2e). Transcript profiling between DMG and DT revealed that more genes were overexpressed in DT compared to DMG (Fig. 2f). In the comparison between QO and WO as the representative female reproductive organ, most transcripts were overexpressed in QO than in WO (Fig. 2f).
Differentially expressed genes in the brain, gut, and reproductive organs
To understand the honeybee’s different caste-specific behaviors several categories of genes related to social communication were investigated. The expression patterns of genes were compared among the worker, drone, and queen. Genes from the brain were classified genes into four categories; OR, neuronal genes, photoreceptors, and hormones (Fig. 3a and Supplementary Table S2). A comparison of the OR genes between WB and DB showed remarkably high expression profiles in WB. However, it remains to be determined how each OR affects the honeybee’s behavior. In the other two groups (neuronal genes group and photoreceptor group), the genes related with each group were expressed more in DB than in WB. Of the hormone group, prohormone-1 (ACSNU02044T0) was the only hormone gene that was overexpressed in WB.

Heat map of transcripts. (a) Expression of 26 selected genes among worker and drone brains. 12 genes were related to the odorant receptor family. Six genes were neuronal genes. Five genes were related to vision. Three genes were related to hormone synthesis or hormone peptides. (b) Expression of 41 selected genes among worker and drone guts. 19 genes were related the digestive enzyme. Nine genes were related to carbohydrate synthesis. 13 genes were related to lipids. (c) Expression of 37 selected genes among worker, drone and queen of reproductive organs. Nine of the major royal jelly protein (MRJP) family. Six genes were related to the juvenile hormone or hormone receptor. 22 genes were related to the growth factor. Each selected gene presented a log2 fold change. (d) Relative expression of brain related genes and reproductive organ related genes exhibiting a significant change. See Supplementary Table S5 for primers. WB worker brain, WV worker ventriculus, WR worker rectum, WO worker ovary, WVG worker venom gland, DB drone brain, DV drone ventriculus (midgut), DR drone rectum, DMG drone mucus gland, DT drone testis, QO queen ovary.
We classified gut genes into three categories digestive enzyme genes, carbohydrate biosynthesis genes, and lipid transport genes. In a previous study of the honey bee protein atlas19, major digestive enzymes were highly expressed in workers among castes. Comparing these to our data, we found that transcripts of the digestive enzyme were expressed mostly in the rectum of workers (Fig. 3b). The differential profiles of digestive enzyme genes between the worker and drone suggest that there are differences between the diet composition of the two castes.
To understand the difference between haploid and diploid, we collected and examined the gene expressions of four different reproductive organs of worker, drone and queen (Fig. 3c). Vitellogenin is a precursor protein of egg yolk that is used as a biomarker in female46,47,48. In reproductive organs, we found that vitellogenin was expressed in all four reproductive organs. It was most highly expressed in QO, followed by WO, DMG, and DT (Fig. 3c). In a previous data, vitellogenin was first detected in the queen at the mid-late pupal stage, in the worker at the late pupal stage, and in the drone at the adult emergence stage49. We classified the genes of reproductive organs into three categories of major royal jelly protein (MRJP) family, hormone-related genes, and growth factors (Fig. 3c and Supplementary Table S2), which are key regulatory factors during early development. When comparing QO with WO, DMG, and DT, transcripts were mostly expressed in QO (Fig. 3c). To validate the result of RNA-seq data, we randomly selected six genes from the brain and reproductive organs and determined their transcription levels using qRT-PCR. The expression patterns of the six genes based on qRT-PCR showed the same patterns (Fig. 3d), which validated the reliability and reproducibility of the RNA-seq data.
Distribution of the immune system across organ types under natural conditions
To understand the innate immune system of A. cerana, we analyzed the differences in the expression patterns of immune-related genes between castes and displayed them in a heat map (Fig. 4a and Supplementary Table S3). We compared 61 immune-related gene expressions in several specific castes. We found that six immune-response peptides (apidaecin, apidermin, hymenoptaecin, hexamerin, defensin, and T-cell immunomodulatory protein) were highly expressed in all the examined organs. It is well-known that apidaecin, apidermin, hymenoptaecin, hexamerin, and defensin are typical AMPs that correspond to the Toll pathway or Imd pathway. Genes related to the Wnt-signal pathway and RNAi pathway showed very low expression patterns of immune-response genes in the reproductive organs of drones and in the guts of workers and drones (Fig. 4a).

Differences in immune system transcripts across castes. (a) Heat map of individual immune genes. (b) Distribution of immune-related transcripts in the organs of each caste shown on a pink-scale. Expression ratio showing the proportion of immune-related transcripts expressed among organs in each caste. (c) Immune genes as a fraction of all transcripts across castes. The collected organs are shown. WB worker brain, WV worker ventriculus, WR worker rectum, WO worker ovary, WVG worker venom gland, DB drone brain, DV drone ventriculus (midgut), DR drone rectum, DMG drone mucus gland, DT drone testis, QO queen ovary.
Among the entire gene expressions of different organs from each caste, we investigated the expression ratio of immune-response genes relative to whole transcripts. In comparing the gene expressions, which are displayed in Fig. 4a, the rate of the expression of immune-related genes was different in each organ, which implies that these genes were mainly abundant in the intestinal organs (Fig. 4b). In comparing the transcriptome profiles of whole organs, queen exhibited weakest innate immune-related genes expressed in natural conditions (Fig. 4c).
Expression of detoxification machinery
We investigated the expression profile of detoxification machinery in unstressed honeybees. In the wild, forager bees collect pollen and transport them to the colony and nurse bees serve nectar as their nutrient resource to feed the brood. Therefore, it is important to detoxify toxic substances such as pesticides that can contaminate the dietary source of the colony. As the detoxification mechanism is assumed to differ among worker, drone, and queen, we compared the detoxification-related expression patterns of five gene families between different castes under natural conditions. First, there were 12 genes in the glutathione-S-transferase family. In order of high expression ratios were 24% in DT, 19% in WB, and 13% in WR (Fig. 5a). Second, there were 39 genes corresponding to oxidation-related biomolecules (peroxiredoxin, superoxide, superoxide and thioredoxin transcripts). We observed the highest expression ratio in WB was 21% (Fig. 5b). Third, there were two genes in catalase. For which the expression ratios were 23% and 22% in WR and WV, respectively (Fig. 5c). Fourth, there were seven genes in the multidrug resistance-associated protein family. In order of high expression, the ratios were 28% and 24% in WR and QO, respectively (Fig. 5d). Fifth, there were 25 genes in the cytochrome P450 transcripts family. The high expression ratio was 37% in WR, then 22% in WV (Fig. 5e). Genes related to detoxification generally showed high expression rates in WV and WR, which may be closely related to the behavior of the worker acting as nurse, server, and pollinator. Compared to the entire transcripts of each organ, genes encoding peroxiredoxin, superoxide, and thioredoxin were expressed ubiquitously with no organ or caste-specific bias (Fig. 5f), which is a similar pattern to the previous proteome report on A. mellifera19.

Expression differences in detoxification machineries among castes. Caste and organ distribution of detoxification machineries genes (a) Glutathione-S-transferases (GST) transcripts (shown in gray). (b) Peroxiredoxin, superoxide, superoxide and thioredoxin transcripts (shown in blue). (c) Catalase (shown in brown). (d) Multidrug resistance-associated protein (shown in purple). (e) Cytochrome P450 (shown in green). Expression ratio showed the proportion of detoxification-related transcripts expressed among organs in each caste (a-e). The collected organs are shown. Modified illustration of the honeybees was adapted with permission from Professor Leonard J. Foster, the corresponding author of Genome Research (2013), 23, 1951–1960. (f) The percent of all quantified detoxification machineries in each caste.
Differences in the haplodiploid sex-determination system between female ovaries
To understand haplodiploid development more comprehensively, we compared transcriptome data between fertile QO and unfertile WO. We found a total of 463 genes and displayed them on a heat map (Fig. 6a). These genes were grouped into three clusters. We classified the genes up-regulated in WO as Cluster 1, genes with no difference in expression between QO and WO as Cluster 2, and genes up-regulated in QO as Cluster 3 (Supplementary Table S4). To obtain information about the biological processes, cellular components, and molecular function of each gene cluster, we analyzed the gene ontology (GO) in three clusters using BLAST2GO program. GO analysis indicated that genes in Cluster 1 were related to the development process, genes in Cluster 2 were related to the development process and immune system process, and genes in Cluster 3 were related to the development process and reproduction (Fig. 6b). Our results showed that both QO and WO were related to the development process, but only QO was related to reproduction process.

Differences in female ovary transcripts. (a) Heat map of WO and QO. Clustering transcripts of ovary (b) GO enrichment graph of clusters: biological process (shown in green bars), cellular component (shown in blue bars), and molecular function (shown in red bars). (c) The percent expression of ovary development factors in WO (shown in red bars) and QO (shown in blue bars). (d) Select nine genes related to the major factors of ovary development. (e) Relative expressions of ovary development related genes exhibiting significant change. WO worker ovary, QO queen ovary.
In molecular biology, transcription factors and elongation factors are key regulators in the transcription of genetic processes50. Serine/threonine protein kinase plays a significant role in post-translational modification51. The proteins that are encoded by these genes are essential for the differentiation and proliferation of germ cells as ovary development factors. In the comparison between QO and WO, QO exhibited extremely large proportions of these factors (Fig. 6c). The transcription levels of nine selected genes encoding for major factors of ovary development can be seen in Fig. 6d. Two of the major factors were up-regulated in WO but others were up-regulated in QO (Fig. 6d). To determine the fold changes in the expression levels of two selected genes of maelstrom and piwi between QO and WO, the results from qRT-PCR analysis matched well with the RNA-seq results (Fig. 6e), which validated the reliability and reproducibility of RNA-seq data.
Expression patterns of venom encoding genes in organs
There were 105 genes related to venom processes. Initially, high expression patterns were expected for the 105 venom-related genes in WVG, but this was not the case. The expressions of the 105 venom encoding genes in the reproductive organs of the drone and queen were compared with those of the WVG. As expected, genes related to venom protease and phospholipase were generally highly expressed in WVG. However, some venom genes, including phospholipase, prepromelittin gene (which is required to produce melittin), and the venom allergen 5-like (ACSNU08074T0), which is a toxic peptide, showed higher expressions in the drone and queen reproductive organs (Fig. 7a). The mRNA-seq results using qRT-PCR revealed the same patterns for four randomly selected genes (Fig. 7b). These results validate the reliability and reproducibility of RNA-seq data.

Expressions differences of venom-specific genes among reproductive organs. (a) Values above zero indicated greater relative transcript abundance in reproductive organs compared that of to the venom gland. Genes were differentially expressed at a p-value < 0.05. (b) Relative expressions of venom-specific genes exhibiting significant change. DMG drone mucus gland, DT drone testis, QO queen ovary.
Discussion
Here, we presented high-quality transcriptome profiles of organs from different caste members of the A. cerana. In the data set, our results showed differences in transcripts between female and male honeybees in natural conditions. Furthermore, our transcriptome analysis of A. cerana, provides an important resource for future molecular studies of Asian honeybees, particularly to elucidate the mechanisms of the innate immune system, social behavior, genetic difference between females (worker and queen) and male (drone), and the potential function of haplodiploid development.
AMPs are effective against pathogens that are frequently encountered in honeybees hive33. Our data demonstrate that some AMPs were highly expressed in DR, and DMG. The results indicate that AMPs and detoxification-related genes were expressed differently among castes. The expression of AMPs-related genes was mainly expressed, while. detoxification genes were low expression in drones. However, both of the AMPs-related genes and detoxification genes were highly expressed in workers. Therefore, we presume that workers possess immunity against bacteria that are introduced from the outside while foraging outside as a pollinator or as nursing brood to feed pollen. Previous studies showed the workers infected by bacteria had a strong immune response38.
It has been reported that the nutrition and dietary habits of bees affect their physiology52. Therefore, in the present study, we compared gene expression patterns between worker guts and drone guts. Detoxification mechanisms in honeybee guts, which help bees develop tolerate to a variety of potentially toxic secondary metabolites and pesticides that they encounter in floral nectars and pollen, remain largely unknown53. The latest study on the protein atlas of honey bee organs19 represented the robust detoxification machinery of worker organs. However, our expression data shows that the detoxification-related transcriptome expressed more in the drone, than in the worker. In this study, we provide information on these observed transcriptional changes, which are not reflected in data that was obtained on the protein level19.
The honeybee has a unique sex-determination system that is also known as haplodiploidy. Unfertilized eggs are haploid, whereas fertilized eggs are diploid54. Haploid eggs develop into males, whereas diploid eggs develop into females. Vitellogenin is a precursor protein of egg yolk that is used as a biomarker in female46,47,48 and is also expressed in the fat bodies of workers and queens55. In a previous study, the vitellogenin protein played an important role in ovary development, immunity, stress response, and sex-determination46,47,48,56. In our results, vitellogenin was expressed in both QO and WO, but transcripts related with ovary development were highly expressed in QO.
Interestingly, the maelstrom transcript and piwi protein transcript were found in both QO and WO. Mael and Piwi proteins are required for transcriptional silencing, which is induced by the piRNA pathway. Our results showed that different expressions of mael and piwi were observed in QO and WO. The Mael protein repressed canonical RNA polymerase II transcription and inhibits germline transposon transcription57,58. Based on previous papers and our recent findings, we propose that mael and piwi play an important role in the development of QO and WO.
Among all organs, venom-producing organs give honeybees the ability to protect their colonies from enemies by using venom as a weapon59. The WVG produces several components of honeybee venom that cause allergic responses in humans60 and other vertebrates. Honeybee venom contains the major allergens of Api m 1 (phospholipase A2), Api m 2 (hyaluronidase), Api m 3 (acid phosphatase), Api m 4 (melittin), and Api m 7 (CUB serine protease). Melittin, the main constituent of honeybee venom, is one of the major proteins that composes venom toxicity61 and is derived from promelittin62,63. However, it was not unknown how prepromelittin is converted into promelittin. In our results, prepromelittin was proven to be transcribed in the reproductive organs of drones and queen. Therefore, it is speculated that cleavage occurs with melittin after the gene is transferred and that biosynthesis of melittin from prepromelittin might occur after the prepromelittin gene is transferred from the parent to the worker bee. Our data do not clearly demonstrate this hypothesis, but we provide a clue that indicates the synthesis process of the melittin, which guides future research.
In this study, we investigated the transcriptome profiles in several organs from different castes to determine unique traits. The results revealed that immune-related genes and detoxification mechanisms were generally highly expressed in workers in natural conditions. Thus, we provide a foundation for understanding the physiology, morphology, and caste formation of A. cerana at the molecular level. We hope that this study will stimulate future research on the Asian honeybee, which is less understood compared to Western honeybees.
Methods
Sample collection
Samples from each developmental stage of A. cerana were collected from colonies kept in Cheonan, South Korea. To obtain for the samples, one colony of a mated egg-laying queen was used. Bees were collected from the colony during swarm season, and placed in a 1.5 ml microfuge tube. Prior to dissection, the bees were exposed to CO2 in order to aid handling. Each organ was dissected using forceps and microdissection scissors. All dissected organs were rinsed with PBS solution64. Each dissected organ sample was flash-frozen in liquid nitrogen and then stored at − 80 °C. Organs were obtained from three castes of the A. cerana i.e., worker, drone, queen. Worker organs included worker brain (WB), worker ventriculus (WV), worker rectum (WR), and worker venom gland (WVG). Drone organs include drone brain (DB), drone ventriculus (DV), drone rectum (DR), drone testis (DT), and drone mucus gland (DMG). Queen organ includes queen ovary (QO).
Preparation of the mRNA-sequencing library
Total RNA was extracted from the organ samples using TRIzol reagent (Invitrogen). An equal amount of 5 μg total RNA from each sample was used to construct the mRNA library according to manufacturer instructions (Lexogen, Austria). The libraries were sequenced at a high-throughput sequencing facility (Macrogen, South Korea) on an Illumina HiSeq 2500 platform. The sequencing data were obtained with two biological replicates of each organ.
Computational analysis
The raw Illumina sequence reads were filtered to remove low quality sequences by using fastp65 prior to bioinformatic analysis. The filtered Illumina short reads were then mapped to the A. cerana reference genome (ACSNU-2.0)66 using Bowtie2 software67. The mapping results of each sample to the reference mRNA sequences obtained by Bowtie2 were then quantified by Kallisto software68. The gene expression profiles of each sample obtained by Kallisto were calculated as TPM value to compare the gene expression levels between samples.
Validation of differentially expressed genes by quantitative real-time PCR
The relative mRNA levels were quantified according to the manufacturer’s instruction using an AccuPower 2X GreenStar qPCR Master Mix (Bioneer, South Korea). Results were normalized to actin mRNA. Gene expression levels were calculated using the comparative Ct method. All experiments were carried out at least three times for each of three biological replicates. The gene-specific primers used for qRT-PCR are listed in Supplementary Table S5.
Data availability
The datasets generated for this study can be found in the NCBI SRA repository, https://www.ncbi.nlm.nih.gov/sra/, with the GEO Accession No.: GSE164333.
References
Garibaldi, L. A. et al. Wild pollinators enhance fruit set of crops regardless of honey bee abundance. Science 339, 1608–1611 (2013).
Southwick, E. E. & Southwick, L. Jr. Estimating the economic value of honey bees (Hymenoptera: Apidae) as agricultural pollinators in the United States. J. Econ. Entomol. 85, 621–633 (1992).
Magrach, A., González-Varo, J. P., Boiffier, M., Vilà, M. & Bartomeus, I. Honeybee spillover reshuffles pollinator diets and affects plant reproductive success. Nat. Ecol. Evol. 1, 1299–1307 (2017).
Corbet, S. A., Williams, I. H. & Osborne, J. L. Bees and the pollination of crops and wild flowers in the European Community. Bee World 72, 47–59 (1991).
Herrera, C. M., de Vega, C., Canto, A. & Pozo, M. I. Yeasts in floral nectar: A quantitative survey. Ann. Bot. 103, 1415–1423 (2009).
Hossen, M. S. et al. Beneficial roles of honey polyphenols against some human degenerative diseases: A review. Pharmacol. Rep. 69, 1194–1205 (2017).
Cheng, P. C. & Wong, G. Honey bee propolis: Prospects in medicine. Bee World 77, 8–15 (1996).
Lerrer, B., Zinger-Yosovich, K. D., Avrahami, B. & Gilboa-Garber, N. Honey and royal jelly, like human milk, abrogate lectin-dependent infection-preceding Pseudomonas aeruginosa adhesion. ISME J. 1, 149–155 (2007).
Alvarez-Suarez, J. M., Tulipani, S., Romandini, S., Bertoli, E. & Battino, M. Contribution of honey in nutrition and human health: A review. Mediterr. J. Nutr. Metab. 3, 15–23 (2010).
Wu, J. et al. Caffeic acid phenethyl ester (CAPE), derived from a honeybee product propolis, exhibits a diversity of anti-tumor effects in pre-clinical models of human breast cancer. Cancer Lett. 308, 43–53 (2011).
Viuda-Martos, M., Ruiz-Navajas, Y., Fernández-López, J. & Pérez-Álvarez, J. Functional properties of honey, propolis, and royal jelly. J. Food Sci. 73, R117–R124 (2008).
Tsuruda, J. M., Amdam, G. V. & Page, R. E. Jr. Sensory response system of social behavior tied to female reproductive traits. PLoS ONE 3, e3397 (2008).
Greenberg, J. K. et al. Behavioral plasticity in honey bees is associated with differences in brain microRNA transcriptome. Genes Brain Behav 11, 660–670. https://doi.org/10.1111/j.1601-183X.2012.00782.x (2012).
Tan, K. et al. A neonicotinoid impairs olfactory learning in Asian honey bees (Apis cerana) exposed as larvae or as adults. Sci Rep 5, 10989. https://doi.org/10.1038/srep10989 (2015).
Ambros, V. et al. A uniform system for microRNA annotation. RNA 9, 277–279. https://doi.org/10.1261/rna.2183803 (2003).
Ament, S. A. et al. The transcription factor ultraspiracle influences honey bee social behavior and behavior-related gene expression. PLoS Genet 8, e1002596 (2012).
Okosun, O. O., Pirk, C. W., Crewe, R. M. & Yusuf, A. A. Glandular sources of pheromones used to control host workers (Apis mellifera scutellata) by socially parasitic workers of Apis mellifera capensis. J. Insect Physiol. 102, 42–49 (2017).
Keeling, C. I., Slessor, K. N., Higo, H. A. & Winston, M. L. New components of the honey bee (Apis mellifera L.) queen retinue pheromone. Proc. Natl. Acad. Sci. 100, 4486–4491 (2003).
Chan, Q. W. et al. Honey bee protein atlas at organ-level resolution. Genome Res. 23, 1951–1960 (2013).
Ashby, R., Forêt, S., Searle, I. & Maleszka, R. MicroRNAs in honey bee caste determination. Sci. Rep. 6, 18794 (2016).
Collins, D. H. et al. MicroRNAs associated with caste determination and differentiation in a primitively eusocial insect. Sci. Rep. 7, 1–9 (2017).
Pires, C. V., Freitas, F. C. D. P., Cristino, A. S., Dearden, P. K. & Simões, Z. L. P. Transcriptome analysis of honeybee (Apis Mellifera) haploid and diploid embryos reveals early zygotic transcription during cleavage. PLoS ONE 11, e0146447 (2016).
Shi, Y. Y. et al. Diet and cell size both affect queen-worker differentiation through DNA methylation in honey bees (Apis mellifera, Apidae). PLoS ONE 6, e18808 (2011).
Diao, Q. et al. Genomic and transcriptomic analysis of the Asian honeybee Apis cerana provides novel insights into honeybee biology. Sci. Rep. 8, 1–14 (2018).
Forêt, S. & Maleszka, R. Function and evolution of a gene family encoding odorant binding-like proteins in a social insect, the honey bee (Apis mellifera). Genome Res. 16, 1404–1413 (2006).
Smith, B. H. The olfactory memory of the honeybee Apis mellifera: I. Odorant modulation of short-and intermediate-term memory after single-trial conditioning. J. Exp. Biol. 161, 367–382 (1991).
Calvello, M. et al. Expression of odorant-binding proteins and chemosensory proteins in some Hymenoptera. Insect Biochem. Mol. Biol. 35, 297–307 (2005).
Grozinger, C. M., Sharabash, N. M., Whitfield, C. W. & Robinson, G. E. Pheromone-mediated gene expression in the honey bee brain. Proc. Natl. Acad. Sci. 100, 14519–14525 (2003).
Sasaki, K., Akasaka, S., Mezawa, R., Shimada, K. & Maekawa, K. Regulation of the brain dopaminergic system by juvenile hormone in honey bee males (Apis mellifera L.). Insect Mol. Biol. 21, 502–509 (2012).
Kamakura, M. Royalactin induces queen differentiation in honeybees. Nature 473, 478–483 (2011).
Corona, M. et al. Vitellogenin, juvenile hormone, insulin signaling, and queen honey bee longevity. Proc. Natl. Acad. Sci. 104, 7128–7133 (2007).
DeGrandi-Hoffman, G. & Chen, Y. Nutrition, immunity and viral infections in honey bees. Curr. Opin. Insect Sci. 10, 170–176 (2015).
Danihlík, J., Aronstein, K. & Petřivalský, M. Antimicrobial peptides: A key component of honey bee innate immunity: Physiology, biochemistry, and chemical ecology. J. Apic. Res. 54, 123–136 (2015).
Kim, W. J. et al. Differential gene expressions of innate immune related genes of the Asian honeybee, Apis cerana, latently infected with sacbrood virus. J. Asia-Pac. Entomol. 20, 17–21. https://doi.org/10.1016/j.aspen.2016.08.009 (2017).
Goode, K., Huber, Z., Mesce, K. A. & Spivak, M. Hygienic behavior of the honey bee (Apis mellifera) is independent of sucrose responsiveness and foraging ontogeny. Horm. Behav. 49, 391–397 (2006).
Harwood, G., Salmela, H., Freitak, D. & Amdam, G. Social immunity in honey bees: Royal jelly as a vehicle in transferring bacterial pathogen fragments between nestmates. J. Exp. Biol. 224, 7 (2021).
Raymann, K. & Moran, N. A. The role of the gut microbiome in health and disease of adult honey bee workers. Curr. Opin. Insect Sci. 26, 97–104 (2018).
Bull, J. C. et al. A strong immune response in young adult honeybees masks their increased susceptibility to infection compared to older bees. PLoS Pathog. 8, e1003083 (2012).
Bastin, F., Cholé, H., Lafon, G. & Sandoz, J.-C. Virgin queen attraction toward males in honey bees. Sci. Rep. 7, 1–11 (2017).
Nunes, T. M. et al. Queen signals in a stingless bee: Suppression of worker ovary activation and spatial distribution of active compounds. Sci. Rep. 4, 1–7 (2014).
Pirk, C. W., Neumann, P., Hepburn, R., Moritz, R. F. & Tautz, J. Egg viability and worker policing in honey bees. Proc. Natl. Acad. Sci. 101, 8649–8651 (2004).
Cowan, D. P. & Stahlhut, J. K. Functionally reproductive diploid and haploid males in an inbreeding hymenopteran with complementary sex determination. Proc. Natl. Acad. Sci. 101, 10374–10379 (2004).
Han, B. et al. Quantitative neuropeptidome analysis reveals neuropeptides are correlated with social behavior regulation of the honeybee workers. J. Proteome Res. 14, 4382–4393 (2015).
Townson, S. M. et al. Honeybee blue-and ultraviolet-sensitive opsins: Cloning, heterologous expression in Drosophila, and physiological characterization. J. Neurosci. 18, 2412–2422 (1998).
Ellegaard, K. M. & Engel, P. Genomic diversity landscape of the honey bee gut microbiota. Nat. Commun. 10, 1–13 (2019).
Amdam, G. V., Simões, Z. L., Guidugli, K. R., Norberg, K. & Omholt, S. W. Disruption of vitellogenin gene function in adult honeybees by intra-abdominal injection of double-stranded RNA. BMC Biotechnol. 3, 1–8 (2003).
Awde, D. N., Skandalis, A. & Richards, M. H. Vitellogenin expression corresponds with reproductive status and caste in a primitively eusocial bee. J. Insect Physiol. 127, 104113 (2020).
Guidugli, K. R. et al. Vitellogenin regulates hormonal dynamics in the worker caste of a eusocial insect. FEBS Lett. 579, 4961–4965 (2005).
Piulachs, M. et al. The vitellogenin of the honey bee, Apis mellifera: Structural analysis of the cDNA and expression studies. Insect Biochem. Mol. Biol. 33, 459–465 (2003).
Yang, H., Basquin, D., Pauli, D. & Oliver, B. Drosophila melanogaster positive transcriptional elongation factors regulate metabolic and sex-biased expression in adults. BMC Genomics 18, 1–13 (2017).
Préat, T. et al. A putative serine/threonine protein kinase encoded by the segment-polarity fused gene of Drosophila. Nature 347, 87–89 (1990).
Sarma, M. S., Whitfield, C. W. & Robinson, G. E. Species differences in brain gene expression profiles associated with adult behavioral maturation in honey bees. BMC Genomics 8, 202 (2007).
Esther, E. et al. Detoxification mechanisms of honey bees (Apis mellifera) resulting in tolerance of dietary nicotine. Sci. Rep. 5, 1–11 (2015).
Charlesworth, B. Sex determination in the honeybee. Cell 114, 397–398 (2003).
Kocher, S. D., Richard, F.-J., Tarpy, D. R. & Grozinger, C. M. Genomic analysis of post-mating changes in the honey bee queen (Apis mellifera). BMC Genomics 9, 232 (2008).
Zhang, W. et al. Molecular cloning, expression and oxidative stress response of the vitellogenin Gene (AccVg) from Apis cerana cerana. Apidologie 48, 599–611 (2017).
Chang, T. H. et al. Maelstrom represses canonical polymerase II transcription within bi-directional piRNA clusters in Drosophila melanogaster. Mol. Cell 73, 291–303 (2019).
Pek, J. W., Lim, A. K. & Kai, T. Drosophila maelstrom ensures proper germline stem cell lineage differentiation by repressing microRNA-7. Dev. Cell 17, 417–424 (2009).
Nouvian, M., Reinhard, J. & Giurfa, M. The defensive response of the honeybee Apis mellifera. J. Exp. Biol. 219, 3505–3517 (2016).
Sobotka, A. K., Kochoumian, L. & Lichtenstein, L. M. Allergens of honey bee venom. Arch. Biochem. Biophys. 172, 661–671 (1976).
Fletcher, J. E. & Jiang, M.-S. Possible mechanisms of action of cobra snake venom cardiotoxins and bee venom melittin. Toxicon 31, 669–695 (1993).
Hoffman, D. R. Hymenoptera venom allergens. Clin. Rev. Allergy Immunol. 30, 109–128 (2006).
Mollay, C., Vilas, U. & Kreil, G. Cleavage of honeybee prepromelittin by an endoprotease from rat liver microsomes: Identification of intact signal peptide. Proc. Natl. Acad. Sci. 79, 2260–2263 (1982).
Carreck, N. L. et al. Standard methods for Apis mellifera anatomy and dissection. J. Apic. Res. 52, 1–40 (2013).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890 (2018).
Park, D. et al. Uncovering the novel characteristics of Asian honey bee, Apis cerana, by whole genome sequencing. BMC Genomics 16, 1 (2015).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357 (2012).
Bray, N. L., Pimentel, H., Melsted, P. & Pachter, L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 34, 525–527 (2016).
Acknowledgements
We are grateful to the members of the Shin laboratory for helpful manuscript. We also thank Professor Si Hyeock Lee and Dr. Woongjoon Moon for their critical comments on this manuscript. This work was supported by the National Research Foundation of Korea (NRF) Grant funded by the Korea government (MSIT) (No. NRF-2019R1A2C1086369).
Author information
Authors and Affiliations
Contributions
C.S. conceived the research and designed the experiments. I.K. and Y.L. collected the samples. I.K. performed the experiments and analyzed the data. J.Y.L. and W.K. contributed to RNA-seq data analysis. I.K. and C.S. prepared the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kang, I., Kim, W., Lim, J.Y. et al. Organ-specific transcriptome analysis reveals differential gene expression in different castes under natural conditions in Apis cerana. Sci Rep 11, 11267 (2021). https://doi.org/10.1038/s41598-021-90635-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-90635-3
- Springer Nature Limited