
Abstract
Dengue fever is a mosquito-borne disease caused by the dengue virus. Aedes aegypti (Ae. Aegypti) is considered the primary vector of Dengue virus transmission in Yunnan Province, China. With increased urbanization, Ae. aegypti populations have significantly increased over the last 20 years. Despite all the efforts that were made for controlling the virus transmission, especially on border areas between Yunnan and Laos, Vietnam, and Myanmar (dengue-endemic areas), the epidemic has not yet been eradicated. Thus, further understanding of the genetic diversity, population structure, and invasive strategies of Ae. aegypti populations in the border areas was vital to uncover the vector invasion and distribution dynamic, and essential for controlling the infection. In this study, we analyzed genetic diversity and population structure of eight adult Ae. Aegypti populations collected along the border areas of Yunnan Province in 2017 and 2018. Nine nuclear microsatellite loci and mitochondrial DNA (mtDNA) sequences were used to achieve a better understanding of the genetic diversity and population structure. One hundred and fourteen alleles were found in total. The polymorphic information content value, together with the expected heterozygosity (He) and observed heterozygosity (Ho) values showed high genetic diversity in all mosquito populations. The clustering analysis based on Bayesian algorithm, the UPGMA and DAPC analysis revealed that all the eight Ae. aegypti populations can be divided into three genetic groups. Based on the mtDNA results, all Ae. aegypti individuals were divided into 11 haplotypes. The Ae. aegypti populations in the border areas of Yunnan Province presented with high genetic diversity, which might be ascribed to the continuous incursion of Ae. aegypti.
Similar content being viewed by others

Introduction
Dengue fever (DF) is an acute infectious disease transmitted by Aedes mosquitoes and one of the major public health problems worldwide. More than 3.9 billion people across 128 countries are at risk of DF infection, while some 100–400 million infections occur each year1. Since the first outbreak of DF in China, which occurred in Guangdong province in 1978, DF has been causing epidemic cycles every 4–7 years in China2.
Over recent years, an increase in human cases of DF has been observed in China3, especially in the Yunnan Province. This southeast Chinese province comprises 16 prefectures and 129 counties and is characterized by a tropical and subtropical climate, which is conducive to dengue virus infection4. Besides, Yunnan Province shares a 4,060-km border with Laos, Vietnam, and Myanmar, all of which are dengue-endemic areas2. Since 2004, sporadic imported cases of DF have almost been reported annually in Yunnan Province, China, while the first cases of local infection occurred in 2008 in Dehong prefecture and Lincang City5,6. In 2013, a large-scale dengue outbreak occurred in Dehong and Xishuangbanna prefectures.
DF prevention and control depend on effective vector control measures. However, DF has four different serotypes that may affect epidemic control. Aedes aegypti and Ae. Albopictus mosquitos are the main vectors that transmit the virus to humans7. These mosquitoes are widely distributed in tropical regions, especially in Southeast Asia8. Ae. aegypti is mainly found in densely populated urban areas9. In Yunnan province, the first case of Ae. aegypti-borne viral infection was reported in Jiegao Port, Ruili, in 200210. At present, the distribution of Ae. aegypti is still limited to border port areas and has not spread to the inland area of Yunnan Province. However, it has been gradually adapting the climate and environment of Yunnan Province. It has been confirmed by field monitoring that a large number of breeding sites has been found in Dehong, Longchuan County and other ports in nearby areas, and it has become a new member of mosquito population in this area4,11,12,13.
Shi et al.14 indicated a population invasion of Ae. aegypti in the border areas of Yunnan Province. Over the last few years, Yunnan Province has taken measures to comprehensively control Ae. aegypti populations in this area. For example, the “Patriotic Health Movement” and “Building a civilized city” campaigns were carried out in the border cities. The ports of Yunnan Province adopted integrated prevention and control methods to control the vectors, using environmental governance and vector monitoring so as to cut off the transmission channels and prevent large-scale invasion and spread of vectors in the region. However, the DF epidemic has become a serious public health threat, which kept being among the top 5 reported cases of the infectious diseases in Yunnan Province. Therefore, it is necessary to continue implementing the analysis of Ae. aegypti population and the tracking of invasive events.
Genetic techniques could be used to infer the pathways and sources of invasive species, inform the genetic composition and demographic history of founding populations15,16. Microsatellites, also known as simple sequence repeats (SRSs) or short tandem repeats (STRs) are short, tandemly repeated DNA motifs of 1–6 nucleotides distributed throughout eukaryotic genomes17. The microsatellite markers show the advantages of high polymorphism, co-dominant expression, and extensive genomic distribution, and have been proven useful for detecting the subtle population structure18. Mitochondrial DNA (mtDNA) is commonly used in molecular evolution studies of insects19. The mtDNA is more sensitive to genetic drift and has greater genetic differentiation. It is particularly useful for detecting genetic differences and reconstructing the transmission history of invading mosquitoes20,21. Previously, nuclear microsatellite markers and mtDNA sequences were used to investigate the genetic diversity and population structure of Ae. aegypti across the Yunnan Province region14,22. The whole genetic information can inform us on the invasion routes, the colonizing capacity, adaptability, and behaviors of invading mosquito lineages23.
In this study, we analyzed genetic diversity and population structure in eight populations of adult Ae. Aegypti those were collected along the border area of Yunnan Province in 2017 and 2018.
Results
Microsatellite genetic diversity
The polymorphic information content (PIC) value is one of the indicators used to measure allele richness of genes. A total of 114 alleles were found for the nine genetic markers; all alleles were sequenced. As shown in Table 1, the microsatellite locus SQM 6 had the highest number (20) of alleles, while marker SQM 1 had the lowest number (7) of alleles. The PIC values were high, ranging from 0.392 to 0.886, and the average value was 0.672, which indicated that sites were highly polymorphic and can reflect the genetic characteristics of all Ae. aegypti populations24. The microsatellite locus SQM 6 had the highest PIC value (0.886), whereas the locus SQM 1 the lowest PIC value (0.392) (Table 1).
Normally, the genetic diversity of the mosquito population is positively related to the expected heterozygosity (He) and observed heterozygosity (Ho) value of the population. He and Ho value of all Ae. aegypti populations ranged from 0.385 to 0.605, which suggests that the genetic diversity of all mosquito populations is relatively high (Fig. 1). Except for CY, the Ho value in other regions is lower than the He value, which indicates that there may be inbreeding within the species. In addition, CY may have a large amount of individual external supplements or may experience bottlenecks.

The Ho and He values of all Ae. aegypti populations.
Nearly all the FIS values in all Ae. aegypti populations, except population YJ, were positive, ranging from 0.05882 to 0.24657 (Table 2), which further indicated that these populations contained different degrees of inbreeding and Heterozygote deficiency that also may be the reason for the deviation of all populations from HWE.
Microsatellite genetic structure
The FIT value over all populations was 0.309, which indicated that there were significant population differences among the individuals screened. The AMOVA results indicated that the largest proportion of genetic variation in Ae. aegypti population existed in individuals and individuals within populations, accounting for 69.14% and 15.69% of the variation, respectively (Table 3). Although the sampling site is an important factor (P < 0.0001), the ratio was relatively low (Table 3). The Bottleneck effect analysis of all populations revealed that nearly all populations, except population ML and RL, are mutation-drift equilibrium (Table 4).
In order to analyze the specific genetic structure of all eight Ae. aegypti populations, based on the Bayesian algorithm, a clustering analysis was carried out (Fig. 2). Combining with The UPGMA and DAPC analyses, revealed that all eight Ae. aegypti populations can be divided into three genetic groups of the populations from Lincang city, Xishuangbanna prefecture and Dehong prefecture were genetically correlated, except for RL population, which was highly related to the population Xishuangbanna prefecture (Figs. 3, 4).

The clustering analysis of all Ae. aegypti populations based on the Bayesian algorithm. Structure bar plot for all Ae. aegypti populations used in this study. The height of each color represents the probability of assignment to a specific cluster. Subdivision of all the individuals into K = 2 and K = 3 clusters.

The UPGMA analysis of all Ae. aegypti populations. UPGMA cluster analysis of 8 sampling locations based on the genetic distance, the evolutionary distances were computed using the maximum composite likelihood method.

Population structure described by discriminant analysis of principal components (DAPC) based on nine nuclear microsatellite loci. Each color corresponds to a single population, and ellipses with dashed lines represent individuals. (a) All eight populations of Ae. aegypti (a total of 96.6% of the variation was explained by 30 PCs in the DAPC analysis); (b) Four closely related populations of Ae. aegypti (a total of 96.4% of the variation was explained by 25 PCs in the DAPC analysis).
The IBD analysis displayed that the genetic distance of all eight Ae. aegypti populations were positively related to geographic distance, which meant the geographical isolation was the primary cause of genetic diversity of Ae. aegypti (Fig. 5). While the Ae. aegypti can only move hundreds of meters around their larval habitats, which suggests that the transmission of Ae. aegypti in Yunnan Province does not depend on its own activities, but on other factors, such as human activities.

The isolation by distance analysis results among all eight Ae. aegypti populations. (a) The original value of the correlation between the distance matrices is represented by the red dot, while histograms represent permuted values (i.e., under the absence of spatial structure). The original value being out of the reference distribution represents the significant spatial structure. (b) The red line represents the positive relationship between genetic distance and geographic distance among all individuals.
The pairwise FST values of Ae. aegypti ranged from 0.061 to 0.220 (Table 5), showing significant genetic differences. All P values were significant (P < 0.05) after Bonferroni corrections were applied.
Haplotype networks and diversity
Based on the mitochondrial COI, ND4, and ND5 areas of Ae. aegypti, all Ae. aegypti individuals from nine populations were divided into 11 haplotypes, among which H1, 2, 3, 7, and 8 were the main haplotypes. The distribution of these haplotypes in a specific population is shown in Fig. 6. All the new sequences generated in this study are available from GenBank (Table 6). Among the eleven haplotypes, H1, 2, 3, 7, and 8 were the main haplotypes in all populations. The haplotype H1 had the most individuals, mostly from MD, MH, CY, LC, and RL, while H3 and H8 were mainly distributed at Xishuangbanna prefecture and H2 and H7 were only distributed at YJ and JH, respectively (Fig. 6). The vast majority of individuals have shared haplotypes, and sampling sites had individuals that belonged to haplotypes. Neutral test and mismatch analysis based on the mitochondrial genes ND4 and ND5 of Aedes aegypti showed that the distribution of nucleotide mismatches in this population had a single peak structure, indicating that the population of Ae. aegypti had experienced at least one significant population expansion (Fig. 7).

The Haplotype analysis of all Ae. aegypti populations based on mitochondria COI and ND4. The network of haplotypes. Each nonsolid black colored circle represents observed haplotype with greater circle size indicating a greater number of individuals with that haplotype. The black line with numbers represents the position of mutant bases. The colors correspond to the different sampling areas.

The Neutral test and mismatch analysis of all Ae. aegypti populations based on mitochondria COI and ND4. The mismatch distributions showed a smooth and main unimodal curve peaks, which coincide with the population expansion model.
Discussion
The information on invasion and spread of mosquito vectors is essential for understanding vector-borne disease outbreak, transmission dynamics among human populations and implementing effective mosquito control programs25. These are all important factors influencing the mosquito population dynamics, genetic structure patterns, and pathogen transfer through vector populations26. Ae. aegypti is the most important epidemic vector that can cause DF and dengue hemorrhagic fever (DHF) in human and is mainly distributed in southeast China. The most suitable habitats of Ae. aegypti include Hainan, Guangdong, Guangxi, the western and southern border areas of Yunnan, and parts of the southern Guizhou region27. Yet, due to climate changes and increased urbanization, a significant northward shift occurred in the northern Chinese region over recent years28.
Ae. aegypti is an invasive species and potential vector of disease agents in China, which has a significant impact on public health. Ae. aegypti-associated infection was first reported in Yunnan Province (Jiegao Port, near Ruili City, Dehong prefecture) in 200229. In 2009, the Ae. aegypti was detected for the first time in Guanlei Port, Mengla city, Xishuangbanna prefecture30, and later on (2014) in Lincang, Mengding county31. The distribution range and abundance of Ae. aegypti species have significantly increased, and were established in at least eight cities in Yunnan Province. Therefore, the monitoring of Ae. aegypti species is essential for preventing and controlling vector-borne infectious diseases. In our study, all the Ae. Aegypti samples were collected from eight sampling places in three prefectures of Yunnan Province (Xishuangbanna prefecture, Lincang city, and Dehong prefecture). The DF cases in Yunnan Province mainly originated from these prefectures.
Our population genetics analyses of the populations from Yunnan border area that were based on two types of genetic markers (Microsatellites and mtDNA) revealed the genetic structure and the population distribution within this region. The PIC value, He and Ho value were important parameters for measuring the genetic diversity of a population; the higher the value, the more complex the population structure is. Our results revealed that Ae. aegypti species in the Yunnan border region had a great allelic variation. The Ae. aegypti mosquitoes may easily transmit the virus to humans and usually find shelter in indoor habitats. Their flight range is limited, which means they can only move hundreds of meters around their larval habitats25,32. The relatively high genetic diversity of all mosquito populations is most likely caused by invasion events and human activities33. The results of IBD analysis support this conclusion that the dispersal of Ae. aegypti species is aided by human activities and transportation in Yunnan Province.
Bayesian algorithm-based population analysis showed that all Ae. aegypti populations could be divided into three genetic groups. The first group had four populations in the Xishuangbanna prefecture (JH, MH, and ML) and RL city, which might be related to the close tourism and commercial trade exchanges between these two regions. The second group represented two populations from Lincang City. The third group was composed of LC and YJ. Contrary to other six populations, YJ population is closely related with LC population, which is significantly different from the UPGMA result. The differences may come from the sample size and range.
Except for Yingjiang, the inbreeding coefficient (FIS) values of eight other Ae. aegypti populations were positive. Combined with the UPGMA and DAPC analysis, the results indicated that there may have a recent invasion and colonization of the Ae. aegypti in YJ city. Due to the limited flight range, this phenomenon is common for Ae. aegypti population on a small spatial scale33. In 2016, the Xishuangbanna Prefecture established a “Spring Patriotic Health Movement” with the scope to provide the integrated control for infectious disease vectors. This may explain the bottleneck effect observed in ML and RL.
MtDNA markers have been widely used to evaluate the genetic diversity of Ae. aegypti populations34,35. In our study, the degree of polymorphism found in the COI and ND4 sequences were relatively high (eight populations were divided into eleven haplotypes). The H1, which was the dominant haplotype, was found in five places. The analysis of all mosquito samples from two localities in Lincang City (MD and CY) showed only one haplotype (H1) for each gene. Dehong Prefecture is close to Myanmar border, and the intensive personnel activities have led to a large number of invasion events. The abundant waters and commercial activities in Xishuangbanna prefecture have also contributed to many invasion events. This idea was supported by the high levels of polymorphism detected in Xishuangbanna and Dehong prefectures (six haplotypes and seven haplotypes, respectively), which may be the main entry points of Ae. aegypti in Yunnan Province. The H2 haplotype was only distributed at YJ independent from other regions. Combined with the negative FIS value of population YJ, the Ae. aegypti species likely invaded Yunnan Province from this region over recent years.
Our research shows that in Dehong and Xishuangbanna prefectures, Ae. aegypti population invade these areas because of the continuous tourist and business activities. Inspection and quarantine need to be strengthened at the border ports and further investigation and research on mosquito vectors should be carried out. The government needs to designate effective prevention and control measures, strengthen environmental governance in the border areas and implement mosquito control measures.
Conclusion
The nuclear microsatellite markers and mtDNA sequences (COI, ND4, and ND5) were used to uncover the population genetics of the Ae. aegypti in the border area of Yunnan Province. Although several attempts have been made by the government of Yunnan Province to control the mosquito vectors, the Ae. aegypti populations in this region showed high genetic diversity and genetic structure due to the continuous invasion, and increased urbanization. Our research confirms that, over recent years, a significant Ae. aegypti invasive event occurred in YJ City; and that the Xishuangbanna and Dehong prefectures were important areas for the Ae. aegypti invasion.
In summary, our results suggest that the control of Ae. aegypti in Yunnan Province is still a demanding task that needs to be taken seriously. Thus, monitoring of suspected cases of DF and the vectors should be enhanced.
Materials and methods
Mosquito sampling and DNA isolation
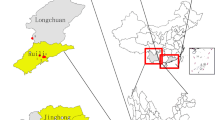
All the adult Ae. aegypti samples were collected from following eight locations along the border area of Yunnan Province between May 2017 and September 2018 (Fig. 8, Table 7). Each collection site covered an area of approximately 500 m in diameter. According to the Surveillance Methods for Vector Density-Mosquito (GB/T 23797–2009), a hand-held aspirator was used to collect the adult mosquitoes (intercepted before biting). All the samples were identified through the analysis of morphological characteristics in the wild field36 and preserved in 100% ethanol at 4 °C for the isolation of genomic DNA37.

The geographical location of collection sites of Ae. aegypti specimens. 1: Mengla (ML); 2: Jinghong (JH); 3: Menghai (MH); 4: Cangyuan (CY); 5: Mengding (MD); 6: Ruili (RL); 7: Longchuan (LC); 8 Yingjiang (YJ).
According to the standard DNA extraction procedure, genomic DNA was isolated from individual mosquito sample with the TaKaRa Mini-BEST Universal Genomic DNA Extraction Kit (Takara, Dalian, China); the quality and quantity of extracted DNA were analyzed using NANOdrop1000, after which samples were stored at − 20 °C until further analysis.
PCR amplification and microsatellite genotyping
Nine microsatellite polymorphic loci were screened from 58 loci, which were described in previous studies by denaturing polyacrylamide gel electrophoresis38,39. The primer sequences and information are summarized in Table 8, forward primers were labeled with a fluorescent dye (FAM, HEX, or TAMRA). All the samples were amplified in the 25 μL reaction, which consisted of 2.5 μL 10 × PCR Buffer, 1 μL 1:10 DNA template, each primer at 0.3 μM, dNTPs at 0.25 μM, MgCl2 at 1.5 mM, TaqDNA polymerase 1.25U and ddH2O. The PCR conditions were as follows: 94 °C for 5 min; 35 cycles at 94 °C for 45 s (for each locus, different annealing temperature was used (Table 8) for 45 s) and 72 °C for 45 s. The final extension was performed at 72 °C for 10 min.
All PCR amplification products were verified by electrophoresis of 3 μL on a 1.5% agarose gel. The formamide was mixed with LIZ 500-labeled size standard using a ratio of 100:1, and 15 μL mixture was added into the sample plate. The PCR amplification products were diluted at 1:10, 1 μL was added into the reaction, and then run on an ABI3730XL (Applied Biosystems, Foster City, USA) capillary sequencer. All microsatellite alleles were evaluated using GeneMapper software (Applied Biosystems)14.
Microsatellite data analysis
Genetic diversity
The PIC values of all nine loci were calculated with PIC-Calc 0.614. The genetic diversity of all Ae. Aegypti populations were characterized by expected heterozygosity (He) and observed heterozygosity (Ho), using POPGENE version 1.32. The FIS value of each mosquito population was also calculated. The statistical significance test was performed with the exact tests available in POPGENE40.
Genetic structure
The genetic variation was tested by the AMOVA test with Arlequin (version 3.5.2.2) for the interpretation of genetic variability and structure among different locations, mosquito populations. The AMOVA was evaluated at four different hierarchical levels: (1) all samples (non-grouped) were analyzed as a single group to test the overall genetic differences between samples; (2) the samples in one region were analyzed as a unique group; (3) the interregional populations were analyzed as an unique group; (4) the individual sample within population was analyzed as an unique group. Based on the stepwise mutation model (SMM), the recent genetic bottleneck in each mosquito populations was calculated by the software BOTTLENECK 1.2.02. The data were analyzed with the recommended settings: an index statistic closer to 1 indicates that the population is in a stable state, while a very low value indicates that the population has experienced a genetic bottleneck in the past41. The sign test implemented in the software was used to test for significant heterozygosis excess. The isolation by distance (IBD) was estimated with Mantel's test in R, using the correlation between genetic distance and geographic distances by the regression of pairwise FST/(1 − FST) on the natural logarithm (Ln) of straight-line geographic distance.
For the determination of real genetic clusters (K) within all mosquito samples, a Bayesian clustering algorithm-based software STRUCTURE 2.2 was employed. All mosquitos were divided into different populations represented by a specific number (K = 8), under the assumption of Hardy–Weinberg equilibrium and linkage equilibrium42. The software parameters were set as follows: the assumed populations ranged from 1 to 8, and the calculation model was set as admixture ancestry and independent allele frequency models 100,000 burn-in steps followed by 1,000,000 MCMC replicates, and each population was calculated for 10 runs. The optimum K value was estimated with Evanno’s △k method based on the second-order rate of change in the log probability of the △k among 10 runs of each assumed K43, and all the results were uploaded to a web-based utility Harvest for the calculation of the optimum K value (https://taylor0.biology.ucla.edu/struct_harvest/). Furthermore, the software CLUMPP 1.1.1 and DISTRUCT 1.1 (Rosenberg 2004 and 2007) were also used to calculate the average coefficients of membership across the 10 replicates of real K value, and the final results were displayed. In order to further explore the genetic structure, UPGMA trees were computed with NTsys, based on microsatellite Nei’s genetic distance. The DAPC analyses were conducted under the R (vision 3.6.2) condition with the R package “adegenet 2.1.0”.
mtDNA analysis and PCR amplification
The mitochondrial genes ND4 and COI of Ae. aegypti were chosen as the examination sites for exploring sequence polymorphism. At least ten mosquito individuals from each population were selected from the previous microsatellite analysis and identified by the amplification and sequencing of the COI gene. All the mosquito DNAs were amplified with the ND4 primers, which were composed of ND4 forward-(5′-TGATTGCCTAAGGCTCATGT-3′) and ND4 reverse-(5′-TTCGGCTTCCTAGTCGTTCAT-3′) primers targeting 344 bp fragment. The following PCR procedure was used: 5 min of initial denaturation (94 °C) followed by thirty cycles of 94 °C, 40 s of denaturation at 56 °C, 40 s of annealing at 72 °C, 1 min of extension and a final extension at 72 °C for 5 min. The 50 μL PCR solution was composed by 5 μL 10 × PCR buffer, 0.3 μL TaqDNA polymerase (5 U/L), 5 μL dNTP (2 mmoL/L), 2 μL of each of the forward and reverse primers (10 μM), 5 μL template DNA and ddH2O. The PCR amplification products were confirmed by agarose gel electrophoresis. All the PCR products were purified with the PCR Purification Kit (OMEGA D6492-02) and cloned to the pMD18-T vector (TaKaRa, Japan) for the final sequencing with ABI 3730XL automatic sequencer (Applied Biosystem).
mtDNA data analysis
All the sequences were analyzed with the software Sequencer 5.0 and BioEdit v7.0.570, and aligned with Clustal W. For the ND4 gene, haplotype diversity (Hd), nucleotide diversity (π) and the Tajima and Fu and Li neutrality tests of all mosquito populations were computed by DNA Sequence Polymorphism v6.12.01. A haplotype network of all mosquito populations was also constructed with the software NETWORK 5.0.1.1 for inferring the relationships of all haplotypes and the distribution of all haplotypes at different locations.
Ethics approval and consent to participate
Pre-permission (May 2017 to September 2018) was granted for adult mosquito observation, adult mosquito collection, and studies in Yunnan province. All the studies were authorized by the Committee for Animal Welfare and Animal Ethics in the Institute of Parasitic Diseases of Yunnan province, China (address: Yunnan province, People’s Republic of China).
Abbreviations
- AMOVA:
-
Analysis of molecular variance
- CDC:
-
Centers for Disease Control and Prevention
- DF:
-
Dengue fever
- DHF:
-
Dengue hemorrhagic fever
- FIS :
-
Inbreeding coefficient
- Hd:
-
Haplotype diversity
- He:
-
Expected heterozygosity
- Ho:
-
Observed heterozygosity
- IBD:
-
Isolation by distance
- mtDNA:
-
Mitochondrial DNA
- PCR:
-
Polymerase chain reaction
- PIC:
-
Polymorphic information content
- SMM:
-
Stepwise mutation model
- SRSs:
-
Simple sequence repeats
- STRs:
-
Short tandem repeats
- WHO:
-
World Health Organization
References
Brady, O. J. et al. Refining the global spatial limits of dengue virus transmission by evidence-based consensus. PLoS Negl. Trop. Dis. 6, e1760. https://doi.org/10.1371/journal.pntd.0001760 (2012).
Wu, J. Y., Lun, Z. R., James, A. A. & Chen, X. G. Dengue fever in mainland China. Am. J. Trop. Med. Hyg. 83, 664–671. https://doi.org/10.4269/ajtmh.2010.09-0755 (2010).
Chen, B. & Liu, Q. Dengue fever in China. Lancet 385, 1621–1622. https://doi.org/10.1016/S0140-6736(15)60793-0 (2015).
Zhang, H. et al. Mosquitoes of Western Yunnan Province, China: seasonal abundance, diversity, and arbovirus associations. PLoS ONE 8, e77017 (2013).
Wang, B. et al. The distinct distribution and phylogenetic characteristics of dengue virus serotypes/genotypes during the 2013 outbreak in Yunnan, China: phylogenetic characteristics of 2013 dengue outbreak in Yunnan, China. Infect. Genet. Evol. 37, 1–7 (2016).
Guo, X. et al. Molecular characterization and viral origin of the first dengue outbreak in Xishuangbanna, Yunnan Province, China, 2013. Am. J. Trop. Med. Hyg. 93, 390–393 (2015).
Powell, J. R. & Tabachnick, W. J. History of domestication and spread of Aedes aegypti-a review. Memórias do Inst. Oswaldo Cruz 108, 11–17 (2013).
Mayer, S. V., Tesh, R. B. & Vasilakis, N. The emergence of arthropod-borne viral diseases: a global prospective on dengue, chikungunya and zika fevers. Acta Trop. 166, 155–163 (2017).
Scott, T. W. et al. Longitudinal studies of Aedes aegypti (Diptera: Culicidae) in Thailand and Puerto Rico: blood feeding frequency. J. Med. Entomol. 37, 89–101 (2000).
Dong, X. et al. Investigation of mosquitoes at frontier ports in Yunnan Province. Chin. J. Vector Bio Control 15, 142–147 (2004).
Shuwei, P. U. et al. Primary study on mosquito community in Mengzi City, Honghe Prefecture, Yunnan. China Trop. Med. 17, 1044–1046 (2017).
Yang, J. et al. Analysis of results of sentinel monitoring of trandmission vector of dengue fever at Dehong State of Yunnan Province in 2016. J. Med. Pest Control 33, 736–738 (2017).
Yunlan, L. et al. Investigation of Aedes aegypti distribution in the road port of Qingshui River at Lincang, Yunnan. Port Health Control 21, 49–51 (2016).
Shi, Q. M. et al. The genetic diversity and population structure of domestic Aedes aegypti (Diptera: Culicidae) in Yunnan Province, southwestern China. Parasites Vectors 10, 292. https://doi.org/10.1186/s13071-017-2213-6 (2017).
Guillemaud, T., Beaumont, M. A., Ciosi, M., Cornuet, J. M. & Estoup, A. Inferring introduction routes of invasive species using approximate Bayesian computation on microsatellite data. Heredity 104, 88–99. https://doi.org/10.1038/hdy.2009.92 (2010).
Maynard, A. J. et al. Tiger on the prowl: invasion history and spatio-temporal genetic structure of the Asian tiger mosquito Aedes albopictus (Skuse 1894) in the Indo-Pacific. PLoS Negl. Trop. Dis. 11, e0005546. https://doi.org/10.1371/journal.pntd.0005546 (2017).
Gadgil, R., Barthelemy, J., Lewis, T. & Leffak, M. Replication stalling and DNA microsatellite instability. Biophys. Chem. 225, 38–48. https://doi.org/10.1016/j.bpc.2016.11.007 (2017).
Huber, K., Mousson, L., Rodhain, F. & Failloux, A. B. Isolation and variability of polymorphic microsatellite loci in Aedes aegypti, the vector of dengue viruses. Mol. Ecol. Notes 1, 219–222 (2001).
Bosio, C. F. et al. Genetic structure of Aedes aegypti populations in Thailand using mitochondrial DNA. Am. J. Trop. Med. Hyg. 72, 434–442 (2005).
Mousson, L. et al. Phylogeography of Aedes (Stegomyia) aegypti (L.) and Aedes (Stegomyia) albopictus (Skuse) (Diptera: Culicidae) based on mitochondrial DNA variations. Genet. Res. 86, 1–11 (2005).
Blacket, M. J., Malipatil, M. B., Semeraro, L., Gillespie, P. S. & Dominiak, B. C. Screening mitochondrial DNA sequence variation as an alternative method for tracking established and outbreak populations of Queensland fruit fly at the species southern range limit. Ecol. Evol. 7, 2604–2616 (2017).
Liu, P. et al. The expanding pattern of Aedes aegypti in southern Yunnan, China: insights from microsatellite and mitochondrial DNA markers. Parasites Vectors 12, 561. https://doi.org/10.1186/s13071-019-3818-8 (2019).
Simberloff, D. et al. Impacts of biological invasions: what’s what and the way forward. Trends Ecol. Evol. 28, 58–66. https://doi.org/10.1016/j.tree.2012.07.013 (2013).
Botstein, D., White, R. L., Skolnick, M. H. & Davis, R. W. Construction of a genetic linkage map in man using restriction fragment length polymorphisms. Am. J. Hum. Genet. 32, 314–331 (1980).
Harrington, L. C. et al. Dispersal of the dengue vector Aedes aegypti within and between rural communities. Am. J. Trop. Med. Hyg. 72, 209–220 (2005).
Kuno, G. Factors influencing the transmission of dengue viruses. Dengue Dengue Hemorrhagic Fever 1, 23–39 (1997).
Liu, B. et al. Modeling the present and future distribution of arbovirus vectors Aedes aegypti and Aedes albopictus under climate change scenarios in Mainland China. Sci. Total Environ. 664, 203–214 (2019).
Xie, H., Zhou, H. & Yang, Y. Advances in the research on the primary dengue vector Aedes aegypti in China. Zhongguo Meijie Shengwuxue ji Kongzhi Zazhi Chin. J. Vector Biol. Control 22, 194–197 (2011).
Gang, W. et al. Using GARP to predict the range of Aedes aegypti in China. Southeast Asian J. Trop. Med. Public Health 45, 290–298 (2014).
Li, Y., Zhu, J. & Li, H. Distribution of the dengue fever vector in Xishuangbanna prefecture of Yunnan. China Trop. Med. 16, 237–265. https://doi.org/10.13604/j.cnki.46-1064/r.2016.03.10 (2016).
Li, H. C. et al. Investigation of distribution of dengue vectors in Lincang border area. China Trop. Med. 15, 186–188 (2015).
Getis, A., Morrison, A. C., Gray, K. & Scott, T. W. Characteristics of the spatial pattern of the dengue vector, Aedes aegypti, in Iquitos, Peru. Am. J. Trop. Med. Hyg. 69, 494–505 (2003).
Hlaing, T. et al. Spatial genetic structure of Aedes aegypti mosquitoes in mainland Southeast Asia. Evol. Appl. 3, 319–339. https://doi.org/10.1111/j.1752-4571.2009.00113.x (2010).
Kamgang, B. et al. Temporal patterns of abundance of Aedes aegypti and Aedes albopictus (Diptera: Culicidae) and mitochondrial DNA analysis of Ae. albopictus in the Central African Republic. PLoS Negl. Trop. Dise. 7, e2590. https://doi.org/10.1371/journal.pntd.0002590 (2013).
Kamgang, B. et al. Geographical distribution of Aedes aegypti and Aedes albopictus (Diptera: Culicidae) and genetic diversity of invading population of Ae. albopictus in the Republic of the Congo. Wellcome Open Res. 3, 79. https://doi.org/10.12688/wellcomeopenres.14659.3 (2018).
Lu, B. & Wu, H. Classification and Identification of Important Medical Insects of China (Henan Science and Technology Publishing House, Henan, 2003).
Reeves, L. E., Holderman, C. J., Gillett-Kaufman, J. L., Kawahara, A. Y. & Kaufman, P. E. Maintenance of host DNA integrity in field-preserved mosquito (Diptera: Culicidae) blood meals for identification by DNA barcoding. Parasites Vectors 9, 503. https://doi.org/10.1186/s13071-016-1791-z (2016).
Paupy, C., Orsoni, A., Mousson, L. & Huber, K. Comparisons of amplified fragment length polymorphism (AFLP), microsatellite, and isoenzyme markers: population genetics of Aedes aegypti (Diptera: Culicidae) from Phnom Penh (Cambodia). J. Med. Entomol. 41, 664–671. https://doi.org/10.1603/0022-2585-41.4.664 (2004).
Slotman, M. et al. Polymorphic microsatellite markers for studies of Aedes aegypti (Diptera: Culicidae), the vector of dengue and yellow fever. Mol. Ecol. Notes 7, 168–171 (2007).
Maitra, A., Cunha-Machado, A. S., Souza Leandro, A., Costa, F. M. D. & Scarpassa, V. M. Exploring deeper genetic structures: Aedes aegypti in Brazil. Acta Trop. 195, 68–77. https://doi.org/10.1016/j.actatropica.2019.04.027 (2019).
Excoffier, L. & Lischer, H. E. Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 10, 564–567. https://doi.org/10.1111/j.1755-0998.2010.02847.x (2010).
Yin, M. et al. Geographical genetic structure of Schistosoma japonicum revealed by analysis of mitochondrial DNA and microsatellite markers. Parasites Vectors https://doi.org/10.1186/s13071-015-0757-x (2015).
Rosenberg, N. A. Distruct: a program for the graphical display of population structure. Mol. Ecol. Notes 4, 137–138. https://doi.org/10.1046/j.1471-8286.2003.00566.x (2003).
Acknowledgements
This work was supported by the grant from the National Natural Science Foundation of China (NO: U1602223) and Biosafety Special Fund (NO: 17SAZ01).
Author information
Authors and Affiliations
Contributions
W.T. and H.Z. conceived and designed the experiments. R.L., C.Z. and L.Y. performed the experiments., C.W., J.A. and P.W. analyzed the data. C.L. and L.A. contributed reagents/materials/analysis tools. W.T., R.L., X.T. wrote the paper. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lv, Rc., Zhu, C.q., Wang, Ch. et al. Genetic diversity and population structure of Aedes aegypti after massive vector control for dengue fever prevention in Yunnan border areas. Sci Rep 10, 12731 (2020). https://doi.org/10.1038/s41598-020-69668-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-69668-7
- Springer Nature Limited