
Abstract
We aimed to construct a novel population pharmacokinetics (PPK) model of doripenem (DRPM) for Japanese patients in intensive care unit, incorporating the clearance of DRPM by continuous renal replacement therapy (CRRT). Twenty-one patients treated with DRPM (0.25 or 0.5 g) by intravenous infusion over 1 h were included in the study. Nine of the 21 patients were receiving CRRT. Plasma samples were obtained before and 1, 2, 4, 6 and 8 h after the first DRPM administration. PPK analysis was conducted by nonlinear mixed effects modeling using a two-compartment model. Total clearance (CLtotal) in the model was divided into CRRT clearance (CLCRRT) and body clearance (CLbody). The final model was: CLtotal (L h−1) = CLbody(non-CRRT) = 3.65 × (Ccr/62.25)0.64 in the absence of CRRT, or = CLbody(CRRT) + CLCRRT = 2.49 × (Ccr/52.75)0.42 + CLCRRT in the presence of CRRT; CLCRRT = QE × 0.919 (0.919 represents non-protein binding rate of DRPM); V1 (L) = 10.04; V2 (L) = 8.13; and Q (L h−1) = 3.53. Using this model, CLtotal was lower and the distribution volumes (V1 and V2) tended to be higher compared to previous reports. Also, Ccr was selected as a significant covariate for CLbody. Furthermore, the contribution rate of CLCRRT to CLtotal was 30–40%, suggesting the importance of drug removal by CRRT. The population analysis model used in this study is a useful tool for planning DRPM regimen and administration. Our novel model may contribute greatly to proper use of DRPM in patients requiring intensive care.
Similar content being viewed by others


Introduction
Population pharmacokinetic (PPK) analysis provides PPK parameters consisting of average and variance for a population using the nonlinear mixed effect model (NONMEM)1 that fits all the drug plasma concentrations of multiple patients2. Individual pharmacokinetic (PK) parameters such as clearance (CL) and distribution volume (Vd) are obtained by Bayesian method using PPK parameters3.
Doripenem (DRPM) is a carbapenem antibacterial agent that has a broad antibacterial spectrum against Gram-positive bacteria, Gram-negative bacteria, and anaerobic bacteria4. Since DRPM shows a time-dependent effect based on the pharmacokinetic and pharmacodynamics (PK/PD) theory, bactericidal effect is greater when the blood concentration is maintained above the minimum inhibitory concentration (MIC) for 40% of the time or more5.
Several PPK analysis models for DRPM have been reported. The PPK model reported by Bhavnani et al.6 was constructed using an intravenous 2-compartment model based on plasma DRPM concentrations obtained in a phase 1 DRPM clinical trial. The PK parameters they reported included total CL (CLtotal), inter-compartmental clearance (Q), distribution volume of central compartment, and distribution volume of peripheral compartment. In addition, Lee et al.7 succeeded to build a good PPK model for patients with sepsis by integrating creatinine clearance (Ccr) as a covariate in CLtotal using an intravenous 1-compartment model. In the PPK analysis reported by Nandy et al.8, covariates such as Ccr, body weight (BW), age and race were incorporated using an intravenous 2-compartment model for a wide range of subjects in phases 1–3 trials. They reported differences in DRPM clearance depending on race. Reports suggested that some degree of uniformity of the population including race may be necessary because racial variation may affect the PK of drugs such as distribution, metabolism, and elimination9,10. Matsuo et al.11 reported a PPK model of DRPM targeting Japanese subjects by incorporating Ccr and age as covariates in CLtotal, using an intravenous 2-compartment model in phase 1 healthy subjects.
A retrospective cohort study conducted by Kumar et al.12 showed that the death rate increased by 7.6% when administration of antibiotics was delayed by 1 h. Other reports13,14,15 also suggested that the risk of death in septic shock patients decreased if antibiotics were administered within 1 h. There is a relationship between the time to start of antibiotic administration and death16, and international guideline has recommended to administer antibiotics within 1 h after diagnosis17. Carbapenem antibiotics are used empirically in the intensive care unit (ICU) for complex and severe infections. Water-soluble drugs with low molecular weight and low protein binding rate, such as carbapenems, are likely to show increased Vd and higher CL in critically ill patients18,19. In critically ill patients in ICU, CRRT is often performed due to declined kidney function such as acute kidney injury and for removal of inflammatory cytokines. Therefore, clearance of DRPM from the body by CRRT should be considered in patients undergoing CRRT. Using an intravenous 2-compartment model that considers the clearance of DRPM by CRRT, Roberts et al.20 constructed a PPK model without incorporating covariates for critically ill patients undergoing CRRT. They reported that it was necessary to consider the removal of DRPM by CRRT, since clearance by CRRT (CLCRRT) contributed to 30–37% of CLtotal. In addition, Monte Carlo simulation using the PK parameters obtained suggested that the change in distribution volume substantially affected the time above MIC.
In this study, we aimed to construct a novel PPK model incorporating optimal covariates including CRRT in each PK parameter for Japanese ICU patients.
Material and methods
Patients
This study was conducted in accordance with the Declaration of Helsinki. The study was started after obtaining approval from the Ethics Committee in Oita University (Approval No. 613). The subjects in this study consisted of 21 inpatients (a total of 97 samples) in ICU treated with DRPM, who gave written informed consent obtained from either the patients or their legally authorized representatives. Nine of the 21 patients were receiving CRRT. Patients who were administered other carbapenem antibacterial agents before administration of DRPM were excluded.
CRRT was conducted by continuous hemodiafiltration. The hemofilter was a cellulose triacetate membrane. The conditions for CRRT were: blood flow rate (QB) = 80–100 mL min−1; dialysate flow rate (QD) = 0.3–0.9 L h−1; replacement fluid flow rate (QS) = 0.3–0.9 L h−1; and filtrate flow rate (QE) = 0.6–1.8 L h−1.
DRPM at a dose of 0.25 or 0.5 g was given by intravenous infusion over 1 h. At the first administration, plasma samples were obtained from blood sampling before DRPM administration and at 1, 2, 4, 6 and 8 h after the start of infusion. Plasma DRPM concentrations were measured by high performance liquid chromatography (HPLC) according to the procedures we reported previously21. In brief, plasma samples were pre-treated by a solid-phase extraction method. The HPLC system (Waters 2695) was used with a Shiseido Capcell Pak C18 MGII column (5 μm, 250 mm × 4.6 mm; Shiseido Co., Tokyo, Japan) and ultraviolet absorbance detection (Waters 2489 UV/Vis). Separation of DRPM and internal standard was satisfactory, and was free of interfering peaks from the plasma matrix. The limit of quantification (LOQ) for the DRPM assay was 0.5 μg mL−1, and the calibration curve was linear from 0.5 to 100 μg mL−1 (r2 = 0.999). Only one sample had a concentration below the LOQ.
Population pharmacokinetics
Analysis of population pharmacokinetics was conducted using nonlinear mixed effects modeling (NONMEM) version 7.3.022. We selected the compartment model using the DRPM plasma concentration‒time curve with log-transform, by comparing the 1-compartment and 2-compartment models using objective function value (OFV) and the Akaike criteria (AIC)23. Since a conventional 2-compartment model would not include the route of DRPM removal by CRRT, we modified the conventional model to include CRRT clearance. Thus, total clearance in the model was divided into CRRT CL (CLCRRT) and body CL (CLbody) using the ADVAN6 subroutines from the NONMEM library. We initially regarded this as the base model (Fig. 1). For each PPK parameter, inter-individual variability was evaluated by the exponential error model, and residual variability was evaluated using the additive error model. Clinically plausible covariates such as Ccr obtained by the Cockcroft–Gault equation24, BW, and albumin (Alb) were screened as PK parameters of DRPM. Screening of covariates was performed by addition of the candidate covariate to the base model. An eligible covariate should have a correlation with one PK parameter (correlation coefficient r > 0.6), and furthermore no correlation with other covariates; that is, no multiple collinearity. After addition of the covariate, a reduction in OFV of more than 2.71 for one degree of freedom was considered a statistically significant improvement (p < 0.10) based on the χ2 test. After selection of the covariates by the above forward addition step, the model was refined by backward elimination step. A covariate was included in the model when the significance level p < 0.05 (a reduction in the OFV of more than 3.84 for one degree of freedom based on the χ2 test) was obtained. This analysis was calculated by the first-order conditional estimation with interaction method.

Base model for this study. V1 distribution volume of central compartment, V2 distribution volume of peripheral compartment, Q distribution clearance between the central and peripheral compartments, CLbody clearance from body, CLCRRT clearance by continuous renal replacement therapy.
Model evaluation
Validity of our model was examined using visual predictive check and goodness-of-fit plots25. Also, evaluation of reliability and stability of the final model was performed by the bootstrap method. A thousand bootstrap data sets were reconstructed by resampling the subjects from the original data set. The average and standard deviation of parameter estimates obtained from the bootstrap were compared to the estimates of parameters for the final model and standard error obtained from the original data set.
Results
Patients
Patient demographics and relevant clinical data are summarized in Table 1. Among 21 patients, 18 patients were males and three were females. The mean ± standard deviation of Ccr was 68.0 ± 33.4 (mL min−1). Nine patients were undergoing CRRT during DRPM treatment, eight of whom had renal indications for CRRT.
Population pharmacokinetics
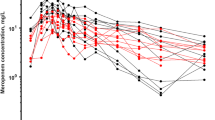
As shown in Fig. 2, a semi-logarithm plot of DRPM plasma concentration versus time showed two phases (distribution phase and elimination phase). Comparisons of 1-compartment and 2-compartment models using OFV and AIC confirmed that both the OFV and AIC of the 2-compartment model were significantly smaller than those of the 1-compartment model (ΔOFV = 89.8, p < 0.05; ΔAIC = 36.3, p < 0.05). Based on this finding, we adopted the 2-compartment model in this study. In addition, comparison with the conventional 2-compartment model without including CRRT clearance confirmed that OFV decreased significantly in our modified 2-compartment model (ΔOFV = 29.8, p < 0.001). Thus, this model that considers removal of DRPM by CRRT was used as the base model in this study.

Semi-logarithm plot of doripenem plasma concentrations. The solid line shows the smooth fitting for doripenem plasma concentrations.
After covariate selection by the forward addition step, models #8 was selected as the full model (Table 2). The full model incorporated Ccr in CLbody, Alb in CLCRRT, and BW in V1. Ccr and BW have been selected as covariates in previous reports7,8,11,26, and Alb has been reported to be removed by continuous hemofiltration using a cellulose triacetate membrane27. Next, at the backward elimination step, when Ccr was excluded from the full model (model #8), OFV increased significantly (Table 2).
The final model was: CLtotal (L h−1) = CLbody(non-CRRT) = 3.65 × (Ccr/62.25)0.64 in the absence of CRRT, or = CLbody(CRRT) + CLCRRT = 2.49 × (Ccr/52.75)0.42 + CLCRRT in the presence of CRRT; CLCRRT = QE × 0.919 (0.919 represents the non-protein binding rate of DRPM)28; V1 (L) = 10.04; V2 (L) = 8.13; and Q (L h−1) = 3.53. Inter-individual variability of CLbody(CRRT), CLbody(non-CRRT), V1, V2, and Q were 7.3%, 22.2%, 13.2%, 24.2%, and 12.7%, respectively; and residual variability was 36.5%. The η shrinkages29 of CLbody(CRRT), CLbody(non-CRRT), V1, V2, and Q were 36.4%, 33.6%, 56.1%, 11.8%, and 56.6%; and ε shrinkage was 23.9%. Figure 3 presents the plot of individual prediction versus time.

Semi-logarithm plot of individual prediction versus time. The solid line shows the smooth fitting for individual predictions.
Model evaluation
The measured DRPM concentrations correlated well with the predicted concentrations both for population and individual predictions (Fig. 4). The conditional weighted residuals (CWRES) and individual weighted residuals (iWRES) distributed uniformly regardless of concentration and time. The above results suggested that the analyses of our model were valid. A comparison of each PPK parameter in the final model with each parameter obtained from 1000 bootstrap samples estimated no large error between the two, and the 95% confidence intervals were relatively small (Table 3). Visual predictive check was performed based on 1000 replicates. As shown in Fig. 5, almost all measured values were within the 95% confidence intervals estimated from our model. Thus, the reliability and stability of each PK parameter are proven and our model is valid.

Goodness-of-fit plots for the final model; population prediction (PRED) versus observation (upper left), Bayesian-estimated individual prediction (IPRED) versus observation (upper right), individual weighted residual versus IPRED (lower left), and conditional weighted residual versus time (lower right). The open circles show observations. The solid black line shows the line of identity. The dotted and dashed gray line shows smooth fitting for observations. |iWRES|, absolute individual weighted residuals.

Visual predictive check for the final model. The open circles show observations for CRRT group and the open triangles for non-CRRT group. The solid line shows the fiftieth percentile of the observations, and the dashed lines show the fifth and ninety-fifth percentiles of the observations. The shaded areas show the 95% confidence intervals of the fifth, fiftieth, and ninety-fifth percentiles of the predictions.
Model simulation
Using our final model, Monte Carlo simulations were performed to evaluate the probability of target attainment (PTA) for targets of 40% and 100% of the time in which free drug concentrations exceed the MIC (40% fT > MIC and 100% fT > MIC, respectively) as well as 100% of the time in which free drug concentrations exceed 4 times the MIC (100% fT > MIC × 4). According to a previous report30, the following dosing regimens were simulated for the above three targets: 1-h intermittent infusion (InI), 4-h extended infusion (ExI), and continuous infusion (CI). Infusion interval was set at 8 h for InI and ExI. Simulated doses per infusion of DRPM were 250 mg, 500 mg, 1000 mg and 2000 mg for InI and Exl, but not for CI (for CI, the dose of DRPM was adjusted so that the daily dose was equal). The PTA was estimated for Pseudomonas aeruginosa (MIC ≤ 2) according to the European Committee on Antimicrobial Susceptibility Testing (EUCAST)31.
As shown in Table 4, PTA when administered by ExI and CI was higher than that when given by InI in all the simulations. For the target of 40% fT > MIC, all three infusion methods of all dosages achieved over 90% PTA regardless of renal function and CRRT.
For the targets of 100% fT > MIC and 100% fT > MIC × 4, however, InI achieved over 90% PTA only in patients with renal function 0 < Ccr ≤ 30 not using CRRT, while all other renal function and CRRT conditions required ExI or CI to achieve 90% PTA. For patients using CRRT, ExI of a higher dosage was needed to achieve over 90% PTA for 100% fT > MIC × 4 versus 100% fT > MIC when renal function was 0 < Ccr ≤ 30 (500 mg vs. 250 mg or above) and 30 < Ccr ≤ 60 (1000 mg vs. 250 mg or above). For patients not using CRRT, the two dosages administered by ExI for the targets of 100% fT > MIC × 4 versus 100% fT > MIC were either not different (250 mg or above for both in 0 < Ccr ≤ 30) or higher for 100% fT > MIC × 4 (500 mg vs. 250 mg or above in 30 < Ccr ≤ 60; 1000 mg vs. 250 mg or above for 60 < Ccr ≤ 90). Note that a similar trend was observed for InI in patients with 0 < Ccr ≤ 30 not using CRRT (100% fT > MIC × 4 vs. 100% fT > MIC: 1000 mg vs. 250 mg or above).
When administered by CI, all dosages (750–6000 mg) achieved over 90% PTA for 100% fT > MIC and doses of 3000 mg or above achieved over 90% PTA for 100% fT > MIC × 4, for all conditions regardless of CRRT or renal function.
Discussion
In our final PPK model for DRPM, CLtot [CLbody(CRRT) and CLbody(non-CRRT)] tends to be lower and V2 is higher compared to previous reports6,8,11. These differences probably arise because the subjects of previous studies included healthy individuals. PK of DRPM is expected to be greatly different between critically ill patients and healthy subjects, since a fluid retention tendency and augmented systemic inflammatory response are observed in the patients32.
The pathological conditions of patients in this study were diverse, including bacteremia, septic shock, infective endocarditis, pneumonia, intraperitoneal infection, urinary-tract infection, and poor infection control after surgery or transplantation. Previous reports on PPK models in patients undergoing CRRT focused on acute infections or sepsis7,26. Our PPK model was not developed targeting specific diseases and included patients using CRRT, which may more realistically reflect the critical clinical setting in which patients have diverse pathological conditions that could change rapidly. Thus this model may complement existing models, especially for disease conditions that change rapidly or for which PPK models have not been established.
In the final model, Ccr was a significant covariate for CLbody. When CRRT is administered, serum creatinine is cleared via two routes: the kidney and CRRT. In the CRRT group, Ccr calculated from serum creatinine is considered inappropriate as an indicator of kidney function for the kidney alone, but appropriate as an indicator of overall renal function for the kidney combined with CRRT. Therefore, Ccr calculated from serum creatinine indicated renal function in different conditions depending on the presence or absence of CRRT. Hence, when used as a covariate in the model, Ccr has to be differentiated depending on the presence or absence of CRRT. That is why different median values of Ccr were used in the final models for the CRRT group and non-CRRT group. Although Ccr is not stable in patients with severe diseases, past reports7,8,26 have indicated that Ccr is a useful marker of renal function in the acute phase. By incorporating Ccr in CLbody into the base model, the inter-individual of CLbody in the final model has become smaller (non-CRRT group: 17.0–7.3%, CRRT group: 29.4–22.2%).
Roberts et al.20 reported that the contribution of CLCRRT to CLtotal was 30–37%. The renal function of the patients undergoing CRRT in this study showed some variations [Ccr; 57.3 ± 8.85 (mean ± SE)]. Therefore, these patients showed CLtotal of 3.81 ± 0.31 (mean ± SE) and CLCRRT of 1.31 ± 0.19, and the contribution of CLCRRT to CLtotal was 40.0 ± 3.92%. Our result of the contribution of CLCRRT to CLtotal was the same as past report, indicating the importance to consider drug removal by CRRT.
Achievement of the maximal bactericidal effect requires at least 40% fT > MIC33. However, it would be necessary to aim for more aggressive exposure of 100% fT > MIC or 100% fT > MIC × 4 in critically ill patients34,35. Hence, we simulated the above three targets for four dosages using three infusion methods (InI, ExI and CI) for each condition of CRRT and renal function. As shown in Table 4, the PTA of InI was inferior to that of ExI and CI, and more than 90% PTA for 100% fT > MIC and 100% fT > MIC × 4 could not be achieved in most conditions. For non-critically ill patients, if the drug exposure target was set at 40% fT > MIC, efficacy against bacteria with MIC 2 or less can be expected by administering 250 mg or more by InI every 8 h, regardless of renal function or CRRT. On the other hand, for critically ill patients not undergoing CRRT, if the drug exposure target was set at 100% fT > MIC, DRPM should be administered by ExI or CI, except for patients with 0 < Ccr ≤ 30 not undergoing CRRT. Using CI, dosages of 3000 mg or above achieved over 90% PTA for even 100% fT > MIC × 4 for all conditions regardless of CRRT or renal function. However, these dosage regimens are expected to be effective only based on simulations. A recent systematic review and meta-analysis of critically ill patients indicated no significant difference in effectiveness between CI and InI36. On the other hand, according to a previous meta-analysis comparing ExI and InI, ExI had a higher clinical success rate and lower mortality than InI37. Considering the possible toxic effect of CI, we recommend that DRPM should be administered by ExI but not by CI to achieve over 90% PTA for 100% fT > MIC and 100% fT > MIC × 4.
Our report had two limitations. First, we were not able to measure DRPM concentrations in the filtrate or plasma concentrations during blood removal and blood returning. Therefore, we were unable to calculate the CLCRRT from measured data. However, DRPM is removed by simple hemodiafiltration process, since it is unnecessary to consider the adsorption process of DRPM at the cellulose triacetate membrane27. Second, this study was conducted in a single facility, and the numbers of subjects and samples were not sufficient. A multicenter joint study is needed to validate the model.
In conclusion, PPK analysis was performed in patients who required systemic management at the ICU and received DRPM for severe infections. Because this population included patients undergoing CRRT, we constructed a model that incorporate the DRPM drug excretion pathway by CRRT. Using this model, Ccr was selected as a significant covariate for CLbody. The contribution rate of CLCRRT to CLtotal was 40 ± 3.92%. Furthermore, the results of Monte Carlo simulation show a possibility that DRPM clearance may be significantly different depending on the presence or absence of CRRT, which may impact the therapeutic effect. The present findings thus suggest that drug removal by CRRT may be important. From the above, our novel model is a useful tool for deciding administration of DRPM and may contribute greatly to further proper use of DRPM in patients requiring intensive care.
References
Sheiner, L. B., Rosenberg, B. & Marathe, V. V. Estimation of population characteristics of pharmacokinetic parameters from routine clinical data. J. Pharmacokinet. Biopharm. 5, 445–479 (1977).
Sheiner, J. & Beal, S. L. Evaluation of methods for estimating population pharmacokinetics parameters. I. Michaelis–Menten model: routine clinical pharmacokinetic data. J Pharmacokinet. Biopharm. 8, 553–571 (1980).
Bayes, T. An essay toward solving a problem in the doctrine of chance. 1763. MD Comput. 53, 370–418 (1991).
Pateron, D. L. & Depestel, D. D. Doripenem. Clin. Infect. Dis. 49, 291–298 (2009).
Drusano, G. L. Antimicrobial pharmacodynamics; critical interactions of ‘bug and drug’. Nat. Rev. Microbiol. 2, 289–300 (2004).
Bhavnani, S. M., Hammel, J. P., Cirincione, B. B., Wikler, M. A. & Ambrose, P. G. Use of pharmacokinetic–pharmacodynamic target attainment analyses to support phase 2 and 3 dosing strategies for doripenem. Antimicrob. Agents Chemother. 49, 3944–3947 (2005).
Lee, D. H. et al. Population pharmacokinetic analysis of doripenem after intravenous infusion in Korean patients with acute infections. Antimicrob. Agents Chemother. 61, e02185–e2216 (2017).
Nandy, P., Samtani, M. N. & Lin, R. Population pharmacokinetics of doripenem based on data from phase 1 studies with healthy volunteers and phase 2 and 3 studies with critically ill patients. Antimicrob. Agents Chemother. 54, 2354–2359 (2010).
Johnson, J. A. Influence of race or ethnicity on pharmacokinetics of drugs. J. Pharm. Sci. 86, 1328–1333 (1997).
Chen, M. L. Ethnic or racial differences revisited: impact of dosage regimen and dosage form on pharmacokinetics and pharmacodynamics. Clin. Pharmacokinet. 45, 957–964 (2006).
Matsuo, Y., Ishibashi, T., Kubota, R. & Wajima, T. Population pharmacokinetics of doripenem in Japanese subjects and Monte Carlo simulation for patients with renal impairment. J. Infect. Chemother. 21, 123–129 (2015).
Kumar, A. et al. Duration of hypotension before initiation of effective antimicrobial therapy is the critical determinant of survival in human septic shock. Crit. Care Med. 34, 1589–1596 (2006).
Ferrer, R. et al. Effectiveness of treatments for severe sepsis: a prospective, multicenter, observational study. Am. J. Respir. Crit. Care. Med. 180, 861–866 (2009).
Ferrer, R. et al. Empiric antibiotic treatment reduces mortality in severe sepsis and septic shock from the first hour: results from a guideline-based performance improvement program. Crit. Care Med. 42, 1749–1755 (2014).
Gaieski, D. F. et al. Impact of time to antibiotics on survival in patients with severe sepsis or septic shock in whom early goal-directed therapy was initiated in the emergency department. Crit. Care Med. 38, 1045–1053 (2010).
Jalili, M. et al. Effect of door-to-antibiotic time on mortality of patients with sepsis in emergency department: a prospective cohort study. Acta Med. Iran. 51, 454–460 (2013).
Andrew, R. et al. Surviving sepsis campaign: international guidelines for management of sepsis and septic shock: 2016. Intensive Care Med. 43(3), 304–377 (2017).
Kitzes-Cohen, R., Farin, D., Piva, G. & Myttenaere-Bursztein, S. A. Pharmacokinetics and pharmacodynamics of meropenem in critically ill patients. Int. J. Antimicrob. Agents 19, 105–110 (2002).
Novelli, A. et al. Pharmacokinetic evaluation of meropenem and imipenem in critically ill patients with sepsis. Clin. Pharmacoknet. 44, 539–549 (2005).
Roberts, J. A. et al. Doripenem population pharmacokinetics and dosing requirements for critically ill patients receiving continuous venovenous haemodiafiltration. J. Antimicrob. Chemother. 69, 2508–2516 (2014).
Tanaka, R. et al. Pharmacokinetic/pharmacodynamic analysis for doripenem regimens in intensive care unit patient. Biol. Pharm. Bull. 40, 1226–1231 (2017).
Beal, S. L. & Sheiner, L. B. NONMEM Users Guides NONMEM Project Group (University of California at San Francisco, San Francisco, 1992).
Akaike, H. A new look at the statistical model identification. IEEE Trans. Autom. Control 19, 716–723 (1974).
Cockcroft, D. W. & Gault, M. H. Prediction of creatinine clearance from serum creatinine. Nephron. 16, 31–41 (1976).
Guidance for Industry: Population Pharmacokinetics. Silver Spring: United States Food and Drug Administration. (Accessed 29 August 2017); http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM072137 (2017).
Abdul-Aziz, M. H. et al. Population pharmacokinetics of doripenem in critically ill patients with sepsis in a Malaysian intensive care unit. Antimicrob. Agents Chemother. 60, 206–214 (2016).
Yasuda, N. et al. Removal of 17 cytokines, HMGB1, and albumin by continuous hemofiltration using a cellulose triacetate membrane: an ex vivo study. J. Surg. Res. 176(1), 226–231 (2012).
Hori, T., Nakano, M., Kimura, Y. & Murakami, K. Pharmacokinetics and tissue penetration of a new carbapenem, doripenem, intravenously administered to laboratory animals. In Vivo. 20, 91–96 (2006).
Savic, R. M. & Karlsson, M. O. Importance of shrinkage in empirical bayes estimates for 493 diagnostics: problems and solutions. AAPS J. 11(3), 558–569 (2009).
Hanberg, P. et al. Population pharmacokinetics of meropenem in plasma and subcutis from patients on extracorporeal membrane oxygenation treatment. Antimicrob. Agents Chemother. 62(5), e02390-e2417 (2018).
The European Committee on Antimicrobial Susceptibility Testing. Breakpoint tables for interpretation of MICs and zone diameters. Version 10.0. (Accessed 28 October 2020); http://www.eucast.org (2020)
Roberts, J. A. & Lipman, J. Pharmacokinetic issues for antibiotics in the critically ill patient. Crit. Care Med. 37, 840–851 (2009).
Drusano, G. L. Antimicrobial pharmacodynamics: criteria interaction of ‘BUG and DRUG’. Nat. Rev. 2, 289–300 (2004).
Tam, V. H., McKinnon, P. S., Akins, R. L., Rybak, M. J. & Drusano, G. L. Pharmacodynamics of cefepime in patients with Gram-negative infections. J. Antimicrob. Chemother. 50, 425–428 (2002).
Li, C., Xiaoli, Du., Kuti, J. L. & Nicolau, D. P. Clinical pharmacodynamics of meropenem in patients with lower. Antimicrob. Agents Chemother. 51, 1725–1730 (2007).
Chen, C.-H. et al. Continuous versus intermittent infusions of antibiotics for the treatment of infectious diseases. Medicine. 98, 1–9 (2019).
Zhenwei, Yu., Pang, X., Xuqi, Wu., Shan, C. & Jiang, S. Clinical outcomes of prolonged infusion (extended infusion or continuous infusion) versus intermittent bolus of meropenem in severe infection: a meta-analysis. PLoS ONE 13(7), 1–11 (2018).
Acknowledgements
The authors thank Kazue Ogata of the Department of Clinical Pharmacy, Oita University Hospital, for technical assistance. This study was supported by the Japan Society for the Promotion of Science (16H00527), and in part by a Grant-in-Aid for Early-Career Scientists from the Japan Society for the Promotion of Science (20K16048).
Author information
Authors and Affiliations
Contributions
K.N., Y.S. and R.T. planned and conducted the analyses. T.K., Y.O., N.Y., and K.G. contributed to obtaining patient consent form by proxy, and patient blood samples. Y.S. managed the programs required for modeling and analysis. T.K. and H.I. supervised the study. All authors were involved with drafting and revising this article. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Nonoshita, K., Suzuki, Y., Tanaka, R. et al. Population pharmacokinetic analysis of doripenem for Japanese patients in intensive care unit. Sci Rep 10, 22148 (2020). https://doi.org/10.1038/s41598-020-79076-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-79076-6
- Springer Nature Limited