
Abstract
DNA barcoding can be useful for species identification and phylogenetic analysis, but its effectivity has not been verified in most neotropical cloud forest plants. We tested three plastid barcodes, rbcLa, matK, and trnH-psbA, in selected pteridophytes, a well-represented group in these forests, from a little-explored area in Oaxaca, Mexico, applying the CBOL criteria for barcoding. We used BLASTn, genetic distance, and monophyly tree-based analyses employing neighbor-joining (NJ), maximum likelihood (ML), and Bayesian inference methods. Universal primers for rbcLa and trnH-psbA were successfully amplified and bi-directionally sequenced, but matK could not be amplified for most species. rbcLa showed the highest species discrimination in BLASTn (66.67%). trnH-psbA exhibited higher significant interspecific divergence values than rbcL and rbcLa + trnH-psbA (two-sample sign test, P value < 2.2e−16). Using NJ and ML phylogenetic trees, monophyletic species were successfully resolved (100%), differing only in support values and displaying full agreement with the most recent fern classification. ML trees showed the highest mean support value (80.95%). trnH-psbA was the only barcode that could detect the Elaphoglossoideae subfamily. Species discrimination did not increase using rbcLa + trnH-psbA. rbcLa is useful for fern barcoding, trnH-psbA is most helpful for phylogenetic analyses, and matK may not work as a universal barcoding marker.
Similar content being viewed by others

Introduction
Monitoring local biodiversity is fundamental for the development of conservation and sustainable strategies. This task requires a trustable species database, which is often lacking or incomplete for many regions on earth, particularly in the tropics, considered the richest in biodiversity1. Based on a recent estimation, there are about 8.7 million eukaryotic species on earth, of which more than 80% of plants remain to be described2. Classical morphological species identification often requires specimens in good conditions and reproductive structures, which are not always easy to obtain in field studies. Also, the higher phenotypic plasticity of plants makes it difficult to obtain an accurate identification, which frequently should be performed by a specialist of the taxon involved. DNA barcoding, a new molecular approach for species identification, overcomes these drawbacks. This technique requires only a piece of tissue of the specimen for species identification. DNA is extracted from a sample of tissue and amplified using universal primers. Then, the short fragment of amplified DNA is sequenced. The sequence is compared with those already published in a DNA database, such as the GeneBank at the National Center of Biotechnology Information (NCBI), or in the Barcode of Life Data system (BOLD) designed explicitly for DNA barcoding3. If the specimen has a DNA sequence that matches ≥ 99% of that already published in the database, then it is concluded that both sequences belong to the same species. Hebert et al. (2003) generated this technique4.
DNA barcoding using cytochrome oxidase I (CO1) has been successfully applied for animal species; but, in plants, CO1 did not work, and more research is required. A sine qua non requirement for species identification using DNA barcoding is the existence of a published trustable sequence. The Consortium for the Barcode of Life's (CBOL) plant working group evaluated seven-candidate plastid DNA regions based on universality, sequence quality, and species discrimination. CBOL recommended using a core of a 2-locus combination of rbcL + matK as the plant barcode5. Other studies suggest using additional loci, including non-coding plastid regions, such as the intergenic spacer trnH–psbA6,7,8 and the nuclear marker ITS9,10. However, such universality has not been found in plants. Therefore, several authors propose a regional barcode for a wide range of ecological and conservation applications since the specimens are most likely to be identified using a restricted reference database11,12.
In Mexico, tropical montane cloud forests (TMCFs) account for 1% of the country's land area but have a higher plant and animal diversity concentration than any other Mexican ecosystem13. Pteridophytes are well represented in TMCFs in Southeast Mexico14. Oaxaca is the state where more diversity of pteridophytes has been observed15, but some places require increased sample collection16. Areas of difficult access, like the Mixteca highlands, had only one reported species15. To our knowledge, no new records were reported since this publication in this area. The purpose of this study is to evaluate the performance of three plastid barcodes: partial gene rbcLa, matK, and the intergenic spacer trnH-psbA, using standard primers and under the three CBOL criteria (universality, quality, and species discrimination)5 to build a barcode library of pteridophytes in the Mixteca highlands, Oaxaca, Mexico.
Material and methods
Study site, determination, and vouchers
Twenty-nine samples of ferns from 11 families and two samples of lycopods were collected at a fragmented cloud forest at San Miguel Cuevas, Santiago Juxtlahuaca Municipality, Oaxaca state, México (17° 15′ 00.96″ N y 98° 02′ 57.34″), with 2187 m asl of mean altitude. Climate is temperate to semi-warm, and soils are rich in organic matter. Local authorities granted permission to visit their forests and to collect parts of the plants. Fresh plant vouchers were determined by Dr. Daniel Tejero Díez from UNAM FES Iztacala following Mickel and Smith17, recent taxonomic monographs18. Scientific names were checked in the Tropicos.org (https://www.tropicos.org/home) website and the Catalogue of Life (https://www.catalogueoflife.org/). The specimen names were compared with the type material in Jstor global plant (https://plants.jstor.org/). The herbarium vouchers were deposited at the National Mexican Herbarium, from Universidad Nacional Autónoma de Mexico (MEXU), and the herbarium from CIIDIR Oaxaca, Instituto Politécnico Nacional (OAX), pending for registration numbers due to the pandemic crisis.
All plant samples were collected in the field under permits issued by the municipal councils of San Miguel Cuevas, Juxtlahuaca. In no case was the full plant collected; the process of collecting the samples did not kill the plants, which were left alive in their original places. None of the samples collected belong to the near-threatened, vulnerable, endangered, critically endangered, extinct in the wild, or extinct of the IUCN red list (accessed September 7, 2021). Of all species studied, only three belong to the category “least concern” according to the IUCN: Asplenium monanthes, Cystopteris fragilis, and Pteridium feei. The rest of the species were not registered in the IUCN red list, probably because of insufficient data. We, therefore, encourage more studies to assess the IUCN status of these plants.
Isolation, amplification, and sequencing of DNA
The number of individuals sampled per taxon was generally one and less frequent 2. Several leaves of each botanical sample were placed in a Ziplock® bag and kept at − 20 °C in a freezer until processed. Genomic DNA was extracted from 2 mg leaf tissue with FastDNA SPIN kit and FastPrep® (MP Biomedicals, USA) equipment. DNA concentration (ng/µl) and purity (260/280A) from total DNA extracted were measured in a Biophotometer (Eppendorf®). Three chloroplast DNA regions were used for amplification: rbcLa, matK, and the intergenic spacer trnH-psbA. We used standard primers from the Canadian Center for DNA Barcoding (CCDB)19 and a second set of primers for MatK20 (see Table 1 for primer DNA sequences). All three chloroplast regions were amplified using a 25 μl volume of reaction mixture: 5 µl MyTaq Buffer reaction (kit MyTaqDNA Polymerase Bioline), 1 μl of forward primer, 1 μl of reverse primer, 0.2 μl of MyTaq Polymerase, 15.8 μl of nuclease-free water, and 2 μl of isolated genomic DNA template. PCR reaction was carried out in an Applied Biosystems Veriti® thermocycler. PCR temperature cycling programs followed Fasekas et al. protocols24. PCR for rbcLa: 94 °C for 4 min; 35 cycles of 94 °C for 30 s, 55 °C for 30 s, 72 °C for 1 min; final extension 72 °C for 10 min. PCR for matK 94 °C for 1 min; 35 cycles of 94 °C for 30 s, 52 °C for 20 s, 72 °C for 50 s; final extension 72 °C during 5 min. PCR for trnH-psbA (for ferns and allies) 94 °C for 4 min; 2 cycles of 94 °C for 45 s, 50 °C for 45 s, 72 °C for 1 min; 35 cycles of 94 °C for 45 s, 45 °C for 45 s, 72 °C for 1 min; final extension 72 °C for 10 min. Amplified PCR products were detected using agarose gel electrophoresis (1.2% agarose gel TBE) under UV light by staining with GelRed Nucleic Acid (Biotium). PCR products were purified using the EZ-10 Spin Column PCR Products Purification Kit (Biobasic). All PCR products were sequenced by Capillary Electrophoresis Sequencing (CES) in an ABI 3730xl System at the Macrogen sequencing facility (Macrogen Inc., Seoul, Korea).
DNA alignment
rbcLa and trnH-psbA sequence chromatograms were manually edited and assembled into contigs using CodonCode Aligner v.9.0.1 http://www.codoncode.com/aligner/. Due to the low amplification frequency, matK was excluded from further evaluations. Consensus sequences were generated and aligned using MUSCLE25. These alignments were examined by eye and corrected when necessary.
BOLD and Genebank
The project was registered under the name “Ferns and allies of a humid temperate forest in Oaxaca, México” project code FERNO (http://www.boldsystems.org) at The Barcode of Life Data System (BOLD), which is an informatics workbench devoted to the acquisition, storage, analysis, and publication of DNA barcode records3. Three files were submitted to BOLD. First, the Specimen data file included detailed voucher information, scientific names of taxa sampled, collection dates, geographical coordinates, elevation, collectors, identifiers, and habitat. Then, an image file was submitted with high-quality specimen images from each fern and lycopod collected. Finally, a trace file was submitted along with primers, the direction of sequences, and the molecular marker. Sequences uploaded to BOLD were edited and aligned in FASTA format and referenced by Sample IDs. Sequences were also submitted to GeneBank.
Species discrimination
To evaluate species discrimination in rbcL and trnH-psbA sequences, we used three approaches: The Basic Local Alignment Search Tool for nucleotide (BLASTN) method26, which searches against the sequence database available online by the National Center for Biotechnology Information (NCBI) https://www.ncbi.nlm.nih.gov, genetic distance and monophyly tree-based analyses using Neighbor-Joining (NJ), Maximum Likelihood (ML) and Bayesian Inference (BI) analysis.
Following previous studies10,27, query sequences having ≥ 99.0% identical sites to sequences in the database were taken as correct assignments. Percentage species resolution was calculated for each plastid region. The combined rbcL + trnH-psbA species resolution was calculated as the cumulative percentage of each molecular marker28.
To determine the best fit model of nucleotide substitution for phylogenetic analyses jModel test v.2.0.29 was used. We found the general time-reversible model plus gamma distribution (GTR + G) as the best fit for rbcLa, which states for variable base frequencies with symmetrical substitution rates. For trnH-psbA, the best fit was achieved with the transversion plus gamma distribution model (TVM + G), with variable base frequencies, equal variable transversion rates, and transition rates. The data set of each plastid region was analyzed alone and in combination. Sequences of rbcL and trnH-psbA were concatenated into a single matrix rbcL + trnH-psbA with Mesquite30.
Genetic distance and NJ bootstrap consensus tree were inferred from 1000 replicates, and the evolutionary distances were computed using the Kimura 2-parameter method with gaps/missing data treatment adjusted using pairwise deletion. Genetic distance and neighbor-joining trees were constructed in MEGAX31 for each plastid barcode alone and in combination. To evaluate which plastid barcode showed more interspecific divergence and checked for any improvement using these barcodes in combination, we conducted two-sample sign tests with the BSDA package in R32.
We ran ML analyses with the IQ-TREE web server (http://iqtree.cibiv.univie.ac.at). Internal node support, bootstrap analyses were calculated using 1000 iterations. Tree inference using Bayesian analysis was run on MrBayes 3.2.2 on XSEDE via the CIPRES supercomputer cluster (www.phylo.org) for 10 million generations. The tree-based methods (NJ, ML, and BI) evaluated which tree produced the greatest species resolution and whether the barcode sequences form monophyletic groups.
Results
Studied species
Table 2 shows the fern and lycopod species determination and that were used for the barcoding analysis.
PCR amplification and sequencing success
Using universal primers from CCDB of rbcL and trnH-psbA, fern DNA was successfully amplified in most cases (96.77%). Nevertheless, we could not get matK amplifications (Table 3). Furthermore, a second set of primers for matK designed specifically for most ferns20 were tested, and we could only get 19.36% amplification. In particular, we could only get amplicons from: Phanerophlebia macrosora, Dryopteris wallichiana, Asplenium monanthes, Lophosoria quadripinnata, Cystopreris fragilis, and Blechnum appendiculatum. Therefore, further evaluations only include rbcL and trnH-psbA.
The sequencing success rate (bidirectional high-quality sequences > 250 bp) was higher for rbcL (93.33%) than for trnH-psbA (80.00%) (Table 3).
Blast discrimination, BOLD, and GeneBank
We found 100% resolution per family and genera of ferns and lycopods using BLASTn in both plasmid barcodes. We contributed to new species in the GeneBank Taxonomy Database for DNA sequences for rbcLa (8 species), and trnH-psbA (16 species). With the accessions already published, we found that rbcLa could discriminate to species level 66.67% of the cases, whereas trnH-psbA discriminates 50%, and rbcLa + trnH-psbA 60.61%. The best BLAST match identification per species for rbcLa plastid barcode is shown in Table 4 and for trnH-psbA, in Table 5.
A specimen data file, image file, and trace file(s) were submitted to BOLD along with edited and aligned sequences for each of our 29 samples of ferns and two samples of lycopods and can be accessed through the BOLD DNA database (http://www.boldsystems.org) under the ‘FERNO’ project. Twenty-nine fern sequences and two lycopod sequences were newly obtained in this study for rbcLa and trnH-psbA and BOLD ID numbers, and GeneBank accession numbers were generated (Table 6).
Genetic distance
The distribution of intraspecific and interspecific K2P distances across all taxon pairs of the ferns of The Mixteca Alta, Oaxaca, cloud forest, obtained from rbcLa, trnH-psbA, and combined DNA sequences of both plastid barcodes are shown in Fig. 1.

Distribution of interspecific and intraspecific K2P distances across all taxon pairs of ferns from Mixteca Alta, Oaxaca, obtained in partial gene rbcLa (a), intergenic spacer (b) and concatenated plastid regions (c).
Based on previous work33, we included only one individual of each species to avoid biases created by an unequal number of sequences of each species. Intergenic spacer trnH-psbA had the highest mean interspecific K2P distance (0.3037 ± 0.1645 s.d.) in contrast to the mean values of rbcLa (0.1275 ± 0.0467 s.d.) and the combined DNA barcodes (0.1959 ± 0.0795 s.d.).
Results from the two-sample sign test in R of single and concatenated DNA sequences of rbcLa and trnH-psbA using tissues from different species of ferns and lycopods of the Mixteca Alta, Oaxaca, Mexico, are shown in Table 7. The intergenic spacer trnH-psbA showed the highest interspecific genetic divergence in comparison to rbcLa (median = − 0.1535, P value < 2.2e−16) and both plastid barcodes concatenated (median = 0.0851, P value < 2.2e−16).
Topology results
Phylogenetic tree-based analysis using neighbor-joining (Supplementary Fig. S1, Supplementary Fig. S2, Supplementary Fig. S3), maximum likelihood (Fig. 2, Supplementary Fig. S4, Supplementary Fig. S5), and Bayesian Inference trees (Supplementary Fig. S6, Supplementary Fig. S7, Supplementary Fig. S8) were reconstructed to evaluate ferns and lycopods species discrimination for the two barcode regions rbcL and trnH-psbA, single and combined (rbcL + trnH-psbA).
In the neighbor-joining trees, samples from Polystichum fournieri FERNO022-20 and Elaphoglossum xanthopodum FERNO030-20 were removed from the analysis of concatenated sequences since there were missing sequences in rbcLa data and trnH-psbA, respectively. The tree-based methods (NJ, ML, and BI) evaluated which tree produced the greatest species resolution and whether the barcode sequences generate monophyletic species (Table 8).

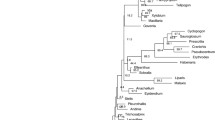
Maximum likelihood cladogram of plastid rbcLa for 27 sequences of ferns and 2 sequences of lycopods from Mixteca Alta, Oaxaca, México, tropical montane cloud forest. Bootstrap values based on 1000 replications are listed as percentages at branching points.
NJ and ML phylogenetic trees resolved 100% of monophyletic species for rbcLa, trnH-psbA, and both barcodes combined (rbcLa + trnH-psbA) with a ≥ 70% clades support using bootstrap of 1000 replicates. The clade support value for rbcLa was higher in ML phylogenetic tree (85.71%) than in the NJ tree (69.23%), whereas the clade support value of trnH-psbA and rbcLa + trnH-psbA was higher in NJ trees (84.61%) than in the ML phylogenetic trees (78.57%). Since the mean clade support of all ML trees was 80.95%, and the mean clade support of all NJ trees was 79.49%, we conclude that the ML and NJ phylogenetic tree satisfactorily resolved the species monophyly of the studied ferns. We present the ML rbcLa phylogenetic tree (Fig. 2) since it yielded the most robust phylogeny: 85.71% of the nodes were supported by a maximum likelihood bootstrap ≥ of 70%.
All Bayesian Inference trees presented polytomies; rbcLa 1 (Supplementary Fig. S6), trnH-psbA 2 (Supplementary Fig. S7), and rbcLa + trnH-psbA 1 (Supplementary Fig. S8). With these polytomies, rbcLa could not resolve 4 monophyletic species, trnH-psbA 18 species, and rbcLa + trnH-psbA 4 species. Unlike the other two phylogenetic methods, BI using concatenated sequences showed an increase in clade support value.
Discussion
Our amplification and sequencing results obtained with rbcLa and trnH-psbA, are very similar to those reported in other ferns studies34,35,36. Contrastingly, matK could not be amplified using two different sets of primers (Table 2). Although matk was proposed with rbcL as the barcode core for plants5,37, ferns appear to be the exception for this common finding. The failure of matK amplification in most leptosporangiate ferns using standard primers is most likely caused by a primer mismatch8,12,20,35,38. In most plants, matK is nested in the trnK intron, but trnK exons are lost in ferns20,39. For detailed studies, highly conserved exons in proximity with variable introns are convenient for phylogenetic analysis, allowing a high amplification efficiency of the primers situated in the exons and intron variability40. Due to the low primer universality of matK in ferns, many studies have designed different matK primers only for local ferns12,20,41. Because of the low amplification rates found in this and other studies34,35, we do not recommend the use of matK in ferns, except for particular situations.
Although we found a successful genera discrimination in these two plastid barcodes using BLASTn analysis, the low results for species discrimination are similar to those observed in ferns of Japan42, in which the rate of BLAST successful species discrimination for rbcLa and trnH-psbA was 70.91% and 65.05%, respectively. We could not find any improvement using both barcodes combined, which differs from results obtained in several studies of land plants6,7,43 and ferns35,42. Low rates of species identification using BLAST in our study are not necessarily caused by low marker performance. Four factors may contribute to explain these results. First, misidentified voucher specimens have been recognized as an increasing problem in public DNA databases, as several authors have acknowledged10,28,44. The rate of specimens correctly identified from the published samples is unknown. Second, online accessions in the GeneBank for our morphological species were limited. We could only find published sequences in 77% of the studied species for rbcLa and 33% for trnH-psbA. Indeed, new 27 rbcLa fern sequences and 27 trnH-psbA fern sequences along two lycopod sequences for each marker were submitted to BOLD along with its metadata.
Third, the widespread existence of hybridization and polyploidy in ferns42,45,46 is another factor that may decrease barcoding species discrimination37. Finally, translocation has been reported in some fern groups41. Other studies found a dramatically reduced trnH-psbA sequence variation for most ferns, probably due to the translocation of this segment into the plastid genome inverted repeat regions41. In our case, however, the intergenic spacer trnH-psbA displayed more interspecific K2P distances than those observed in rbcLa and the combined plastid barcodes (Fig. 1, Table 7). The faster rate of molecular divergence reported in several works5,6,47 for trnH-psbA than that for rbcLa in land plants may account for this result. Our results concur with those found in a recent meta-analysis using five major plant taxonomical groups8, which found a clear barcode gap on trnH-psbA sequences only in the fern group. Our two-sample sign test reveals that the intergenic spacer trnH-psbA offers better species discrimination than rbcLa and both plastid barcode combined for the studied group of ferns (Table 7).
We found similar results in barcode identification performance to those in other fern studies. For instance, higher interspecific variability in trnH-psbA than in rbcLa was also found in a study made in Moorea, French Polynesia with filmy ferns36, a work on Chinese medicinal pteridophytes34, and in studies involving several species of Adiantum35 and Ophioglossum48. However, some exceptions have been found. The mean interspecific divergence values across all taxon pairs (K2P genetic distances) in Japan’s pteridophytes42 did not reveal significant species discrimination between trnH-psbA and rbcLa. The trnH-psbA translocation mentioned above could partly explain these contrasting differences among different ferns studies reported only in certain groups of ferns.
From all topologies obtained in this work, maximum likelihood trees yielded the most robust phylogeny (Table 8). The phylogenetic arrangement found in our study concurs with a recent extant classification of ferns and lycopods49 and with other fern studies42,50. In all of our phylogenetic trees obtained for rbcLa and trnH-psbA, Marattia weinmannifolia is placed near the lycopods. The Marattiacea family is an eusporangiated and ancient group of ferns with fossil records extended back to the Middle Carboniferous51. In a recent study52, results of parsimony dating showed a minimum age estimate of 201–236 Ma, corresponding to late Triassic, for the most recent common ancestor of the extant Marattiaceae. Of all the ferns that we studied, the Marattiaceae is the most primitive, and this explains the higher similarity with the Lycopod outgroup, which is among the oldest groups of vascular plants51.
A paraphyletic clade was observed in the NJ rbcLa tree (Supplementary Fig. S1) and all three phylogenetic trees of psbA-trnH (Supplementary Fig. S2, Supplementary Fig. S4, Supplementary Fig. S7). Elaphoglossum (E. xanthopodum and E. petiolatum) was placed out of the Dryopteridacea family clade. The intergenic spacer trnH-psbA probably was more sensitive to nucleotide substitutions in this genus than rbcLa. A morphological and molecular study of the Elaphoglossum species53, which does not include our studied species, found that the relationship between Elaphoglossum with other fern genera is not clear. This genus was placed within Dryopteridaceae based on its chromosome number (x = 41) and monolete spores. However, in a recent extant fern classification based on new phylogenetic data49, Elaphoglossum was placed in a separate subfamily from the rest of the genera of Dryopteridaceae: Elaphoglossoideae. In agreement with such a decision, our phylogenetic trees using trnH-psbA could also successfully discriminate Elaphoglossum from other members of the Dryopteridaceae family.
Conclusions
Based on the amplification capacity and sequence quality, the partial gene rbcLa and the intergeneric spacer trnH-psbA performed relatively well as barcode markers for ferns in the Mixteca Alta Oaxaca. Our ML phylogenetic trees agree with the recent extant lycophyte and fern phylogeny of the Pteridophyte Phylogeny Group (PPG). rbcLa outperforms in species discrimination and availability of sequences in public databases. However, trnH-psbA outperforms rbcLa in interspecific K2P distances and therefore could be helpful in some phylogenetic analysis involving groups without the inverted sequences translocation that may render low discrimination power. We did not find an increase in species discrimination using both plastid barcodes together in BLASTn, genetic distance, or any topology tree methods. Plastid barcode matK failed to successfully amplify fern and lycopod DNA sequences using universal primers. Our study pinpoints two problems: the low availability of DNA sequences for neotropical fern species and the need for more phylogenetic and polyploidy studies in ferns that clarify the phylogeny of certain groups, such as Elaphoglossum. We hope that the local barcode library that we generated could be the starting point for adding more sequences for a wide range of ecological, conservation, phylogenetic, and medical purposes.
Data availability
All generated ferns and lycopods DNA barcode sequences were registered at the GeneBank Taxonomy Database. Such sequences, the geographic coordinates of the specimens, and pictures of the plants were recorded in The Barcode of Life Data System (BOLD) (Table 6).
References
Gaston, K. J. & Williams, P. H. Spatial patterns in taxonomic diversity. In Biodiversity: A Biology of Numbers and Difference (ed. Gaston, K. J.) 202–229 (Blackwell Science, Limited, 1996).
Sweetlove, L. Number of species on Earth tagged at 8.7 million. Nature https://doi.org/10.1038/news.2011.498 (2011).
Ratnasingham, S., & Hebert, P. D. BOLD: The Barcode of Life Data System (http://www.barcodinglife.org). Mol. Ecol. Notes 7 3, 355–364 (2007).
Hebert, P. D., Cywinska, A., Ball, S. L. & DeWaard, J. R. Biological identifications through DNA barcodes. Proc. R. Soc. Lond. 270(1512), 313–321 (2003).
Group, C. et al. A DNA barcode for land plants. Proc. Natl. Acad. Sci. USA 106(31), 12794–12797 (2009).
Kress, W. J. & Erickson, D. L. A two-locus global DNA barcode for land plants: The coding rbcL gene complements the non-coding trnH-psbA spacer region. PLoS One 26, 508 (2007).
Fazekas, A. J. et al. Multiple multilocus DNA barcodes from the plastid genome discriminate plant species equally well. PLoS One 3(7), e2802 (2008).
Pang, X. et al. Utility of the trnH–psbA intergenic spacer region and its combinations as plant DNA barcodes: A meta-analysis. PLoS One 7(11), e48833 (2012).
China Plant, B. O. L. G., Li, D. Z. et al. Comparative analysis of a large dataset indicates that internal transcribed spacer (ITS) should be incorporated into the core barcode for seed plants. Proc. Natl. Acad. Sci. USA 108, 49, 19641–6 (2011).
Abdullah, M. T. Conserving the biodiversity of Kuwait through DNA barcoding the flora. A thesis submitted for the degree of Doctor of Philosophy, Institute of Molecular Plant Sciences, School of Biological Sciences, University of Edinburgh & Royal Botanic Garden Edinburgh (2017).
Lahaye, R. et al. DNA barcoding the floras of biodiversity hotspots. Proc. Natl. Acad. Sci. USA 105(8), 2923–2928 (2008).
De Groot, G. A. et al. Use of rbcL and trnL-F as a two-locus DNA barcode for identification of NW-European ferns: An ecological perspective. PLoS One 6(1), e16371 (2011).
Ponce-Reyes, R. et al. Vulnerability of cloud forest reserves in Mexico to climate change. Nat. Clim. Change 2(6), 448–452 (2012).
Villaseñor, J. L. Checklist of the native vascular plants of Mexico. Rev. Mex. Biodivers. 87(3), 559–902 (2016).
Tejero-Díez, J. D & Mickel J. T. Pteridofitas. In: García Mendoza, A. J., Ordóñez Díaz M. J. & Briones Salas M. A. (Eds.). Biodiversidad de Oaxaca. Inst. Biol. UNAM, Fondo Oaxaqueño para la conservación de la naturaleza y World Wildlife Found. México D.F. 2004.
Martínez-Salas, E. & Ramos, C. H. Biodiversidad de pteridophyta en México. Rev. Mex. Biodivers. 85, 110–113 (2014).
Mickel, J. T. & Smith, A. R. The Pteridophytes of Mexico. Mem. New York Bot. Garden 88, 1–1030 (2004).
Li, F. W., Pryer, K. M. & Windham, M. D. Gaga, a new fern genus segregated from Cheilanthes (Pteridaceae). Syst. Bot. 37(4), 845–860 (2012).
Kuzmina M.L. Canadian Center for DNA Barcoding ( CCDB) Protocols. Primers sets for plants and fungi. http://www.dnabarcoding.ca/CCDB_DOCS/CCDB_PrimerSets-Plants.pdf (2011).
Kuo, L. Y., Li, F. W., Chiou, W. L. & Wang, C. N. First insights into fern matK phylogeny. Mol. Phylogenet. Evol. 59(3), 556–566 (2011).
Levin, R. A. et al. Family-level relationships of Onagraceae based on chloroplast rbcL and ndhF data. Am. J. Bot. 90(1), 107–115 (2003).
Tate, J. A. & Simpson, B. B. Paraphyly of Tarasa (Malvaceae) and diverse origins of the polyploid species. Syst. Bot. 28(4), 723–737 (2003).
Sang, T., Crawford, D. J. & Stuessy, T. F. Chloroplast DNA phylogeny, reticulate evolution, and biogeography of Paeonia (Paeoniaceae). Am. J. Bot. 84(8), 1120–1136 (1997).
Fazekas, A. J., Kuzmina, M. L., Newmaster, S. G. & Hollingsworth, P. M. DNA barcoding methods for land plants. In DNA Barcodes 223–252 (Humana Press, 2012).
Edgar, R. C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32(5), 1792–1797 (2004).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J. Mol. Biol. 215(3), 403–410 (1990).
Bafeel, S. O. et al. DNA barcoding of arid wild plants using rbcL gene sequences. Genet. Mol. Res. 11(3), 1934–1941 (2012).
Burgess, K. S. et al. Discriminating plant species in a local temperate flora using the rbcL+ matK DNA barcode. Methods Ecol. Evol. 2(4), 333–340 (2011).
Posada, D. & Crandall, K. A. MODELTEST: Testing the model of DNA substitution. Bioinformatics 14(9), 817–818. https://doi.org/10.1093/bioinformatics/14.9.817 (1998).
Maddison, W. P. Mesquite: A modular system for evolutionary analysis. Evolution 62, 1103–1118 (2008).
Kumar, S., Stecher, G., Li, M., Knyaz, C. & Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35(6), 1547–1549 (2018).
Mangiafico, S. S. Summary and Analysis of Extension Program Evaluation in RVol 125 16–22 (Rutgers Cooperative Extension, 2016).
Braukmann, T. W., Kuzmina, M. L., Sills, J., Zakharov, E. V., & Hebert, P. D. Testing the efficacy of DNA barcodes for identifying the vascular plants of Canada. PLoS One 12(1), 1(2017).
Ma, X. Y. et al. Species identification of medicinal pteridophytes by a DNA barcode marker, the chloroplast psbA-trnH intergenic region. Biol. Pharm. Bull. 33(11), 1919–1924 (2010).
Wang, F. H., Lu, J. M., Wen, J., Ebihara, A., & Li, D. Z. Applying DNA barcodes to identify closely related species of ferns: A case study of the Chinese Adiantum (Pteridaceae). PLoS One 11(9), 0160611 (2016).
Nitta, J. H. Exploring the utility of three plastid loci for biocoding the filmy ferns (Hymenophyllaceae) of Moorea. Taxon 57(3), 725–736 (2008).
Hollingsworth, P. M., Graham, S. W., & Little, D. P. Choosing and using a plant DNA barcode. PLoS One 6(5), e19254 (2011).
Li, F. W. et al. Identifying a mysterious aquatic fern gametophyte. Plant. Syst. Evol. 281(1), 77–86 (2009).
Wolf, P. G., Rowe, C. A., Sinclair, R. B. & Hasebe, M. Complete nucleotide sequence of the chloroplast genome from a leptosporangiate fern, Adiantum capillus-veneris L. DNA Res. 10(2), 59–65 (2003).
Cai, M. & Ma, H. Using nuclear genes to reconstruct angiosperm phylogeny at the species level: A case study with Brassicaceae species. J. Syst. Evol. 54, 438–452 (2016).
Li, F. W., et al. rbcL and matK earn two thumbs up as the core DNA barcode for ferns. PLoS One 6 10 e26597 (2011).
Ebihara, A., Nitta, J. H., & Ito, M. Molecular species identification with rich floristic sampling: DNA barcoding the pteridophyte flora of Japan. PLoS One 5 12, e15136 (2010).
Tan, S. L. et al. DNA barcoding herbaceous and woody plant species at a subalpine forest dynamics plot in Southwest China. Ecol. Evol. 8(14), 7195–7205 (2018).
Schneider, H. & Schuettpelz, E. Identifying fern gametophytes using DNA sequences. Mol. Ecol. Notes 6(4), 989–991 (2006).
Grant, V. Plant Speciation (Columbia University Press, 1981).
Schuettpelz, E., Grusz, A. L., Windham, M. D. & Pryer, K. M. The utility of nuclear gapCp in resolving polyploid fern origins. Syst. Bot. 33(4), 621–629 (2008).
Kress, W. J., Wurdack, K. J., Zimmer, E. A., Weigt, L. A. & Janzen, D. H. Use of DNA barcodes to identify flowering plants. Proc. Natl. Acad. Sci. USA 102(23), 8369–8374 (2005).
Patel, M. & Reddy, M. N. Discovery of the World’s smallest terrestrial Pteridophyte. Sci. Rep. 8(1), 1–7 (2018).
Ppg, I. A community-derived classification for extant lycophytes and ferns. J. Syst. Evol. 54(6), 563–603 (2016).
Schuettpelz, E. & Pryer, K. M. Fern phylogeny inferred from 400 leptosporangiate species and three plastid genes. Taxon 56(4), 1037–1050 (2007).
Foster, A. S. & Gifford, E. M. Comparative Morphology of Vascular Plants (W.H. Freeman and Company, 1974).
Lehtonen, S. et al. Exploring the phylogeny of the marattialean ferns. Cladistics 36(6), 569–593 (2020).
Skog, J. E., Mickel, J. T., Moran, R. C., Volovsek, M. & Zimmer, E. A. Molecular studies of representative species in the fern genus elaphoglossum (Dryopteridaceae) based on cpDNA sequences rbcL, trn LF, and rps 4-trn S. Int. J. Plant Sci. 165(6), 1063–1075 (2004).
Acknowledgements
Funding was provided by Instituto Politécnico Nacional (Grant nos. 20171792, 20180889, 20195601, 20201673 and 20211341). Raúl Rivera García and Heladio Luna, assisted in field work. We are thankful to San Miguel Cuevas Community at Oaxaca State, México, for permitting access to their forest. We are most grateful for the comments by two anonymous reviewers.
Author information
Authors and Affiliations
Contributions
S.T.A., R.D.C. Conceived and designed the experiments. S.T.A. Performed molecular experiments. S.T.A. Wrote the paper. A.V.M., R.D.C. Collected plant samples. R.D.C. Reviewing and editing. D.T.D.; A.V.M. Data curation, plant identification. S.T.A. Analysed the data. S.T.A., R.D.C. Discussion of the research. C.A.C.M. Contributed reagents/materials/analysis tools: D.T.D., R.D.C. commented on the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Trujillo-Argueta, S., del Castillo, R.F., Tejero-Diez, D. et al. DNA barcoding ferns in an unexplored tropical montane cloud forest area of southeast Oaxaca, Mexico. Sci Rep 11, 22837 (2021). https://doi.org/10.1038/s41598-021-02237-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-02237-8
- Springer Nature Limited