
Abstract
Currently, few studies are reported on the composition of microbiota in stroke patients and the association with stroke prognosis. This study investigated the differing microbiota composition in stroke patients and confirmed the association of microbiota composition with poor functional outcome. Between January of 2018 and December of 2019, 198 patients with acute cerebral infarction were included in this study. For the case–control study, age and sex-matched normal healthy subjects (n = 200) were included when receiving their health screening examinations. We isolated bacterial extracellular membrane vesicles and extracted DNA from blood samples. Taxonomic assignments were performed by using the sequence reads of 16S rRNA genes following blood microbiota analysis. Statistical analysis was conducted appropriately by using Statistical Analysis System software. The mean age of the stroke patients were 63.7 ± 12.5 years, and the male sex was 58.5%. Of the total enrolled patients, poor functional outcome (modified Rankin Score ≥ 3) was noted in 19.7%. The principal component analysis of microbiota composition revealed significant differences between healthy control subjects and stroke patients. At the genus level, Aerococcaceae(f), ZB2(c), TM7-1(c), and Flavobacterium were significantly increased in stroke patients compared to the healthy controls, whereas Mucispirillum, rc4-4, Akkermansia, Clostridiales(o), Lactobacillus, and Stenotrophomonas were decreased considerably. For the functional outcome after ischemic stroke, Anaerococcus, Blautia, Dialister, Aerococcaceae(f), Propionibacterium, Microbacteriaceae(f), and Rothia were enriched in the group with good outcomes, whereas Ruminococcaceae(f) and Prevotella were enriched in the group with poor outcome. There was apparent dysbiosis of blood microbiota in patients with acute ischemic stroke compared to healthy people. Ruminococcaceae(f) and Prevotella were elevated in stroke patients with poor functional outcome.
Similar content being viewed by others


Introduction
Stroke refers to damage caused by blocked or diminished blood flow in specific brain regions. It can cause permanent neurologic sequelae and death1. Stroke is a global health problem and a major cause of economic and social burden worldwide2,3. Inflammatory and immune reactions occur in ischemic brain tissues after stroke4, which aggravates pre-existing neurologic symptoms and leads to a poor prognosis5. Infections and systemic inflammatory response have a significant impact on mortality after stroke6.
Approximately 50% of stroke patients have gastrointestinal complications such as gastrointestinal bleeding, constipation, diarrhea, and dysphagia7, of which are associated with neurological deterioration, poor prognosis, and mortality8. Interestingly, microbiota are closely related to gastrointestinal complications9. Previous studies suggested that changes in the composition or imbalance of microbiota after a stroke may adversely affect stroke outcome by regulating pro-inflammatory mediators or inducing stress reactions at the injured site10,11. However, few studies have reported on the composition of the microbiota in stroke patients and the association between this microbiota and stroke prognosis.
Meanwhile, extracellular vesicles (EVs) are one way to transfer information from eukaryotic cells, consisting of various substances such as DNA, RNA, proteins, or lipids12,13. Bacteria in our body also generate EVs, which are small in size, unlike EVs from human cells14. Various isolation methods are currently being used, including ultracentrifugation, microfiltration, and gel filtration15. Bacteria-derived EVs are important signaling pathways between the microbiome and host16,17,18. Through this pathway, the microbiome transmits information to the host and affects the occurrence of various diseases, including the gut-brain axis19,20,21. To date, there are few studies analyzing microbiota composition with blood bacteria-derived EVs in stroke patients.
We hypothesized that the microbiota composition is different between acute ischemic stroke patients and non-stroke patients, and the microbiota composition is associated with the poor functional outcome after stroke. This study investigated the difference in microbiota composition in patients with stroke in a case–control study and confirmed the association of microbiota composition with poor functional outcome in acute ischemic stroke by analyzing bacteria-derived EVs in the blood sample.
Results
Comparison of microbiota composition between stroke patients and healthy controls
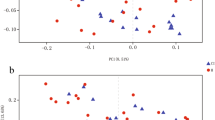
There were no differences based on age and sex between the healthy control subjects and the stroke patients (age: 63.5 ± 12.5 vs. 63.7 ± 12.5, p = 0.888; male sex: 58.6% vs. 58.5%, p = 0.906). The principal component analysis for microbiota composition revealed substantial differences between healthy control subjects and stroke patients (Fig. 1). The demographics of stroke patients are shown in Table 1.

A plot of principal component analysis among patients with stroke and control groups. The relative abundance of operational taxonomic units (OTUs) accounting for > 0.1% of the total bacterial community are shown at the phylum (A), class (B), order (C), family (D), and genus (E) levels.
At the phylum level, the composition of the blood microbiota was frequently in the order of Firmicutes, Bacteroidetes, Proteobacteria, Verrucomicrobia, and Actinobacteria (which accounted for 92% of the total) in the control group. Among those, Verrucomicrobia, Firmicutes, and Deferribacteres were significantly higher in healthy controls than in stroke patients. In acute stroke patients, Proteobacteria, Firmicutes, Bacteroidetes, Actinobacteria, and Verrucomicrobia accounted for 90% of the total. Actinobacteria, Proteobacteria, OD1, and TM7 were significantly higher in stroke patients than in the control group (Fig. 2a). Further comparative analysis regarding class, order, and family is demonstrated in Fig. 2b–d and Supplementary Table 1. At the genus level, Akkermansia, Bacteroides, Lactobacillus, Ruminococcus, and Oscillospira were frequently found in both groups. When comparing the two groups, Enterobacteriaceae(f) (p < 0.001), Pseudomonas (p < 0.001), Flavobacterium (p < 0.001), Staphylococcus (p = 0.038), Prevotella (p < 0.001), Micrococcus (p < 0.001), Corynebacterium (p < 0.001), Enhydrobacter (p < 0.001), Comamonadaceae(f) (p < 0.001), Collinsella (p < 0.001), Faecalibacterium (p < 0.001), Blautia (p = 0.021), Anaerococcus (p < 0.001), Finegoldia (p < 0.001), Dialister (p < 0.001), Neisseriaceae(f) (p < 0.001), ZB2(c) (p < 0.001), Aerococcaceae(f) (p < 0.001), and TM7-1(c) (p < 0.001) were increased in stroke patients, while Bacteroides (p < 0.001), Akkermansia (p < 0.001), Clostridiales(o) (p < 0.001), Ruminococcaceae(f) (p = 0.006), Lachnospiraceae(f) (p = 0.001), Lactobacillus (p < 0.001), Ruminococcus (p < 0.001), Parabacteroides (p = 0.004), [Ruminococcus] (p < 0.001), Oscillospira (p < 0.001), Mucispirillum (p < 0.001), Actinomyces (p < 0.001), Klebsiella (p < 0.001), Stenotrophomonas (p < 0.001), and rc4-4 (p < 0.001) were decreased in stroke patients. Of those, Aerococcaceae(f), ZB2(c), TM7-1(c), and Flavobacterium were drastically increased, with 448.4-fold, 108.7-fold, 73.5-fold, and 68.6-fold elevations, respectively, whereas Mucispirillum, rc4-4, Akkermansia, Clostridiales(o), Lactobacillus, and Stenotrophomonas decreased drastically, with 0.01-fold, 0.06-fold, 0.08-fold, 0.16-fold, 0.17-fold, and 0.20-fold reductions, respectively (Table 2, Fig. 2e).

Composition of microbiota among patients with stroke and control groups. The relative abundance of operational taxonomic units (OTUs) accounting for > 0.1% of the total bacterial community are shown at the phylum (A), class (B), order (C), family (D), and genus (E) levels.
Composition of microbiota for poor functional outcome
Of the total enrolled patients, poor functional outcome was noted in 39 (19.7%). PCA analysis showed no significant differences between the two groups (Supplementary Fig. 1). Proteobacteria, Firmicutes, Bacteroidetes, and Actinobacteria were dominant phyla regardless of functional outcome. Rhodobacterales at the order level and Ruminococcaceae, Prevotellaceae, [Tissierellaceae], Veillonellaceae, Microbacteriaceae, Propionibacteriaceae, and Rhodobacteriaceae at the family level demonstrated significant differences between the functional outcome groups (p < 0.05) (Supplementary Table 2). At the genus level, Ruminococcaceae(f) (p = 0.004) and Prevotella (p = 0.046) in the poor outcome group were significantly higher than in the good outcome group, whereas Anaerococcus (p = 0.018), Blautia (p = 0.039), Dialister (p = 0.002), Aerococcaceae(f) (p = 0.041), Propionibacterium (p = 0.041), Microbacteriaceae(f) (p = 0.021), and Rothia (p = 0.026) were significantly higher in the good outcome group than the poor outcome group (Table 3, Fig. 3).

Composition of microbiota among patients with good outcomes and those with poor outcomes. The relative abundance of operational taxonomic units (OTUs) accounting for > 0.1% of the total bacterial community are shown at the phylum (A), class (B), order (C), family (D), and genus (E) levels.
Discussion
In this study, dysbiosis of blood microbiota in acute ischemic stroke patients was noted compared to normal controls. At the genus level, Aerococcaceae(f), ZB2(c), TM7-1(c), and Flavobacterium were significantly increased in stroke patients, whereas Mucispirillum, rc4-4, Akkermansia, Clostridiales(o), Lactobacillus, and Stenotrophomonas were significantly decreased. Among stroke subtypes, Lactobacillales and Haemophilus were enriched in cases of large artery atherosclerosis, and Pseudomonas was enriched in patients with cardioembolism compared to those with small vessel occlusion. For the functional outcome after ischemic stroke, Anaerococcus, Blautia, Dialister, Aerococcaceae(f), Propionibacterium, Microbacteriaceae(f), and Rothia were significantly higher in the group with a good outcome than the group with the poor outcome. Ruminococcaceae(f) and Prevotella were significantly higher in the poor outcome group than in the good outcome group.
There is little research on microbiota composition and stroke. A small case–control study that analyzed fecal gut microbiota compositions and organic acids, serum interleukin 6 levels, and microbiota showed that Lactobacillus ruminis was higher in stroke patients than controls22. The authors concluded that gut dysbiosis might affect the host metabolism and inflammation, which led to stroke occurrence. In another study using fecal sample analysis, patients with stroke had higher levels of Odoribacter, Akkermansia, Ruminococcaceae_UCG_005, and Victivallis compared to controls, and Christensenellaceae_R-7_group had a positive correlation with clinical outcome23. There was a study investigating the gut microbiota composition in atherosclerotic stroke patients24. In this previous study, stroke patients had higher Enterobacter, Megasphaera, Oscillibacter, and Desulfovibrio and less beneficial microbes, including Bacteroides, Prevotella, and Faecalibacterium24. Our study differs from previous studies in microbiome composition, possibly due to differences in the design, method, and study population of the study. In common with previous studies and the results of our study, there was a significant dysbiosis of microbiota composition in stroke patients, which can lead to disruption of normal metabolism.
Compared to controls, stroke patients in our study were enriched in Enterobacteriaceae(f), Pseudomonas, Flavobacterium, Staphylococcus, Prevotella, Micrococcus, Corynebacterium, Enhydrobacter, Comamonadaceae(f), Collinsella, Faecalibacterium, Blautia, Anaerococcus, Finegoldia, Dialister, Neisseriaceae(f), ZB2(c), Aerococcaceae(f), and TM7-1(c). Enterobacteriaceae, Faecalibacterium, and Blautia are generally considered beneficial bacteria, while Pseudomonas, Flavobacterium, Staphylococcus, Prevotella, Micrococcus, Corynebacterium, Enhydrobacter, Comamonadaceae, Collinsella, Anaerococcus, Finegoldia, Dialister, Neisseriaceae, and Aerococcaceae are opportunistic pathogens. The association between these microbiota and stroke has not been established, except for Prevotella24. We do not know the exact mechanism and meaning of the difference in each microbiome, but this finding can be used in basic research for the microbiome-brain interaction. A detailed comparison of previous results and our study can be found in the Supplementary Table 3.
There are a few studies on the relationship between the microbiome and prognosis after acute ischemic stroke. Li et al. reported a positive correlation of Christensenellaceae_R-7_group and norank_f_Ruminococcaceae with the National Institutes of Health Stroke Scale (NIHSS) and the modified Rankin scale (mRS) score at one month and a negative correlation with Enterobacter23. Our study revealed Ruminococcaceae(f) and Prevotella were higher in the poor outcome group. Ruminococcaceae(f) is a butyrate-producing bacteria, and its depletion is associated with the disease. One human study reported higher stroke risk with reduced butyrate-producing bacteria, which is not in line with our study results25. In our study, Ruminococcaceae(f) decreased in stroke patients compared to healthy controls, whereas Ruminococcaceae(f) was higher in patients with poor outcomes. Although it is difficult to explain the reason for this discrepancy, it may be due to the small number of our patient group, difference in comparison group and different onset time after stroke. Therefore, it is difficult to conclude the benefit or harm of Ruminococcaceae from the results in our study.
Prevotella is associated with mucosal inflammation26, which may adversely affect recovery after stroke. Among the microbiota higher in the good outcome group, Blautia is associated with reduced death in graft-versus-host disease after allogeneic blood/marrow transplantation27. Although a mechanism is not clear, it may be that stroke prognosis results from the immune response. In a previous study, Dialister was enriched in stroke patients28, but its role in the mechanism or prognosis of a stroke is unknown. The difference between previous research and our study may be due to the difference in study design, sample size, and outcome measurement methods. Furthermore, our subjects are Koreans, and this racial difference may affect the discrepancy among the researches. Nevertheless, our research is significant as it suggests an association of microbiota composition with functional outcome in acute stroke patients.
Our study has several limitations. First, we analyzed bacteria DNA from extracellular membrane vesicles in the blood instead of small-intestinal fluid or fecal samples. Bacterial extracellular membrane vesicles contain information about DNA fragments and RNA, as well as immunomodulatory materials such as bacterial toxins and phospholipids29,30. As the extracellular membrane vesicles are very small, they can cross the cellular membrane of the intestine barrier and be distributed via blood throughout the body29,30. Therefore, blood samples could be alternative tissues for metagenome analysis of microbiota31,32. Second, although we enrolled patients with definite stroke subtypes, at least 20% of patients with cerebral infarction were classified as cryptogenic stroke or undetermined subtype. However, because we did not include cryptogenic stroke or undetermined subtype for the analysis, it is difficult to generalize our research results to total stroke patients. Third, patients with stroke have different risk factors and drug usage than healthy control subjects. These various factors such as accompanying risk factors or concomitant medications might have influenced the composition of blood microbiota. An additional analysis of risk factors and drug-matched control is required to confirm the difference in microbiota composition. Fourth, our study had a small sample size and was in a single center. Fifth, because our research had an association design, we could not identify causal relationships. Research on immunomodulation, such as transplanting bacteria or extracellular membrane vesicles, is necessary. Sixth, we aimed determine the impact of microbiota composition on stroke prognosis, but we could not collect samples before the stroke event. Seventh, Trimethylamine N-oxide (TMAO) is an important biomarker for the association between microbiome and cardiovascular disease33. Nevertheless, blood TMAO levels were not measured in our study. Lastly, a serial analysis for the blood microbiota composition could not be completed because blood sampling was done once at admission.
In conclusion, there was an apparent difference in the blood microbiota composition in patients with acute ischemic stroke compared to controls, significantly increased levels of Aerococcaceae(f), ZB2(c), TM7-1(c), and Flavobacterium, and decreased Mucispirillum, rc4-4, Akkermansia, Clostridiales(o), Lactobacillus, and Stenotrophomonas. Regarding stroke patients, Ruminococcaceae(f) and Prevotella were increased in the group with poor functional outcomes. Further research regarding the causal relationship and modification of microbiota is needed.
Materials and methods
Subjects
Our study had two parts. The first part compared the composition of the microbiota between normal healthy controls and acute cerebral infarction patients. The second determined whether the microbiota composition is associated with poor functional outcome in patients with acute cerebral infarction.
Between January of 2018 and December of 2019, a total of 580 acute stroke patients were admitted to our hospital within 24 h of symptom onset. Among those, 249 patients agreed to participate in the study. The subtypes of cerebral infarction were categorized by the Trial of Org 10,172 in the Acute Stroke Treatment (TOAST) classification system at discharge34, patients with small vessel occlusion, cardioembolism, or large artery atherosclerosis were included. Patients with undetermined cause (n = 28) was excluded in this study because this classification consists of heterogeneous etiology with different outcomes35. After excluding 2 patients who withdrew their consent, 219 patients were enrolled. Among those, patients with a history of any malignancy (n = 5), patients who have taken probiotics, prebiotics, or antibiotics within the last 3 months (n = 4), patients with autoimmune disease or vasculitis or on immunomodulating drugs (n = 4), patients with Parkinson’s disease (n = 2), patient with ulcerative colitis (n = 1) were excluded. (Supplementary Fig. 1) A total of 203 blood samples were analyzed for metagenomic analysis. After excluding 5 patients with flawed metagenome analysis, finally, 198 patients were included in our study. All included patients were examined by our routine stroke study protocol36, which includes a chest X-ray, 12-lead electrocardiography, standard blood laboratory tests after 12 h of fasting, and brain imaging studies (brain CT, MRI, CT angiography, MR angiography, or digital subtraction angiography)37,38.
The definition of risk factors was reported in the Supplementary Methods section of a previous study37,39. Neurological severity was determined with the NIHSS score at admission40. High-grade white matter hyperintensities (HWMHs) were a Fazekas score of ≥ 2 in deep and/or periventricular white matter41,42. The Kappa value was 0.92 in the presence of high-grade white matter hyperintensities. Cerebral atherosclerosis was defined as the presence of one or more vessels with more than 50% stenosis/occlusion in the intra- or extracranial cerebral arteries43. Stroke subtypes were grouped by the Trial of Org 10,172 in the Acute Stroke Treatment (TOAST) classification system34. Neurological specialists investigated three months of the mRS score as clinical outcome44. An mRS score of ≥ 3 at three months was considered a poor functional outcome. The Institutional Review Board approved our research (ECT 2018-11-025), and we received informed consent from all subjects and/or their care-givers.
For the case–control study, age and sex-matched healthy subjects (n = 200) were included when receiving health screening examinations at Seoul National University Hospital Healthcare System Gangnam Center (IRB No. 1502–034-647) and Inje University Haeundae Paik Hospital (IRB No. 1297992-2015-064)45. The control subjects had no history of stroke or any conventional vascular risk factors and clinical findings suggestive of gastrointestinal disorders. Moreover, the controls had not taken any medications, probiotics, prebiotics, or antibiotics within three months of blood sampling.
Isolation of bacterial extracellular membrane vesicles and extraction of DNA from the blood sample
In this study, we used ultracentrifugation and microfiltration methods to isolate bacterial EVs46,47,48,49. Blood samples in ethylenediaminetetraacetic acid tubes were collected at admission. The serum was separated by centrifugation (1500×g, 15 min) at 4 °C and stored at − 70 °C until analysis. The serum samples were diluted 1:3 with 1 × PBS (pH 7.4; ML008-01, Welgene Inc., Gyeongsan, Korea) and centrifuged at 10,000×g for one minute at 4 °C. The supernatants were acquired and filtered with a 0.22-μm size to remove foreign particles and bacteria. Separated bacterial extracellular membrane vesicles were boiled at 100 °C for 40 min and then centrifuged at 13,000 rpm for 30 min at 4 °C. The supernatants were then acquired. Bacterial DNA was extracted from the boiled extracellular membrane vesicles with a PowerSoil DNA Isolation Kit (MO BIO Laboratories Inc., Carlsbad, CA USA) according to the manufacturer’s protocols. The DNA from the extracellular membrane vesicles in each sample was quantified with a QIAxpert system (QIAGEN, Hilden, Germany)31,45.
16S rRNA gene sequencing
The method of 16S rRNA gene sequencing was described in a previous study32. The hypervariable region (V3–V4) for genomic bacteria DNA was amplified according to Illumina 16S metagenomic sequencing protocols (Illumina, San Diego, CA, United States). The barcoded fusion primer sequences of 16S_V3_F (50-TCGTCGGCAGCG TCAGATGTGTATAAGAGACAG CCTACGGGNGGCWGCAG-30) and 16S_V4_R (50-GTCTCGTGGGCTCGGAGATGTG TATAAGAGACAGGACTACHVGGGTATCTAATCC-30) were utilized for amplification. To prepare the libraries, PCR products for the MiSeq System guide (Illumina) and QIAxpert (QIAGEN, Hilden, Germany) were used, respectively. After extracting and quantifying the PCR products, equimolar ratios from each mixture were analyzed and sequenced on the MiSeq platform. The data sequencing set included 1,679,505 high-quality gene sequences and a mean of 16,243 reads per sample32. After excluding reads with low-quality and extra-long tails trimming, the remaining representative reads were clustered into operational taxonomic units (OTUs) with a 97% similarity in sequence cut-off at the genus level32.
Taxonomic assignments by sequence reads of the 16S rRNA genes following the blood microbiota analysis
Analysis of all microbiota compositions was blind to the clinical data. The methodology of microbiota analysis of the blood samples has been previously reported32. Based on the barcode and sequences of the primers using MiSeq (Illumina), raw pyrosequencing reads were acquired from the filtration via the sequencer31,32. Taxonomic assignments were performed using the profiling program MDx-Pro ver.1 (MD Healthcare, Seoul, Korea)31,32. High-quality sequencing reads were chosen after filtering based on the quality score (average Phred score 20) and read length (300 bp)32. OTUs were clustered with the sequence clustering algorithm CD-HIT50. The taxonomy assignment was investigated with QIIME51 and UCLUST against the 16S rRNA gene sequence database in GreenGenes 8.15.13 (http://qiime.org/home_static/dataFiles.html)52. Depending on the sequence similarity, 16S rRNA gene sequences were placed at the taxonomic levels. The composition of bacteria at each taxonomic level was plotted as a stack bar. If the case clusters could not be addressed at the genus level due to redundant sequences in the database or a lack of sequences, the taxonomic levels were addressed at higher levels, as demonstrated in parentheses45. Results were normalized to have a mean of 0 and standard deviation of 132. Two-dimensional scatter plots with axes of the first and second principal components were generated using the Matlab 2011a45.
Statistical analysis
Categorical variables were investigated by Fisher's exact test or the Chi-square test. Differences in the beta diversity of bacterial communities were tested using the non-parametric Permutational Multivariate Analysis of Variance (PERMANOVA). The clustering pattern and characteristics were analyzed by the Kruskal–Wallis test based on significant differences in the Shannon index. Statistical analysis was conducted with SAS software (version 9.3; SAS Institute, Cary, NC, United States). A p-value of less than 0.05 indicates statistical significance.
Ethics approval
This study was approved by the institutional research ethics committee of Seoul National University Hospital Healthcare System Gangnam Center (IRB No. 1502-034-647), Inje University Haeundae Paik Hospital (IRB No. 1297992-2015-064), and Ewha Womans University Mokdong Hospital (IRB No. 2018-11-025). The procedures used in this study adhere to the tenets of the Declaration of Helsinki.
Data availability
Data is available at https://doi.org/10.6084/m9.figshare.12813953.
References
Sacco, R. L. et al. An updated definition of stroke for the 21st century: A statement for healthcare professionals from the American Heart Association/American Stroke Association. Stroke 44, 2064–2089. https://doi.org/10.1161/STR.0b013e318296aeca (2013).
Murray, C. J. & Lopez, A. D. Measuring the global burden of disease. N. Engl. J. Med. 369, 448–457. https://doi.org/10.1056/NEJMra1201534 (2013).
Kim, J. Y. et al. Executive summary of stroke statistics in Korea 2018: A report from the epidemiology research council of the Korean Stroke Society. J. Stroke 21, 42–59. https://doi.org/10.5853/jos.2018.03125 (2019).
Shekhar, S. et al. Targeting vascular inflammation in ischemic stroke: Recent developments on novel immunomodulatory approaches. Eur. J. Pharmacol. 833, 531–544. https://doi.org/10.1016/j.ejphar.2018.06.028 (2018).
Jin, R., Liu, L., Zhang, S., Nanda, A. & Li, G. Role of inflammation and its mediators in acute ischemic stroke. J. Cardiovasc. Transl. Res. 6, 834–851. https://doi.org/10.1007/s12265-013-9508-6 (2013).
Teh, W. H. et al. Impact of stroke-associated pneumonia on mortality, length of hospitalization, and functional outcome. Acta Neurol. Scand. 138, 293–300. https://doi.org/10.1111/ane.12956 (2018).
Camara-Lemarroy, C. R., Ibarra-Yruegas, B. E. & Gongora-Rivera, F. Gastrointestinal complications after ischemic stroke. J. Neurol. Sci. 346, 20–25. https://doi.org/10.1016/j.jns.2014.08.027 (2014).
O’Donnell, M. J. et al. Gastrointestinal bleeding after acute ischemic stroke. Neurology 71, 650–655. https://doi.org/10.1212/01.wnl.0000319689.48946.25 (2008).
Singh, V. et al. Microbiota dysbiosis controls the neuroinflammatory response after stroke. J. Neurosci. 36, 7428–7440. https://doi.org/10.1523/JNEUROSCI.1114-16.2016 (2016).
Caso, J. R. et al. Colonic bacterial translocation as a possible factor in stress-worsening experimental stroke outcome. Am. J. Physiol. Regul. Integr. Comp. Physiol. 296, R979-985. https://doi.org/10.1152/ajpregu.90825.2008 (2009).
Liesz, A. et al. DAMP signaling is a key pathway inducing immune modulation after brain injury. J. Neurosci. 35, 583–598. https://doi.org/10.1523/JNEUROSCI.2439-14.2015 (2015).
Yáñez-Mó, M. et al. Biological properties of extracellular vesicles and their physiological functions. J. Extracell. Vesicles 4, 27066. https://doi.org/10.3402/jev.v4.27066 (2015).
Hosseini-Beheshti, E. et al. Exosomes confer pro-survival signals to alter the phenotype of prostate cells in their surrounding environment. Oncotarget 7, 14639–14658. https://doi.org/10.18632/oncotarget.7052 (2016).
Brown, L., Wolf, J. M., Prados-Rosales, R. & Casadevall, A. Through the wall: Extracellular vesicles in Gram-positive bacteria, mycobacteria and fungi. Nat. Rev. Microbiol. 13, 620–630. https://doi.org/10.1038/nrmicro3480 (2015).
Konoshenko, M. Y., Lekchnov, E. A., Vlassov, A. V. & Laktionov, P. P. Isolation of extracellular vesicles: General methodologies and latest trends. Biomed. Res. Int. 2018, 8545347. https://doi.org/10.1155/2018/8545347 (2018).
Shen, Y. et al. Outer membrane vesicles of a human commensal mediate immune regulation and disease protection. Cell Host Microbe 12, 509–520. https://doi.org/10.1016/j.chom.2012.08.004 (2012).
O’Donoghue, E. J. & Krachler, A. M. Mechanisms of outer membrane vesicle entry into host cells. Cell. Microbiol. 18, 1508–1517. https://doi.org/10.1111/cmi.12655 (2016).
Ahmadi Badi, S. et al. Microbiota-derived extracellular vesicles as new systemic regulators. Front. Microbiol. 8, 1610. https://doi.org/10.3389/fmicb.2017.01610 (2017).
van den Elsen, L. W., Poyntz, H. C., Weyrich, L. S., Young, W. & Forbes-Blom, E. E. Embracing the gut microbiota: The new frontier for inflammatory and infectious diseases. Clin. Transl. Immunol. 6, e125. https://doi.org/10.1038/cti.2016.91 (2017).
Kelly, J. R. et al. Breaking down the barriers: The gut microbiome, intestinal permeability and stress-related psychiatric disorders. Front. Cell. Neurosci. 9, 392. https://doi.org/10.3389/fncel.2015.00392 (2015).
Muraca, M., Putignani, L., Fierabracci, A., Teti, A. & Perilongo, G. Gut microbiota-derived outer membrane vesicles: Under-recognized major players in health and disease?. Discov. Med. 19, 343–348 (2015).
Yamashiro, K. et al. Gut dysbiosis is associated with metabolism and systemic inflammation in patients with ischemic stroke. PLoS ONE 12, e0171521. https://doi.org/10.1371/journal.pone.0171521 (2017).
Li, N. et al. Change of intestinal microbiota in cerebral ischemic stroke patients. BMC Microbiol. 19, 191. https://doi.org/10.1186/s12866-019-1552-1 (2019).
Yin, J. et al. Dysbiosis of gut microbiota with reduced trimethylamine-N-oxide level in patients with large-artery atherosclerotic stroke or transient ischemic attack. J. Am. Heart Assoc. 4, 11. https://doi.org/10.1161/jaha.115.002699 (2015).
Zeng, X. et al. Higher risk of stroke is correlated with increased opportunistic pathogen load and reduced levels of butyrate-producing bacteria in the gut. Front. Cell Infect. Microbiol. 9, 4. https://doi.org/10.3389/fcimb.2019.00004 (2019).
Larsen, J. M. The immune response to Prevotella bacteria in chronic inflammatory disease. Immunology 151, 363–374. https://doi.org/10.1111/imm.12760 (2017).
Jenq, R. R. et al. Intestinal blautia is associated with reduced death from graft-versus-host disease. Biol. Blood Marrow Transplant. 21, 1373–1383. https://doi.org/10.1016/j.bbmt.2015.04.016 (2015).
Ji, W. et al. Analysis of intestinal microbial communities of cerebral infarction and ischemia patients based on high throughput sequencing technology and glucose and lipid metabolism. Mol. Med. Rep. 16, 5413–5417. https://doi.org/10.3892/mmr.2017.7227 (2017).
Lee, E. Y. et al. Gram-positive bacteria produce membrane vesicles: proteomics-based characterization of Staphylococcus aureus-derived membrane vesicles. Proteomics 9, 5425–5436. https://doi.org/10.1002/pmic.200900338 (2009).
Deatherage, B. L. & Cookson, B. T. Membrane vesicle release in bacteria, eukaryotes, and archaea: A conserved yet underappreciated aspect of microbial life. Infect. Immun. 80, 1948–1957. https://doi.org/10.1128/IAI.06014-11 (2012).
Park, J. Y. et al. Metagenome analysis of bodily microbiota in a mouse model of Alzheimer disease using bacteria-derived membrane vesicles in blood. Exp. Neurobiol. 26, 369–379. https://doi.org/10.5607/en.2017.26.6.369 (2017).
You, Y.-A., Yoo, J. Y., Kwon, E. J. & Kim, Y. J. Blood microbial communities during pregnancy are associated with preterm birth. Front. Microbiol. 10, 12. https://doi.org/10.3389/fmicb.2019.01122 (2019).
Nam, H. S. Gut microbiota and ischemic stroke: The role of trimethylamine N-oxide. J. Stroke 21, 151–159. https://doi.org/10.5853/jos.2019.00472 (2019).
Adams, H. P. Jr. et al. Classification of subtype of acute ischemic stroke. Definitions for use in a multicenter clinical trial. TOAST. Trial of Org 10172 in Acute Stroke Treatment. Stroke 24, 35–41 (1993).
Nam, H. S. et al. Long-term mortality in patients with stroke of undetermined etiology. Stroke 43, 2948–2956. https://doi.org/10.1161/strokeaha.112.661074 (2012).
Chang, Y. et al. Plasma fibroblast growth factor 23 concentration is associated with intracranial cerebral atherosclerosis in acute ischemic stroke patients. J. Clin. Neurol. 16, 29–36. https://doi.org/10.3988/jcn.2020.16.1.29 (2020).
Song, T. J. et al. Association between aortic atheroma and cerebral small vessel disease in patients with ischemic stroke. J. Stroke 18, 312–320. https://doi.org/10.5853/jos.2016.00171 (2016).
Chang, Y., Kim, J., Kim, M. H., Kim, Y. J. & Song, T. J. Interarm blood pressure difference is associated with early neurological deterioration, poor short-term functional outcome, and mortality in noncardioembolic stroke patients. J. Clin. Neurol. 14, 555–565. https://doi.org/10.3988/jcn.2018.14.4.555 (2018).
Woo, H. G., Chang, Y., Ryu, D. R. & Song, T. J. Plasma Klotho concentration is associated with the presence, burden and progression of cerebral small vessel disease in patients with acute ischaemic stroke. PLoS ONE 14, e0220796. https://doi.org/10.1371/journal.pone.0220796 (2019).
Oh, M. S. et al. Validity and reliability of a korean version of the national institutes of health stroke scale. J. Clin. Neurol. 8, 177–183. https://doi.org/10.3988/jcn.2012.8.3.177 (2012).
Song, T. J., Chang, Y., Kim, A. R., Kim, Y. & Kim, Y. J. High dietary glycemic load was associated with the presence and burden of cerebral small vessel diseases in acute ischemic stroke patients. Nutr. Res. 51, 93–101. https://doi.org/10.1016/j.nutres.2017.12.009 (2018).
Chang, Y., Kim, J., Kim, Y. J. & Song, T. J. Inter-arm blood pressure difference is associated with recurrent stroke in non-cardioembolic stroke patients. Sci. Rep. 9, 12758. https://doi.org/10.1038/s41598-019-49294-8 (2019).
Chang, Y., Choi, G. S., Lim, S. M., Kim, Y. J. & Song, T. J. Interarm systolic and diastolic blood pressure difference is diversely associated with cerebral atherosclerosis in noncardioembolic stroke patients. Am. J. Hypertens. 31, 35–42. https://doi.org/10.1093/ajh/hpx126 (2017).
Song, T. J. et al. High dietary glycemic load is associated with poor functional outcome in patients with acute cerebral infarction. J. Clin. Neurol. 14, 165–173. https://doi.org/10.3988/jcn.2018.14.2.165 (2018).
Lee, Y. et al. Rapid assessment of microbiota changes in individuals with autism spectrum disorder using bacteria-derived membrane vesicles in urine. Exp. Neurobiol. 26, 307–317. https://doi.org/10.5607/en.2017.26.5.307 (2017).
Choi, Y. et al. Gut microbe-derived extracellular vesicles induce insulin resistance, thereby impairing glucose metabolism in skeletal muscle. Sci. Rep. 5, 15878. https://doi.org/10.1038/srep15878 (2015).
Andreu, Z. et al. Comparative analysis of EV isolation procedures for miRNAs detection in serum samples. J. Extracell. Vesicles 5, 31655. https://doi.org/10.3402/jev.v5.31655 (2016).
Théry, C., Amigorena, S., Raposo, G. & Clayton, A. Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr. Protoc. Cell Biol. https://doi.org/10.1002/0471143030.cb0322s30 (2006).
Livshits, M. A. et al. Isolation of exosomes by differential centrifugation: Theoretical analysis of a commonly used protocol. Sci. Rep. 5, 17319. https://doi.org/10.1038/srep17319 (2015).
Fu, L., Niu, B., Zhu, Z., Wu, S. & Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 28, 3150–3152. https://doi.org/10.1093/bioinformatics/bts565 (2012).
Caporaso, J. G. et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336. https://doi.org/10.1038/nmeth.f.303 (2010).
Edgar, R. C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461. https://doi.org/10.1093/bioinformatics/btq461 (2010).
Funding
This work was supported by a grant from the Basic Science Research Program through the National Research Foundation of Korea funded by the Ministry of Education (2018R1D1A1B07040959 to T-JS). The funders had no role in study design, data collection and analysis, the decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. Material preparation, data collection and analysis were performed by Y.C. and T.-J.S. The first draft of the manuscript was written by Y.C. and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript. Conceptualization: Y.C., T.-J.S.; Methodology: Y.C., T.-J.S.; Formal analysis and investigation: Y.C., T.-J.S.; Writing—original draft preparation: Y.C., T.-J.S.; Writing—review and editing: Y.C., H.G.W., J.H.J., G.H.K., K.D.P., T.-J.S.; Funding acquisition: T.-J.S.; Resources: T.-J.S.; Supervision: T.-J.S.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chang, Y., Woo, H., Jeong, J. et al. Microbiota dysbiosis and functional outcome in acute ischemic stroke patients. Sci Rep 11, 10977 (2021). https://doi.org/10.1038/s41598-021-90463-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-90463-5
- Springer Nature Limited