
Abstract
Diagnosis and classification of gliomas mostly relies on histopathology and a few genetic markers. Here we interrogated microarray gene expression profiles (GEP) of 268 diffuse astrocytic gliomas—33 diffuse astrocytomas (DA), 52 anaplastic astrocytomas (AA) and 183 primary glioblastoma (GBM)—based on multivariate analysis, to identify discriminatory GEP that might support precise histopathological tumor stratification, particularly among inconclusive cases with II–III grade diagnosed, which have different prognosis and treatment strategies. Microarrays based GEP was analyzed on 155 diffuse astrocytic gliomas (discovery cohort) and validated in another 113 tumors (validation set) via sequential univariate analysis (pairwise comparison) for discriminatory gene selection, followed by nonnegative matrix factorization and canonical biplot for identification of discriminatory GEP among the distinct histological tumor subtypes. GEP data analysis identified a set of 27 genes capable of differentiating among distinct subtypes of gliomas that might support current histological classification. DA + AA showed similar molecular profiles with only a few discriminatory genes overexpressed (FSTL5 and SFRP2) and underexpressed (XIST, TOP2A and SHOX2) in DA vs AA and GBM. Compared to DA + AA, GBM displayed underexpression of ETNPPL, SH3GL2, GABRG2, SPX, DPP10, GABRB2 and CNTN3 and overexpression of CHI3L1, IGFBP3, COL1A1 and VEGFA, among other differentially expressed genes.
Similar content being viewed by others

Introduction
Diffuse gliomas comprise a variety of tumor entities of different cell lineages and histopathological features which are classified into distinct subtypes by the World Health Organization (WHO)1,2, from which astrocytic lineage tumors (i.e. astrocytomas and glioblastomas) are by far the most common (around 90%)3. Relevant histological and immunohistochemical features together with the presence of codeletion of chromosome 1p/19q and isocitrate dehydrogenase 1 (IDH1) gene mutation, are currently used for the differential diagnosis between oligodendroglial tumors and diffuse astrocytomas1,4. However, further differential diagnoses among the distinct subtypes of diffuse astrocytomas might be challenging and they might even lead to inconclusive results, particularly among grade II and III tumors. For this purpose molecular characteristics of these tumors have been recurrently investigated. However, while genetic alterations are found in the majority of tumors, they are not entirely specific, and thereby they are not considered in the current WHO-2016 classification of gliomas. Thus, astrocytic gliomas are currently divided in non-diffuse (pilocytic astrocytoma grade-I, PA; WHO-I) and diffuse tumors based on conventional histopathology. The latter tumors were further divided on histopathological grounds into three grades of malignancy associated with distinct median survival rates (range: from 1 to > 10 years)5,6: WHO grade-II diffuse astrocytoma (DA), WHO grade-III anaplastic astrocytoma (AA) and WHO grade-IV glioblastoma (GBM). Of note, DA might evolve to AA, and AA might transform to GBM7,8. Because of this, GBM is further subdivided into primary de novo GBM (pGBM) and secondary GBM (sGBM) resulting from progression of a prior lower grade astrocytic tumor (e.g. AA)7.
At present, the IDH1-mutation together with codeletion of chromosomes 1p/19q have become a major criterion for the differential diagnosis between oligodendrocytic and oligoastrocytic tumors vs diffuse astrocytic gliomas1,2. However, DA and AA also show a variable frequency of IDH-mutation, while this mutation is absent in the great majority of GBM9, which limits its diagnostic utility among diffuse astrocytic tumors. Other genetic markers that have been associated with specific subtypes of astrocytomas and diffuse astrocytic tumors could be useful for glioma classification10,11, include gains and losses of specific chromosomal regions together with mutations of the EGFR, MDM4, PTEN, PDGFRA and CDKN2A genes, but they are not considered in the WHO-2016 classification2. Altogether, this highlights the need for deeper genomic analysis of astrocytic tumors to gain further insight in those gene profiles that might help to unequivocally distinguish among the different subtypes of astrocytic tumors and support the differential diagnosis and subclassification of diffuse gliomas, particularly in cases12,13 with an inconclusive histopathological diagnosis.
Despite the expression levels of specific genes, such as CHI3L1 and TOP2A, which have been related to necrosis in GBM14,15, and IGFBP2 and VEGFA involved in tumor progression12, mRNA-based gene expression profiling (GEP) has frequently shown discrepant results in gliomas, hampering application of GEP in clinical practice. To a certain extent such discrepancies are due to: (i) analysis of small patient cohorts16, (ii) focused on individual tumor types such as GBM17, together with (iii) the use of different microarray platforms, and/or iv) diverse mathematical approaches and multivariate data analysis algorithms12. As an example, principal component analysis (PCA)18 has been frequently used for GEP analysis, despite its limitations for the analysis of high-dimensional databases that contain a number of variables that significantly exceeds the number of tumors. For such situations, variable selection techniques together with other matrix factorization algorithms, such as nonnegative matrix factorization (NMF)19, have been proposed for the discovery of clusters that might gather important biological information, as recently demonstrated in pancreatic cancer20.
In this study we aimed at identifying a panel of informative genes for subclassification of a large series of 268 astrocytic diffuse gliomas into their DA, AA and GBM subtypes based on GEP data analyzed with combination of low-rank matrix decompositions, as CUR decomposition21, followed by NMF19 and canonical biplot22. A panel of 27 discriminatory genes were finally identified that efficiently differentiate among the three subtypes of diffuse astrocytic tumors.
Results
Pairwise comparisons of gene expression data from the discovery cohort showed > 800 differentially expressed gene probes among the three subtypes of diffuse astrocytic tumors analyzed (Fig. 1) with significant adjusted P-values. Despite this, no gene probe showed significant differences in gene expression (discriminatory) levels between DA and AA tumors. In contrast, 445 gene probes were differentially expressed (P < 0.05) between DA and GBM (222 were underexpressed in GBM and 223 overexpressed) and 448 gene probes where distinctly expressed (P < 0.05) in AA vs GBM (235 underexpressed in GBM and 213 overexpressed probes); 339 probes corresponding to 283 genes were differentially expressed in common in the two above comparisons (Fig. 1). Comparison between DA vs GBM revealed 33/445 differentially expressed gene probes, corresponding to 27 distinct genes, with fold-change (FC) value differences (vs mean probe intensity) of FC ≥ 4 (Table 1). These included 9/27 genes with increased expression values in DA vs GBM (ETNPPL, FSTL5, SFRP2, SH3GL2, CNTN3, SPX, GABRG2, GABRB2, DPP10) and 18/27 genes with higher expression in GBM vs DA (CHI3L1, COL1A1, COL3A1, POSTN, COL1A2, IGF2BP3, NNMT, SHOX2, XIST, HS3ST3B1, PTX3, VEGFA, IBSP, TOP2A, LOX, IGFBP3, ANXA1, PDPN) (Fig. 1; Table 2). Similarly, the comparison between AA and GBM revealed 13/448 differentially expressed gene probes corresponding to 9 genes to display FC ≥ 4 (vs mean probe intensity). Once compared to GBM, AA showed overexpression of two gene probes (CNTN3 and ETNPPL genes) and underexpression of another 11 gene probes corresponding to 7 genes: CHI3L1, COL1A1, COL3A1, POSTN, NNMT, PTX3, COL1A2 (Fig. 1; Table 1).

Number of differentially expressed gene probes in samples corresponding to distinct tumor subtypes from the discovery cohort. Differentially expressed probes were identified by pairwise comparisons based on p-values < 0.05. The number of overexpressed and underexpressed probes in DA and AA versus GBM are shown. DEGP, differentially expressed gene probes; DA, diffuse astrocytoma; AA, anaplastic astrocytoma; GBM, glioblastoma; FC, fold-change in gene expression levels.
Based on the list of gene probes identified with both approaches, a total of 27 differentially expressed genes were selected for subsequent multivariate analyses (Table 2 and Supplementary Table S1). More detailed analysis of those 27 genes found that XIST was the only gene with the most clearly different expression profile between DA and AA tumors (Table 2). Among the other 26 genes, ETNPPL, SFRP2, SH3GL2, FSTL5, DPP10, SPX, GABRB2, CNTN3 and GABRG2 displayed greater expression levels in DA + AA vs GBM. Of these latter 9 genes, 3 are coded in chromosome 4 (ETNPPL, SFRP2 and FSTL5), 2 in chromosome 5 (GABRB2, GABRG2), 1 in chromosome 2 (DPP10), 1 in chromosome 3 (CNTN3), 1 in chromosome 9 (SH3GL2) and the SPX gene is coded in chromosome 12. Likewise, another 17 genes, including the CHI3L1, COL1A1, IGFBP3 genes, were overexpressed in GBM vs both DA + AA. More than half of these later genes (10/17 genes) are coded in a total of only 4 chromosomes, including chromosomes 7 (COL1A2, IGF2BP3, IGFBP3), 17 (COL1A1, HS3ST3B1, TOP2A), 1 (CHI3L1, PDPN) and 3 (PTX3, SHOX2). The remaining 7 genes are coded each in a distinct chromosome, e.g. chromosomes 2 (COL3A1), 4 (IBSP), 5 (LOX), 6 (VEGFA), 9 (ANXA1), 11 (NNMT) and 13 (POSTN). Interestingly, genes coded in the long arm of chromosome 2q, 4q and 5q appeared to be relevant in all group comparisons (Table 2).
NMF was subsequently applied to the discovery cohort of 155 diffuse astrocytic glioma GEP data about of the 27 genes previously selected, in order to further establish the relevance of the selected genes to classify the 3 subtypes of diffuse astrocytic gliomas. Two different clusters (corresponding to DA + AA and GBM) were detected with NMF, DA and AA tumors being graphically visualized as a single cluster (Fig. 2A). Once we investigated the relevance of each gene to the formation of both clusters (Fig. 2A) we confirmed that the ETNPPL, SH3GL2, GABRB2, CNTN3, SPX, GABRG2, DPP10, SFRP2, FSTL5 genes where those most contributing to the DA + AA cluster, followed by a few genes displaying a lower contribution (XIST, IGFBP3, ANXA1, TOP2A, PDPN and VEGFA) (Fig. 2B). In turn, the most relevant genes to explain the GBM cluster were the CHI3L1, ANXA1, IGFBP3, COL1A2, COL1A1, NNMT, COL3A1, PDPN, POSTN, VEGFA, PTX3, TOP2A, HS3ST3B1, IGF2BP3, LOX, SHOX2, XIST and IBSP genes. Of note, the ETNPPL gene among DA + AA tumors and the CHI3L1 gene in GBM were those genes ranking the highest for each cluster. Interestingly, several of these differentially expressed genes identified in our study have also emerged as genes relevant for discriminating distinct subtypes of diffuse astrocytomas in other studies12,15,23, including the GBM-associated CHI3L1, COL1A1, VEGFA and ANXA genes (Supplementary Table S2). In turn, new gene associated here for the first time with DA + AA included the DPP10 gene. The variability encountered among the 155 samples of the discovery cohort was then visualized in a low-dimensional space using canonical biplot representation.

Multidimensional relationship between the 27 genes selected and the histological subtypes of diffuse astrocytic tumors. NMF analysis revealed two gene clusters based on the coordinates of distinct sets of genes to each cluster shown in the bar charts and heatmap (Upper graphics in panel A, left graphic in panel B). Scores obtained by NMF (corresponding to the grade of membership of each sample to the defined clusters) detected two major subsets of gliomas among DA (dipicted green), AA (blue) and GBM (red) (panel A): one group consisting mainly of GBM and a few samples of AA cases and the other group included both DA and AA. The canonical biplot representation is shown in the right of Panel B, where the mean values of each subgroup of astrocytic tumors is plotted with a star colored (individual DA, AA and GBM tumors are labeled as green, blue and red points) and the discriminatory genes are plotted as vectors. The discriminatory genes found to distinguish among the three WHO subtypes of diffuse astrocytic tumors are listed in panel C in a color code defined by their known functions. R software for statistical computing and graphics (v3.5.2) was used. DA, diffuse astrocytoma; AA, anaplastic astrocytoma; GBM, glioblastoma.
Canonical biplot based on the 27 selected genes showed differential GEP for the samples visualized in a two-dimensional space (Fig. 2B). In this biplot representation most genes contributed substantially to the formation of the factorial horizontal axis 1, which provides a clear distinct structure between GBM and both DA + AA samples, the most discriminatory genes being the ETNPPL, SH3GL2, GABRG2, SPX, DPP10, TOP2A, SHOX2, IGF2BP3, ANXA1, VEGFA and CHI3L1 genes. Some of these axis 1 genes showed higher expression values among DA + AA tumors (ETNPPL, DPP10, SH3GL2, GABRG2, SPX) while others were found to be overexpressed in GBM vs both DA + AA (e.g. IGFBP3, IGF2BP3, SHOX2, VEGFA). In turn, vertical axis 2 was relevant to differentiate DA from AA gliomas. This later axis was mostly explained by differences in the expression levels of XIST together with differential expression levels of genes contributing both to axis 1 and 2, such as SFRP2, FSTL5, TOP2A and SHOX2, principally. Of note, XIST showed lower expression levels in DA tumors (versus some AA and GBM), while FSTL5 and SFRP2 expression was greater in DA vs AA gliomas. Similarly to NMF, canonical biplot confirmed the low contribution of TOP2A and XIST to the DA + AA cluster (and SFRP2 to the GBM cluster), which was due to the fact that some AA samples had similar expression levels for both genes to those observed among GBM (or to the DA + AA cluster, in case of SFRP2) (Fig. 2B).
From the functional point of view, genes found to discriminate among the different subtypes of diffuse astrocytic gliomas corresponded to: (i) genes involved in development (HOXD10, CNTN3, LOX, SFRP2, XIST); (ii) cell adhesion (IBSP, PDPN, POSTN); (iii) cell metabolism (ETNPPL, HS3ST3B1, NNMT, DPP10, SHOX2, SPX); (iv) proliferation (COL1A1, COL1A2, COL3A1, FSTL5, IGFBP3, IGF2BP3, TOP2A); (v) angiogenesis (VEGFA); (vi) neurotransmission (GABRB2, GABRG2, SH3GL2); and (vii) the inflammatory response (ANXA1, CHI3L1, PTX3). Of note, DA was associated with greater expression of genes related to development and cell adhesion, AA to genes involved in metabolism and neurotrasmission and GBM to genes associated with angiogenesis and inflammatory responses (Fig. 2C).
In order to confirm the discriminatory power of the combination of the 27 genes identified to discriminate among distinct tumor subtypes, validation in an independent cohort of 113 gliomas was performed (Fig. 3). In this validation series, NMF and canonical biplot clearly identified the same two clusters. The DA + AA cluster was mostly explained by the SH3GL2, DPP10, GABRG2 and GABRB2 genes, while the GBM cluster (including a few AA tumors) was characterized by the VEGFA, POSTN, CHI3L1 and SHOX2, among other genes (Fig. 3A, B). SFRP2, FSTL5 and ETNPPL were the genes mostly contributing to the separation between DA and AA. These series distinct GEP of different subtypes of diffuse astrocytic glioma that were also confirmed using unsupervised nonhierarchical cluster in discovery, validation and total cohorts (Fig. 3C). These results showed the same overall behavior with two clearly different clusters, where some AA samples showed the same genetic patterns as DAs, and others showed clear genetic similarities with GBM.

Multivariate analysis of the association between the 27 genes found to discriminate among different histopathological subtypes of diffuse gliomas in the validation set of 113 diffuse astrocytic gliomas. Nonnegative matrix factorization plots of contribution scores of each sample to the formation of each cluster (A) and the importance of each gene to the two clusters formed (B, left) are shown together with canonical biplot (B, right), that confirmed classification of diffuse astrocytic gliomas around two leading molecular groups: a group including mostly GBM tumors and another group mostly composed of both DA and AA tumors. Dendrograms obtained via non-hierarchical NMF clustering for each (discovery, validation and total cohort) sample are shown in panel C. For the distinction of sample scores in panel A see legend to Fig. 2. R software for statistical computing and graphics (v3.5.2) was used. DA, diffuse astrocytoma; AA, anaplastic astrocytoma; GBM, glioblastoma.
Finally, to quantify how well this same combination of 27 genes separates DA, AA and GBM samples, LDA analysis was performed for the validation cohort. Overall, LDA showed an accuracy of 87% (98/113 tumors were properly classified; 12/14 DA, 17/24 AA, 69/75 GBM) to predict for the tumor histopathological diagnosis based on GEP was observed (Fig. 3C). The remaining 15/113 (15%) tumors (2/14 DA, 7/24 AA and 6/75 GBM) were not accurately classified and they corresponded to: (i) 2 DA classified as AA; (ii) 1 AA classified as DA; (iii) 6 AA identified as GBM; (iv) 1 GBM classified as DA; and (v) 5 GBM classified as AA.
Discussion
At present it is well established that multiple signaling pathways and cell functions are altered in astrocytic gliomas in association with a great number of recurrent genetic and microenvironmental changes17. In order to dissect the link between these genetic alterations and their functional consequences, GEP of DA, AA and GBM have been investigated in the last decade12,14,16,24,25,26,27. However, in only a few of these studies attempts have been made to identify genes differentially expressed between distinct histopathological glioma grades14,24, other studies focusing on the value of distinct GEP to help predicting tumor progression and patient survival12,25 or for identifying new targets for more effective therapy27. In turn, these studies have frequently been based on small patient cohorts16,25 with admixtures of tumors of all glioma subtypes12,14,28, they have used different microarray platforms12,29,30 and data analysis algorithms12,31, which have (frequently) led to controversial or even contradictory results.
Microarray GEP technologies provide massive data on the expression levels of thousand genes simultaneously but require the use of mathematical algorithms to capture the multidimensional nature of RNA expression data, in order to extract the critical information they might contain31. So far, these analyses have been based on distinct (frequently suboptimal) mathematical models which might introduce disturbing levels of variability on the study conclusions. Thus, some studies16,31 have used traditional multivariate statistical techniques, particularly PCA, which are not appropriate for data sets with few patients analyzed for thousands of variables, because of the variability overlooked in a high-dimensional space32. Because of these limitations of PCA and other traditional multivariate analysis algorithms, several new techniques have emerged for selection of the most informative variables under these circumstances31. Here we used CUR decomposition and univariate analysis (pairwise comparisons) to select for the most relevant genes to define diffuse astrocytic glioma subtypes in our discovery cohort. In order to further overcome analysis of samples that have a priori structured groups we used NMF, a method previously applied in glioma samples, which proved to provide useful conclusions to identify dynamic immune profiles during tumor evolution17. Based on this approach we identified unique GEP associated within the different histopathological subtypes of diffuse astrocytic gliomas. As expected, the largest GEP differences were found between DA and AA on one side, and GBM on the other side. In contrast, no differentially expressed genes were found to clear discriminate between DA and AA gliomas in the gene selection step (univariate analysis). Overall, these results are consistent with previous observations highlighting the remarkable difficulty to distinguish DA and AA based on mRNA GEP data33. Similarly, multivariate analysis based on NMF also revealed (only) two clearly different groups of diffuse astrocytic tumors, one including lower-grade astrocytomas (DA + AA) and the other GBM together with a few AA. Of note, these later AA might represent those AA tumors that might evolve to grade-IV glioma, further studies being required to confirm this hypothesis.
Lack of discrimination between DA and AA gliomas is not surprising and confirms that both tumor types display similar GEP and that they might potentially represent a single molecular subtype of astrocytomas. Even though, DA tumors showed lower expression of the XIST gene which together with overexpression of the FSTL5 and SFRP2 genes, once compared to AA tumors. Interestingly, XIST is a gene whose expression varies substantially with sex, that has been claimed to be a key gene in the oncogenesis of gliomas34. Since similar male/female distribution was observed in DA and AA tumors (data not shown) our results suggest that differences in XIST expression among DA and AA tumors might probably be due to its potential role on cell proliferation and invasion and requires further investigation. At the same time FREM3 has been previously found to be overexpressed in both DA and AA vs oligodendroglioma tumors35, and SFRP2 has been recently reported to contribute to the discrimination between DA and GBM30, in line with our findings. While XIST and SFRP2 were the best discriminatory genes between DA and AA gliomas, additional differences between these tumor subtypes and GBM were found, including overexpression of the DPP10, ETNPPL and SH3GL2 genes and underexpression of CHI3L1, VEGFA and IGF genes in the former two tumor types, in close association with unique chromosomal location profiles.
Overall, neither IDH1/2-mutations nor other chromosomal alterations showed the potential for being a discriminatory marker for distinct subtypes of diffuse astrocytomas in our own tumor cohort due to the low and variable IDH-mutational frequency and the heterogeneous cytogenetic profiles observed in GBM36 and other subtypes of diffuse astrocytomas, in line also with previous findings by others37. Thus, in the absence of other discriminatory markers, overexpression of DDP10, ETNPPL and SH3GL2 in DA + AA vs GBM might be considered in distinguishing these tumor subgroups, particularly for unclassifiable tumors or in case of small biopsy samples. In line with previous findings for the ETNPPL gene30, this gene together with SH3GL2, and the DPP10 gene identified here for the first time as relevant discriminatory gene, might represent a comprehensive panel of genes, mainly coded in chromosomes 4 and 2, to differentiate DA and AA gliomas. DPP10 is a gene associated with cell development and inhibition of cell growth. Altogether, these findings further highlight the potential relevance of the loss of function of these genes during tumor progression to more advance diffuse astrocytic gliomas.
In this regard, GBM and some AA showed higher levels of expression of TOP2A compared to DA and to most AA, suggesting that this gene might be involved in determining a high proliferation rate among AA tumors, that might potentially progress to sGBM since gene is involved in promoting cell growth signals38.
Similarly, IGF genes, a family of genes previously associated with malignant astrocytomas26,39 and progression to sGBM12,15, were found here to progressively increase its expression from DA to AA and GBM. Of note, IGFBP3 overexpression in GBM was associated in our study with overexpression of VEGFA, several collagen family genes (COL1A1, COL1A2 and COL3A1), and CHI3L1, among other genes. Since VEGFA interacts with IGFBP2 during angiogenesis40, overexpression of VEGFA and IGFBP3 genes might play an important role in tumor growth and expansion through promoting the formation of new blood vessels, in line with the increased angiogenesis observed in GBM41 vs other diffuse astrocytomas. In addition, the close association observed here between increased VEGFA and COL1A2, COL3A1 and COL1A1 expression in GBM, might also contribute to explain the effect of VEGFA on inducing collagenase expression and remodeling the tumor microenvironment in malignant astrocytomas (i.e. GBM). At the same time, these results support previous observations suggesting that COL3A1 is a reliable biomarker of GBM42. Similarly, CHI3L1, a gene that encodes for a protein involved in the inflammatory response, found here to be associated with GBM, has been related to a poorer outcome of GBM, due to a greater invasion and shorter patient survival23.
Of note, the 27 gene panel here identified also showed a high accuracy to distinguish between the distinct histopathological subtypes of diffuse astrocytic tumors in our validation cohort. These results support its potential utility in the subclassification of diffuse astrocytic tumors in routine clinical practice, particularly for cases with inconclusive histopathological diagnosis. In addition, they might provide further prognostic information among AA. Further prospective studies in large series of astrocytoma patients are necessary to confirm our results and extend these findings to other subtypes of glioma, prior to their translation into routine laboratory diagnostics.
Materials and methods
Diffuse astrocytic tumor datasets
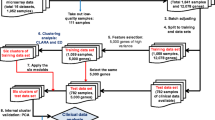
Data from a total of 10 glioma patient cohorts was downloaded from the Gene Expression Omnibus (GEO, https://www.ncbi.nlm.nih.gov/geo/) public functional genomic repository (Fig. 4). The data search strategy included the “astrocytoma”, “gene expression” and “humans” medical subject headings (MeSH). Based on these terms, a total of 1,330 studies were identified between the year 2000 and 2017. From these studies, only those referred to “expression profiling by array” were further selected, resulting in 10 datasets that fulfilled the inclusion criteria: i) usage of the HG-U133Plus2 array platform for GEP analyses; and, ii) human tumor samples investigated. Cell line and animal model studies, as well as human tumor series containing secondary GBM and/or tumor samples of only one diffuse astrocytic histopathological grade were excluded from the analysis (Fig. 4). From the 10 series that fulfilled all inclusion criteria, 5 series (our own series14 and 4 additional cohorts28,43,44,45) consisting of a total of 155 diffuse gliomas were randomly selected to be used as discovery cohort with the following distribution according to the WHO-2016 diagnostic criteria: DA, 19 cases (11%); AA, 28 cases (16%) and GBM, 108 cases (73%) (Fig. 4; Supplementary Table S3). The remaining 5 patient series 16,25,26,46 were used for the validation cohort (Fig. 4; Supplementary Table S3), for a total of 113 diffuse astrocytic gliomas consisting of 14 DA (12%), 24 AA (21%) and 75 GBM (67%). The two (discovery and validation) cohorts also showed a similar distribution per age (median of 60 vs 56 years, respectively; p = 0.591, by the Mann–Whitney U) and sex (male/female ratio of 1.94 and of 1.63, respectively; p = 0.705 by the Fisher exact test) for patients for whom data on these features were publicly available (108/155 and 64/113 had data on age and 47/155 and 79/113 patients had data on sex, respectively).

Flowchart summarizing sample data collection, selection criteria and steps, and the gene expression bioinformatics pipeline used in this study. Based on the filters and exclusion criteria, a total of 155 plus 113 diffuse astrocytic tumors grouped into 3 diffuse astrocytic tumor subtypes were included in the discovery and validation cohorts, respectively. For data analysis, individual variability was first removed by applying RMA, followed by the ComBat method. Then, those probes showing the highest differences among the groups of tumors were selected via CUR decomposition and pairwise comparisons. Based on the selected gene probes and multivariate analysis via nonnegative matrix factorization and canonical biplot graphical representation, those genes providing the highest discrimination among the distinct subtypes of astrocytic gliomas were identified.
Normalization of gene expression data and data analysis tools
For data analysis summarized in Fig. 4, genomic tools from the Bioconductor and R environment for Statistical Computing and Graphics free software47 (v3.5.2; www.cran.r-project.org) were used. For data normalization, the robust multi-array average (RMA) expression measure was used. Subsequently, variability due to each GEO database was removed using the ComBat procedure included in the sva R-package, which shrinks the variance of independent studies48 initially detected by data analysis techniques. PCA score plots were used to visualize the stabilized series patterns (Supplementary Fig. S1A). Gene symbols of the 54,675 probes were annotated and those without associated information, as well as those corresponding to Affymetrix control probes, were excluded from further analyses. Multiple probes of the same gene were kept in the analysis in order to minimize loss of differences between glioma subtypes. Finally, a total of 44,723 probe sets corresponding to RNA expression probes of 21,336 genes were included in the analysis (Supplementary Table S4). Bioinformatics analyses were implemented using the R-package rCUR to perform CUR decomposition49, limma to detect significant genes with differential gene expression levels between groups -providing models to compare many RNA targets simultaneously50, NMF to conduct NMF factorization19 and MultBiplotR to perform Canonical Biplot analysis and representation51. The R-code to reproduce the analyses in this work is available in https://github.com/ananieto/Scientif-Reports-Gonzalez-Garcia_et_al.
Gene selection
Data analysis for selection of differentially expressed genes was performed in two sequential steps. First, the 1,000 probes showing the greatest variability were selected based on factors of influence (called leverages) of CUR decomposition21 (Fig. Supplementary S1B). Subsequently, supervised analysis was performed to identify differentially expressed genes, based on FC values. For this purpose, univariate linear modelling was first performed to identify those gene probes that showed significant differences in expression levels between different astrocytic tumor subtypes in pairwise comparisons (i.e. DA vs AA, DA vs GBM, AA vs GBM). P-values obtained were adjusted by applying the Benjamin-Hochberg correction -also termed BH or False Discovery Rate (FDR) as the most widely used for genomics studies. FDR controls the expected value for the proportion of false positive cases among the null hypotheses rejected. Those genes with significant adjusted p-values which also differed in gene expression levels by FC ≥ 4 between the tumor grade groups, were selected.
CUR decomposition
CUR is defined as a low-range approximation of a matrix \({{\varvec{X}}}_{I x J },\) expressed in a small subset of rows and/or columns21. In this work, we select the variables (i.e. probes) that mostly contributed to the model in terms of variability. Leverage is defined as the amount of variance contributed by each variable to the factorial model, similar to the explanatory power of a variable in regression analysis. Since our goal was to select the probes with greater variability, for each of them we defined their leverage (\({l}_{j}\)) as
where \({v}_{j} (j=1,\dots ,44723)\) is the right singular vectors obtained in Singular Value Decomposition (SVD) of \(X\), and \(R\) the number of latent variables in the dimension reduction process. In our analysis, \(R\) was equal to the number of PCs needed to absorb all the variability.
Multivariate analysis based on NMF and canonical biplot representation
All differentially expressed gene probes displaying FC > 4 in pairwise comparisons were subsequently included in multivariate analysis based on NMF19. Briefly, NMF is an unsupervised clustering method defined as a matrix factorization technique that decomposes the original dataset (\(X\in {\mathbb{R}}_{+}^{J x I}\)) into two positive matrices whose product closely approximates \(X (X\approx WH\), where \(I\) and \(J\) refer to the number of samples and genes, respectively, \(W\in {\mathbb{R}}_{+}^{J x K}\) and \(H\in {\mathbb{R}}_{+}^{K x I}\) are nonnegative coefficients matrices and \(K\) is the number of clusters retained). The \(H\) matrix can be used to group the I samples into K clusters. Its columns represent the membership of each sample to the clusters. \(W\) rows define the physical meaning of clusters in terms of gene expression, where an \({w}_{jk}\) element symbolizes the expression level of gene \(j\) in cluster \(k\); i.e., \(W\) denotes the contribution of each gen to the cluster, in such a way that the higher the contribution, the more important that gene is in the formation of its cluster. Finally, canonical biplot22 was used as a visualization tool of the multivariate data matrix. For this purpose, a priori structure of groups in a low dimensional space with maximum discriminatory power between classes was used, in which the discriminatory genes involved in separation of the three histological tumor subtypes are shown. To facilitate its interpretation, an example is provided in Supplementary Figure S2. In order to validate the power of the contribution of the discriminatory genes selected above through NMF analysis, to classify individual samples into the distinct WHO 2016 tumor subtypes, unsupervised NMF was applied to the discovery cohort (n = 155), the validation set (n = 113) and the entire cohort (n = 268) of diffuse astrocytic gliomas.
Linear discriminant analysis (LDA)
To validate the power of the contribution of the discriminatory genes selected above through NMF analysis and quantify how well the gene signature separates DA and AA gliomas, LDA was applied to the validation set of 113 diffuse astrocytic gliomas and the percentage of correct tumor classification recorded.
Data availability
The datasets analysed during the current study are available in the GEO repository, https://www.ncbi.nlm.nih.gov/geo/.
References
Louis, D. N. et al. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta Neuropathol. 131, 803–820 (2016).
Weller, M. et al. European Association for Neuro-Oncology (EANO) guideline on the diagnosis and treatment of adult astrocytic and oligodendroglial gliomas. Lancet Oncol. 18, e315–e329 (2017).
Ostrom, Q. T. et al. CBTRUS Statistical Report: primary brain and other central nervous system tumors diagnosed in the United States in 2010–2014. Neuro. Oncol. 19, v1–v88 (2017).
Shirahata, M. et al. Novel, improved grading system(s) for IDH-mutant astrocytic gliomas. Acta Neuropathol. 136, 153–166 (2018).
Reuss, D. E. et al. IDH mutant diffuse and anaplastic astrocytomas have similar age at presentation and little difference in survival: a grading problem for WHO. Acta Neuropathol. 129, 867–873 (2015).
Morshed, R. A. et al. Molecular features and clinical outcomes in surgically treated low-grade diffuse gliomas in patients over the age of 60. J. Neurooncol. 141, 383–391 (2019).
Ding, Y., Yu, S., Bao, Z., Liu, Y. & Liang, T. CDC20 with malignant progression and poor prognosis of astrocytoma revealed by analysis on gene expression. J. Neurooncol. 133, 87–95 (2017).
Wesseling, P. & Capper, D. WHO 2016 classification of gliomas. Neuropathol. Appl. Neurobiol. 44, 139–150 (2018).
Ceccarelli, M. et al. Molecular profiling reveals biologically discrete subsets and pathways of progression in diffuse glioma. Cell 164, 550–563 (2016).
Verhaak, R. G. W. et al. Integrated Genomic Analysis Identifies Clinically Relevant Subtypes of Glioblastoma Characterized by Abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 17, 98–110 (2010).
Phillips, H. S. et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell 9, 157–173 (2006).
Xu, Y. et al. Screening critical genes associated with malignant glioma using bioinformatics analysis. Mol. Med. Rep. 16, 6580–6589 (2017).
Khani, P. et al. Genetic and epigenetic contribution to astrocytic gliomas pathogenesis. J. Neurochem. 148, 188–203 (2019).
Vital, A. L. et al. Gene expression profiles of human glioblastomas are associated with both tumor cytogenetics and histopathology. Neuro. Oncol. 12, 991–1003 (2010).
Phillips, L. M. et al. Glioma progression is mediated by an addiction to aberrant IGFBP2 expression and can be blocked using anti-IGFBP2 strategies. J. Pathol. 239, 355–364 (2016).
Liu, Z., Yao, Z., Li, C., Lu, Y. & Gao, C. Gene expression profiling in human high-grade astrocytomas. Comp. Funct. Genom. 20, 11. https://doi.org/10.1155/2011/245137 (2011).
Wang, Q. et al. Tumor evolution of glioma-intrinsic gene expression subtypes associates with immunological changes in the microenvironment. Cancer Cell 32, 42-56.e6 (2017).
Jolliffe, I. T. & Cadima, J. Principal component analysis: a review and recent developments. Philos. Trans. R. Soc. A 374, 20150202 (2016).
Gaujoux, R. & Seoighe, C. A flexible R package for nonnegative matrix factorization. BMC Bioinform. 11, 367 (2010).
Mishra, N. & Guda, C. Genome-wide DNA methylation analysis reveals molecular subtypes of pancreatic cancer. Oncotarget 8, 28990–29012 (2017).
Mahoney, M. W. & Drineas, P. CUR matrix decompositions for improved data analysis. PNAS 106, 697–702 (2009).
Iñigo, A. C., García-Talegón, J., Vicente-Palacios, V. & Vicente-Tavera, S. Canonical Biplot as a tool to detect microclimates in the inner and outer parts of El Salvador Church in Seville, Spain. Meas. J. Int. Meas. Confed. 136, 745–760 (2019).
Steponaitis, G. et al. High CHI3L1 expression is associated with glioma patient survival. Diagn. Pathol. 11, 42 (2016).
Melin, B. S. et al. Genome-wide association study of glioma subtypes identifies specific differences in genetic susceptibility to glioblastoma and non-glioblastoma tumors. Nat. Genet. 49, 789–794 (2017).
Kawaguchi, A. et al. Gene expression signature-based prognostic risk score in patients with glioblastoma. Cancer Sci. 104, 1205–1210 (2013).
Donson, A. M. et al. Increased immune gene expression and immune cell infiltration in high-grade astrocytoma distinguish long-term from short-term survivors. J. Immunol. 189, 1920–1927 (2012).
Shankar, G. M. et al. Genotype-targeted local therapy of glioma. Proc. Natl. Acad. Sci. USA 115, E8388–E8394 (2018).
Grzmil, M. et al. MAP kinase-interacting kinase 1 regulates SMAD2-dependent TGF-β signaling pathway in human glioblastoma. Cancer Res. 71, 2392–2402 (2011).
Toedt, G. et al. Molecular signatures classify astrocytic gliomas by IDH1 mutation status. Int. J. Cancer 128, 1095–1103 (2011).
Xu, Y. et al. Identification of differentially expressed key genes between glioblastoma and low-grade glioma by bioinformatics analysis. PeerJ 7, e6560 (2019).
Ang, J. C., Mirzal, A., Haron, H. & Hamed, H. N. A. Supervised, unsupervised, and semi-supervised feature selection: a review on gene selection. IEEE/ACM Trans. Comput. Biol. Bioinforma. 13, 971–989 (2016).
Sill, M., Saadati, M. & Benner, A. Applying stability selection to consistently estimate sparse principal components in high-dimensional molecular data. Bioinformatics 31, 2683–2390 (2015).
Iorgulescu, J. B. et al. The Misclassification of Diffuse Gliomas: Rates and Outcomes. Clin. Cancer Res. 25, 2656–2663 (2019).
Cheng, Z. et al. Long non-coding RNA XIST promotes glioma tumorigenicity and angiogenesis by acting as a molecular sponge of miR-429. J. Cancer 8, 4106–4116 (2017).
Weller, M. et al. Molecular classification of diffuse cerebral WHO grade II/III gliomas using genome- and transcriptome-wide profiling improves stratification of prognostically distinct patient groups. Acta Neuropathol. 129, 679–693 (2015).
Ballester, L., Huse, J., Tang, G. & Fuller, G. Molecular classification of adult diffuse gliomas: conflicting IDH1/IDH2, ATRX, and 1p/19q results. Hum. Pathol. 69, 15–22 (2017).
Chen, N. et al. IDH1/2 gene hotspot mutations in central nervous system tumours: analysis of 922 Chinese patients. Pathology 48, 675–683 (2016).
Wiedemeyer, W. R. et al. Pattern of retinoblastoma pathway inactivation dictates response to CDK4/6 inhibition in GBM. Proc. Natl. Acad. Sci. USA. 107, 11501–11506 (2010).
Han, S., Li, Z., Master, L. M., Master, Z. W. & Wu, A. Exogenous IGFBP-2 promotes proliferation, invasion, and chemoresistance to temozolomide in glioma cells via the integrin β1-ERK pathway. Br. J. Cancer 111, 1400–1409 (2014).
Turkowski, K. et al. VEGF as a modulator of the innate immune response in glioblastoma. Glia 66, 161–174 (2018).
Huang, L. et al. IGFBP2 expression predicts IDH-mutant glioma patient survival. Oncotarget 8, 191–202 (2017).
Gao, Y.-F. et al. COL3A1 and SNAP91: novel glioblastoma markers with diagnostic and prognostic value. Oncotarget 7, 70494–70503 (2016).
Sun, L. et al. Neuronal and glioma-derived stem cell factor induces angiogenesis within the brain. Cancer Cell 9, 287–300 (2006).
Turkheimer, F. E. et al. Chromosomal patterns of gene expression from microarray data: methodology, validation and clinical relevance in gliomas. BMC Bioinform. 7, 526 (2006).
Auvergne, R. M. et al. Transcriptional differences between normal and glioma-derived glial progenitor cells identify a core set of dysregulated genes. Cell Rep. 3, 2127–2141 (2013).
Zhou, J. et al. MicroRNA-326 functions as a tumor suppressor in glioma by targeting the nin one binding protein (NOB1). PLoS ONE 8, e68469 (2013).
Team, R. C. R: A language and Environment for Statistical Computing (R Core Team, Vienna, 2019).
Leek, J. T., Johnson, W. E., Parker, H. S., Jaffe, A. E. & Storey, J. D. The SVA package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics 28, 882–883 (2012).
Bodor, A., Csabai, I., Mahoney, M. W. & Solymosi, N. rCUR: an R package for CUR matrix decomposition. BMC Bioinform. 13, 103 (2012).
Ritchie, M. E. et al. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47 (2015).
Vicente-Villardón, J. L. MultBiplotR: MULTivariate analysis using biplots. Arthritis Res. Ther. https://doi.org/10.1186/s13075-019-2066-9 (2019).
Acknowledgements
MG-T, AO and MDT were supported by the following Grants: CB16/12/00400 (CIBER-ONC, Instituto de Salud Carlos III, Ministerio de Economía y Competitividad, Madrid, Spain and FONDOS FEDER), ISCIII PI16/0476 (Instituto de Salud Carlos III, Ministerio de Economía y Competitividad, Madrid, Spain) and GRS2049/A/19 (Consejería de Sanidad Junta de Castilla y León, Gerencia Regional de Salud, Spain).
Author information
Authors and Affiliations
Contributions
Study design, A. Orfao and M.D.T.; Methodology, N.G.G., A.B.N.L., P.G.V., A. Orfao and M.D.T.; Samples and clinical data collection: A.L.V., H.J.T. and M.G.T.; Statistical and computational analyses, N.G.G., A.B.N.L. and P.G.V.; Writing—original draft preparation, A. Orfao, A. Otero and M.D.T.; Project administration, M.D.T.; Funding acquisition, M.G.T., A. Orfao, A. Otero and M.D.T. All authors have contributed significantly to this article and all have read and approved its final version.
Corresponding author
Ethics declarations
Competing interest
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
González-García, N., Nieto-Librero, A.B., Vital, A.L. et al. Multivariate analysis reveals differentially expressed genes among distinct subtypes of diffuse astrocytic gliomas: diagnostic implications. Sci Rep 10, 11270 (2020). https://doi.org/10.1038/s41598-020-67743-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-67743-7
- Springer Nature Limited
This article is cited by
-
Screening of co-expressed genes in hypopharyngeal carcinoma with esophageal carcinoma based on RNA sequencing and Clinical Research
Scientific Reports (2024)
-
Genetic Discrimination of Grade 3 and Grade 4 Gliomas by Artificial Neural Network
Cellular and Molecular Neurobiology (2024)