
Abstract
Pathogenicity islands (PAIs) represent horizontally acquired chromosomal regions and encode their cognate integrase, which mediates chromosomal integration and excision of the island. These site-specific recombination reactions have to be tightly controlled to maintain genomic stability, and their directionality depends on accessory proteins. The integration host factor (IHF) and the factor for inversion stimulation (Fis) are often involved in recombinogenic complex formation and controlling the directionality of the recombination reaction. We investigated the role of the accessory host factors IHF and Fis in controlling the stability of six PAIs in uropathogenic Escherichia coli strain 536. By comparing the loss of individual PAIs in the presence or absence of IHF or Fis, we showed that IHF specifically stabilized PAI I536 and that in particular the IHFB subunit seems to be important for this function. We employed complex genetic studies to address the role of IHF in PAI I536-encoded integrase (IntI) expression. Based on different YFP-reporter constructs and electrophoretic mobility shift assays we demonstrated that IntI acts a strong repressor of its own synthesis, and that IHF binding to the intI promoter region reduces the probability of intI promoter activation. Our results extend the current knowledge of the role of IHF in controlling directionality of site specific recombination reactions and thus PAI stability.
Similar content being viewed by others


Introduction
Urinary tract infection is the most frequent type of nosocomial, but also community-acquired bacterial infection and in general the most frequent type of bacterial infections in women1. The vast majority of these infections is caused by uropathogenic Escherichia coli (UPEC) and are therefore from the perspective of general health, but also due to economic reasons, the most important problem associated with this bacterial species2,3,4. E. coli factors that facilitate the colonization of the urinary tract, e.g. fimbrial adhesins as well as toxins are often encoded on large mobile genomic regions designated pathogenicity island (PAIs). Seven archetypal PAIs have been described in uropathogenic model strain E. coli 536 and for six of these PAIs experimental data is available showing that they differ in their genetic stability5,6,7,8. These PAIs carry virulence and fitness-associated genes that contribute to the bacterial pathogenesis and/or provide a survival advantage in the host5,9,10. PAIs form a distinct class of integrative genetic elements11,12,13, which exhibit several defining features, including the presence of a P4-type integrase of the tyrosine recombinase family14,15,16. A comprehensive understanding of the mechanisms that result in the mobilization of PAIs is necessary for the development of strategies to prevent the generation of new bacterial strains with unknown pathogenic potential and for approaches that aim at the attenuation of bacterial virulence.
The chromosomal integration and excision of PAI I536-VI536 of E. coli strain 536 was shown to be dependent on PAI encoded P4-like integrases17. P4-like integrases belong to the family of tyrosine recombinases that are characterized by the presence of a conserved C-terminal Tyr residue18. The integrase of the bacteriophage P4 operates by integrating the phage DNA into the host chromosome via site-specific recombination19,20, which requires the P4 integrase, the phage attachment site attP, and the chromosomal attachment site attB19,21. The site-specific recombination reaction between the attP and attB sites involves DNA cleavage, formation of a Holliday junction, and rejoining of DNA fragments22,23,24. In the case of bacteriophage P4, the integration site resides within a leucyl-tRNA locus21,25. After integration of the bacteriophage genome into the bacterial chromosome, the P4 int gene is located in the proximity of the attL site23. In addition to the integrase itself, bacteriophages such as ʎ, P2 and P4 require accessory host proteins, such as the integration host factor (IHF) and the factor for inversion stimulation (Fis) for integrative recombination. These accessory DNA bending proteins bridge between distinct DNA sites, thereby implementing integrative or excisive recombination26,27,28.
IHF is a small hetero- and homo dimer forming DNA binding protein composed of two subunits IHFA and IHFB29,30. Upon binding, IHF is known to bend DNA29,31. It is generally involved in a variety of DNA-depended processes including site-specific DNA recombination, transposition, transcriptional regulation and DNA replication32,33,34. The bacteriophage-encoded integrase and IHF are known to form a complex during bacteriophage genome integration, the so-called intasome32,35. After integration, expression of the integrase gene must be tightly regulated, as the integrase is also catalyzing the excision reaction25. For the bacteriophage P4, it was shown that the integrase is functioning as a repressor of its own promoter25. In addition, the P4-encoded excisionase (Vis) was shown to be involved in the repression of the integrase promoter (PintP4)27. The PintP4 contains also an IHF binding site, but if IHF plays a role in the regulation of intP4 expression is not clear27. In addition to IHF, the host protein Fis appears to be involved in the integrative process of temperate bacteriophages36. We therefore decided to test if these proteins are involved in the excision process of PAI I536-VI536 and constructed isogenic ihfA/B and fis mutants of E. coli 536 reporter strains for the measurement of PAI I536-VI536 stability37. We found that Fis only plays a minor stabilizing role on individual PAIs. Likewise, IHF played a minor role in stabilizing PAI II536-VI536. However, the stability of PAI I536 was severely compromised in cells lacking IHF.
Results
Role of Fis and IHF in chromosomal stability of PAI I536-VI536 of E. coli strain 536
In order to address the question whether Fis and/or IHF play a role in stabilizing PAI I536-VI536 of E. coli strain 536, we constructed isogenic fis and ihfA/B mutants in our previously described reporter strains that allow for the measurement of the stability of PAI I536-VI53637. Briefly, we inserted a Pdps-yfp module in intergenic regions of the six PAIs of E. coli strain 536. This module allows expression of YFP (yellow fluorescent protein) when the cells enter stationary phase and is inactive during logarithmic growth. In addition, we inserted the module in the conserved chromosomal control position TR. As we have shown previously, the control position TR was functionally equivalent to five other tested chromosomal positions throughout the E. coli K-12 chromosome with respect to the expression of the Pdps module38. Here, we used the yfp-based module in order to distinguish cells that carry the module from those that have lost it simply by analyzing the YFP signal by flow cytometry. We found that whereas Fis might play a minor, but significant role in the stabilization of PAI IV536 and PAI VI536 under standard laboratory growth conditions (Fig. 1), IHFA/B did not negatively affect the stability of PAIs of E. coli strain 536, except for PAI I536 (Fig. 1). We found that almost 40% of the bacteria had lost PAI I536 in the ihfA/B mutant as judged by the absence of YFP signal in a large part of the population. To validate that the YFP-negative cells had lost PAI I536, we plated aliquots of an overnight (ON) culture on LB plates, incubated overnight and examined the obtained colonies by fluorescence microscopy. We subsequently analyzed the colonies by PCR and Sanger sequencing. In addition, we sorted 100.000 YFP-negative cells and used their genomic DNA as PCR template. Sequencing of the PCR product revealed exactly the same “scar sequence” resulting from chromosomal PAI I536 deletion as the sequencing of the PCR product of individual YFP-negative colonies, which indicated that the PAI excision process was very precise (Fig. S1). As PAI I536 is also encoding the toxin α-haemolysin, one important virulence factors of E. coli strain 536, we decided to analyze the IHF-dependent regulation of the stability of PAI I536 in more detail.

Impact of Fis and IHF on the stability of six archetypal PAIs of E. coli strain 536. Fraction (%) of YFP-negative cells in cultures of E. coli strain 536 and its 536 fis−, ihfA−, ihfB− and ihfAB− mutants. Each of the columns represents the average of three biological replicates, in which 107 cells per sample were analyzed (*P < 0.05; **P < 0.01; ***P < 0.001).
Lack of ihfB is sufficient to destabilize PAI I536
Next, we analyzed the stability of PAI I536-VI536 in cells lacking either functional ihfA or ihfB. We found that loss of functional ihfA increased the number of YFP-negative cells for all reporter strains, including the chromosomal control position TR (Fig. 1)37,38. In contrast, the absence of ihfB was specifically destabilizing PAI I536. Notably, the number of YFP-negative cells in the ihfB mutant was in the same range as observed with the ihfA/B double knockout mutant, suggesting that IHFB could play a greater role than IHFA in IHFAB-dependent stabilization of PAI I536 (Fig. 1). Importantly, complementation with ihfB restored the stability of PAI I536 (Fig. 2).

The deletion of ihfB is sufficient to destabilize PAI I536. Fraction (%) of YFP-negative cells in cultures of wild type E. coli strains 536 and 536 ihfB− containing the vector control and the ihfB complemented E. coli 536 ihfB−. Each of the columns represents the average of three biological replicates in which 107 cells per sample were analyzed (*P < 0.05; **P < 0.01; ***P < 0.001).
An IHF binding site in the promoter region of intI is required for the stabilization of PAI I536
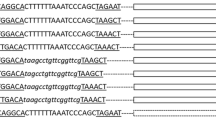
An IHF binding site in the promoter region of intP4 with unknown function has previously been described27,39. We therefore performed a computational analysis in order to screen for potential high affinity binding sites for IHF in the upstream region of the coding sequences of integrase I of E. coli strain 536 (Fig. 3A).

IHF binds to a high affinity binding site in the intI promoter region. (A) Sequence logo used to computationally predict IHF binding sites in the vicinity of intI-VI (up) and nucleotide sequence upstream of the intI gene (down). The binding site sequence logo was created using MEME39 and the experimentally identified IHF binding sites listed in RegulonDB38. Annotated are the main features of the intI gene: predicted IHF binding site (box), start codon of the coding sequence of intI (black arrow). (B) Analysis of scIHF binding to the intI upstream region by electrophoretic mobility shift assay (EMSA). Single-chain IHF bound specifically to the promoter fragment (PintI) only in the presence of the IHF binding site (left), but not when the binding site was absent (right). M, DNA size marker. For this figure, the original gel has been cropped and the arrangement of the two parts of the gel representing either pUC19PintI or pUC19PintI ΔIHFbs has been reversed. The original agarose gel is provided in Supplementary Figure S6. (C) Digoxygenin-labeled probes comprising the intI upstream region with or without IHF binding site (25 ng) were incubated with or without purified scIHF (80 µg, 40 µg, 20 µg; 0 µg). Single-chain IHF bound specifically to the promoter fragment (PintI) only in the presence of the IHF binding site (left), but not when the binding site was absent (right).
In order to test, if IHF is binding to the predicted site in the promoter region of intI, we overexpressed and purified scIHF, a genetically engineered and well-characterized single-chain IHF variant that functions like the wild-type heterodimer in vitro and in vivo29,40,41 (Fig. S2). We cloned the upstream region of intI with or without the potential IHF binding site into the plasmid pUC19, digested the plasmid and tested for in vitro binding of scIHF using electrophoretic mobility shift assays. As shown in Fig. 3B, the mobility of restriction fragments of the plasmid including the intI promoter (PintI) fragment lacking the IHF binding site, was not affected at the scIHF concentrations tested. In contrast, the wild type intI promoter fragment showed a scIHF concentration-dependent diffuse shift indicating a specific and stable scIHF-intI promoter interaction. Additionally, we investigated the interaction of scIHF with shorter DNA probes comprising the intI upstream region with the predicted IHF binding site. In one of the probes this binding site was destroyed. These 3′ digoxygenin-labeled probes were incubated with different amounts of purified scIHF, and a clear shift of the DNA band was only observed with the probe carrying the IHF binding site. In case of the probe with the mutated IHF binding site, no DNA shift was visible (Fig. 3C).
The IHF binding site is affecting the stability of PAI I536 in trans
The presence of the IHF binding site in the upstream region of intI suggested that IHF might stabilize PAI I536 by regulating intI expression. However, it was also possible that IHF as part of the intasome was stabilizing PAI I536 by inhibiting the excision process itself. If the latter would be the case, the presence or absence of the IHF binding site should not have affected PAI I536 stability, when intI is expressed in trans. We therefore cloned intI with IHF binding site (pMC1) and without the IHF binding site (pMC2) into a low copy vector and measured the stability of PAI I536 in E. coli strain 536 intI−. As shown in Fig. 4, the deletion of intI stabilized the island as judged by the almost undetectable YFP-negative cells when compared to the wild type as described previously17,37. Complementation of strain 536 intI− with pMC1 decreased PAI I536 stability back to wild type levels. However, complementation of E. coli 536 intI− with pMC2 decreased the stability of PAI I536 in the same order of magnitude as the ihfB mutation. This indicated that the major function of IHF in this system was indeed the regulation of intI expression (compare Fig. 4 to Fig. 1).

The IHF binding site is operational in trans. Fraction of YFP-negative cells in cultures of E. coli strain 536, its isogenic intI− mutant as well as in strain 536 intI− complemented with a plasmid-encoded copy of intI containing the IHF binding site (pMC1) or complemented with a plasmid-encoded copy of intI lacking the IHF binding site (pMC2) in percent. Each of the columns represents the average of three biological replicates in which 107 cells per sample were analyzed. (*P < 0.05; **P < 0.01; ***P < 0.001; schematic representation of the main features of pMC1 and pMC2 are shown below the graph).
Integrase I is repressing its own promoter
We therefore decided to analyze a chromosomal intI promoter-yfp transcriptional fusion (536 intI::yfp) in more detail37. Fluorescence measurements revealed an overall relatively high level of YFP expression (Fig. 5A) and a flow cytometric analysis of the bacterial population showed that this signal was overall normally distributed within the population (Fig. 5B). Despite the fact that the bacterial population also contained a very small fraction of highly fluorescent cells, the chromosomal 536 intI::yfp fusion was unique with respect to the other chromosomal integrase promoter-yfp fusion constructs (PintII to PintVI-yfp) of E. coli strain 536, as none of these showed such a basal, but readily detectable fluorescence signal37. As all chromosomal int promoter-yfp fusions were constructed in an identical way, i.e. by precisely replacing the open reading frame of the integrase genes by the reporter gene yfp, this suggested that the regulation of intI expression was distinct from that of intII to intVI. However, if the stability of PAI I536 was, at least in part, regulated at the level of intI expression, it was not clear how an overall normally distributed integrase I expression could result in the observed rare event of PAI I536 excision. We therefore speculated that an important factor for the regulation of intI expression was still missing in our reporter system for intI expression. As the integrase of bacteriophage P4 was shown to repress its own transcription, we decided to test the function of IntI in intI regulation by re-introducing intI into the system27. In order to do so, we first constructed an artificial operon with the reporter gene yfp transcriptionally fused downstream of the coding region of intI (536 intI-yfp). Afterwards we cloned the artificial operon into a low copy vector (pMC5).

Integrase I of E. coli 536 is subject to autoregulation and represses its own promoter. (A) YFP fluorescence signal of E. coli strains 536 intI::yfp (chromosomal promoter fusion), 536 intI-yfp (chromosomal operon), and E. coli strain 536 (pMC5) that encodes an artificial plasmid-encoded intI-yfp operon, and E. coli strain 536 (pMC3) with a plasmid-encoded intI::yfp fusion normalized to the cell density (OD595). The columns represent the average of three biological replicates (*P < 0.05; **P < 0.01; ***P < 0.001). (B) YFP fluorescence signal distribution within the corresponding bacterial populations (107 cells per sample were analyzed).
The comparison of the integrase promoter activity of the artificial operon in its native chromosomal position with pMC5 did not show any significant differences, both at population average as well as on overall fluorescence signal distribution within the population (Fig. 5A,B). However, when compared to the direct 536 intI::yfp fusion, the artificial operon showed a significant reduction of the population average fluorescence signal indicating that expression of the integrase I protein is necessary for the repression of its own synthesis (Fig. 5A). The comparison of the population average fluorescence signal when intI::yfp was localized on a low copy vector (pMC3; Fig. 5A, black bar) with pMC5 (Fig. 5A, light grey bar) showed as well a significant and even more pronounced reduction for the artificial operon. As for the chromosomal 536 intI::yfp fusion, flow cytometric measurements showed that the relatively strong fluorescence signal produced by pMC3 was normally distributed (Fig. 5B). In contrast the vast majority of cells containing pMC5 did not show a detectable fluorescence signal (Fig. 5B light grey).
IHFB is reducing the probability of intI promoter activation
The artificial operon containing intI now showed characteristics that were similar to what we had observed earlier for PintII to PintVI, i.e. an almost non-detectable population average fluorescence signal and a very small subpopulation of highly fluorescent cells37. We therefore decided to investigate the role of the IHF binding site in the artificial operon containing intI and cloned the artificial operon lacking the IHF binding site into a low copy plasmid (pMC7) and analyzed the sample by flow cytometry. As shown in Fig. 6A,B, the overall number of fluorescent bacteria was low and both samples showed a similar signal distribution. However, E. coli strain 536 containing pMC7 that lacks the IHF binding site showed an approximately 20-fold higher number of cells with high fluorescence signal than E. coli 536 containing pMC5 (Fig. 6A). Notably, the same analysis of pMC5 in E. coli strain 536 ihfB- resulted in very similar numbers of cells with high fluorescence signal indicating that one of the main functions of IHF in regulating intI expression is to reduce the probability of full promoter activation (compare Fig. 6A to Fig. S3B).

Impact of the presence of the IHF binding site on intI promoter activity in E. coli strains 536 (pMC5) and 536 (pMC7). (A) Number of YFP-positive cells in the populations of E. coli strains 536 intI-yfp, 536 (pMC5) and 536 (pMC7). The columns represent the average of three biological replicates, in which 107 cells per sample were analyzed (*P < 0.05; **P < 0.01; ***P < 0.001). (B) Distribution of the YFP fluorescence signal in the populations of E. coli strains 536 (pMC5) (grey) and 536 (pMC7) (black), which lacks the IHF binding site upstream of intI. In both populations 107 cells were analyzed per sample.
Discussion
The PAIs of UPEC strain 536 are an example for horizontally acquired genetic material that has equipped the host strain with virulence and fitness traits8,10,22,42,43,44. A better understanding of the mechanisms involved in integration and stabilization of newly acquired genetic material is necessary to design strategies for the prevention or reversion of these processes. Especially in the management of milder bacterial infections, drugs that attenuate a pathogen during an infection, e. g. by stimulating the excision and loss of PAIs, might be a more sustainable approach than classical antibiosis, as such drugs would be expected to impose much less selective pressure on the bacteria. For E. coli strain 536 it is known that PAI I536-VI536 display different levels of stability and that the difference is, at least in part, dependent on the activity of the P4-like integrase genes8,17,37.
In this study we investigated in detail the effect of the two nucleoid-associated proteins Fis and IHF in the stabilization process of PAI I536-VI536, as these proteins are known to be important host factors for temperate bacteriophages. We found that Fis might play a minor role in the stabilization of individual PAIs (Fig. 1), while IHFA/B showed a strong effect specifically on the chromosomal stability of PAI I536. Deletion of ihfB was sufficient to destabilize PAI I536 (Fig. 1). Our observation, which suggests that IHFB, most likely in the form of a homodimer, was sufficient to stabilize PAI I536, is consistent with previous studies that indicated a more important in vivo role for IHFB homodimers than for IHFA homodimers30,45,46.
We provide experimental evidence that IHF was stabilizing PAI I536 via regulation of intI expression by our observation that the IHF binding site mediated the observed phenotype in trans (Fig. 4). IHF can regulate transcription by different mechanisms. Usually, IHF binding modulates the DNA architecture and can thus affect the accessibility of promoter structures. Transcription can be induced when IHF binding leads to DNA bending, which allows the RNA polymerase to interact directly with a transcriptional regulator bound in a distant DNA region47. IHF-mediated DNA bending can also facilitate the interaction of the C-terminal domain of the RNA polymerase α subunit with upstream promoter elements, thereby inducing transcription48. In addition, open complex formation can be facilitated due to IHF binding49. If in case of PAI I536 the IHF binding site would have had a function exclusively in the excision process, its episomal presence or absence would not have affected the stability of PAI I536 to the same extent as the ihfB deletion. We showed that the IntI protein is repressing its own expression (Fig. 5). The regulation of intI by IntI in conjunction with IHF can therefore explain the observed increased frequency of the loss of PAI I536 in cells lacking IHF (Fig. 7): In wild type cells, IntI is repressing its own synthesis and a functional collaboration with IHF is further reducing the probability of the activation of the intI promoter. In a small subpopulation of cells, IntI may not form a stable complex with IHF on the promoter, and if additional IntI is produced, PAI I536 is excised. In the ihfB mutant, the repression complex involving DNA, IHFAB heterodimer and IntI cannot be formed, which results in an overall higher frequency of intI promoter activation, and production of additional IntI is resulting in PAI I536 excision. Over time, this results in the observed substantial fraction of cells lacking PAI I536 within the population. If our model depicted in Fig. 7 is correct, it has several interesting implications. First, if the stability of PAI I536 is so strictly regulated by the amount of IntI, the model would predict that there is virtually no unbound IntI available. This means that an extra copy of the intI promoter would be deregulated in the E. coli 536 wild type strain as well as in mutants lacking intI, which is exactly what we observe (Fig. S5). In contrast, if an extra copy of intI is reintroduced into the transcription unit itself, the promoter is again under strict autorepression (Fig. 5). Such a tight repression also indicates a very precisely ordered sequence of events for the activation of this promoter, which may be further elucidated by time-resolved in vitro footprinting techniques in the future.

Increased frequency of intI promoter activation in E. coli strain 536 ihfB− results in a rapid loss of PAI I536. Model for the regulation of intI expression by IntI and IHFB. In E. coli strain 536 (left) the expression of intI is strictly regulated by IntI and IHFB. In E. coli strain 536 ihfB− (right) the probability for intI activation is higher than in the wild type. This results over time in a significantly higher fraction of cells that have lost PAI I536. For details see text.
Why is it so important for genome plasticity and PAI stability that integrase expression is strictly regulated? Lysogenic bacteriophages employ site-specific recombination in order to integrate into or excise from the genome of their host. The underlying mechanism has been resolved for several model phages and is considered to be identical for all temperate bacteriophages23,25,26. The architecture of the intasome directs integration or excision of the phage genome24. Accordingly, the correct stoichiometry of the different components of the recombinogenic complex, including the integrase, recombination directionality factor (RDF), and accessory factors such as IHF, is important for determining the direction of the recombination event50. As the att sites, which flank chromosomal islands or prophage genomes differ from each other in their organization and number of protein binding sites20,51, it can be expected that the assembly process and overall structure of the recombinogenic complex differs between individual phage families and PAIs. A tightly balanced integrase level necessary for recombination may result from structural requirements of the intasome assembly50. This may explain why expression of the integrases is tightly, but not uniformly regulated in all islands in UPEC strain 536. Panis and co-workers suggested that many integrase genes of prophages are negatively autoregulated by itself and by a RDF due to overlapping attL and integrase promoter regions. RDF binding to the attL site prevents binding of the RNA polymerase to the −35 region of the integrase promoter thus controlling the integrase gene expression52. In case of PAI I536, the IHF-dependent regulation of intI expression may represent a similar mechanism to modulate the ratio of proteins involved in intasome formation. In addition to the integrase, which may be expressed at different levels, intasome formation could also be controlled via the expression level of other proteins involved, such as IHF. The fact that we identified an IHF binding site upstream of intI and demonstrated that this site is specifically recognized by scIHF in vitro (Fig. 3) supports this hypothesis. A similar role of IHF was also described for bacteriophage HP1 in Haemophilus influenzae Rd, where IHF binding to the attB and attP sites as well as to the upstream region of the integrase gene promoted integrase expression and the rate of integrative recombination53. Also the regulatory region of the bacteriophage P4 integrase gene was shown to contain a high affinity IHF binding site27,54.
Could the role of IHF in PAI I536 instability reflect unique aspects of the regulation of PAI I536-associated genes? Although IHF was discovered because of its requirement for bacteriophage λ recombination, it also plays an important role as a transcription factor and is itself regulated such that its intracellular concentration increases upon entry of the stationary phase. In E. coli and S. enterica serovar Typhimurium, IHF is involved in controlling the transcription of more than 100 genes of various functions55,56,57. Comparison of the transcriptome of ihfA, ihfB and ihfAB mutants of S. Typhimurium indicated that the single and double deletion mutants had overlapping, but not identical expression profiles suggesting that IHFA and IHFB homodimers can have differential functions56. Mangan and colleagues reported that IHF contributes to the coordinated and synchronized expression of main virulence factors of S. Typhimurium including the type three secretion systems encoded on SPI-1 and SPI-2 or of flagella56. Horizontally acquired DNA is often subjected to “xenogenic silencing” mediated by H-NS58,59. In S. Typhimurium IHF reduces H-NS-mediated silencing when cells enter the stationary phase, thereby modulating the topology of repressed promoters and the accessibility for other regulators60. Whether the specific importance of IHF for intI expression and PAI I536 stability represents an aspect of a fine-tuned bacterial regulatory network accompanied with improved fitness or pathogenicity requires further analyses. PAI I536 encodes among other gene products important UPEC virulence factors, such as α-haemolysin, F17- and CS12-like fimbrial adhesins6. These virulence factors are not known to be expressed in an IHF-dependent manner. Studies on the influence of IHF on (PAI-associated) gene expression as well as on the time-resolved, growth phase-specific regulation of gene expression on individual PAIs in UPEC 536 are not yet available, which could explain the unique regulatory mechanism controlling PAI I536 stability.
Many PAIs share features with lysogenic bacteriophage genomes and it has been discussed that PAIs may have evolved from former lysogenic bacteriophages by gradually losing some of their phage characteristics. Thus IHF-dependent regulation of integrase gene expression, together with the role of both, integrase and IHF, as members of the recombinogenic complex involved in site-specific recombination may be a relic of the “former life style” of PAI I536 as a selfish mobile genetic element. Clearly, the seven PAIs in E. coli strain 536 evolved from different ancestral phages. According to the amino acid sequence, the PAI I536-encoded integrase is more closely related to the P4 integrase than the other PAI-encoded integrases in UPEC strain 536 (Figure S4), which may explain the specific role that IHF plays for the stability of PAI I536.
Methods
Bacterial strains and plasmids used in this study
The bacterial strains used in this study are listed in Table S2. The plasmids used in this study are listed in Table S3.
Construction of strains and plasmids
Construction of fis, ihfA, ihfB and ihfA/B mutants
The fis, ihfA, ihfB and ihfA/B deletion mutants of our previously described reporter strains of E. coli strain 536 enabling the analysis of PAI I536-VI536 stability and of control strain 536 TR tagged with the Pdps module37 were constructed using the Red/ET recombineering method61. For the replacement of fis and the ihfA by a zeocin resistance cassette, the template pKD8 and primer pairs MC38/MC39 and MC46/MC47 were used, respectively. For the replacement of ihfB with a gentamicin resistance cassette, the template pKD11 and primer pair MC48/MC49 were used. Double mutants for ihfA/B were constructed by first replacing ihfA with a zeocine resistance cassette and afterwards ihfB with a gentamicin cassette as described above. The replacement was afterwards controlled by PCR using primer pairs MC60/MC61, MC68/MC69 and MC70/MC71 as listed in Table S1. The obtained PCR products were additionally Sanger sequenced.
Construction of E. coli 536 intI-yfp
For the construction of a chromosomal int-yfp operon the yfp-ble segment was PCR amplified with Q5 High-Fidelity DNA Polymerase using primers MC138 and MC139 and pBAD24yfp-ble as template. The chromosomal fusion to intI was afterwards done by RedE/T recombineering61. The 5′and 3′ junctions were afterwards verified with PCR using the primer pairs MC140/MBP206 and 1720/MC14, respectively. The nucleotide sequence of the obtained PCR products was additionally determined by Sanger sequencing.
Construction of pWKS30ihfB
The plasmid pWKS30ihfB was constructed by amplifying ihfB with Q5 High-Fidelity DNA polymerase using primer pairs MC123/MC71 and chromosomal DNA of E. coli strain 536 as template. The PCR product was then cloned into EcoRV-HF digested low copy plasmid pWKS3062. Verification of pWKS30ihfB was done by PCR using primer pair MC111/MC112 and Sanger sequencing.
Construction of pBAD24yfp-ble
Construction of the intermediate pBAD24yfp. The yfp fragment was PCR amplified with Q5 High-Fidelity DNA polymerase using the primer pair 69_s/385. The PCR product was then digested with NcoI/XbaI and the 758-bp fragment cloned into the previously NcoI/XbaI digested vector pBAD24. The resulting plasmid was isolated and verified by analytical digestion, PCR using the control primers 733 and 734 to amplify the whole insert, and by Sanger sequencing (Table S1). The plasmid was named pBAD24yfp. The plasmid pBAD24yfp-ble was constructed by subcloning the ble cassette from the XbaI digested pKD8. The 748-bp ble fragment was subcloned into the XbaI digested pBAD24yfp. The plasmid was isolated and verified by analytical digestion, PCR using the control primers 733 and 734 to amplify the whole insert, and by Sanger sequencing (Table S1).
Construction of pKD8 and pKD11
The plasmid pKD8 was constructed by subcloning the ble cassette from the EcoRV-HF/PvuII-HF digested plasmid pEM7/Zeo into the PvuII-HF/NaeI digested pKD4 backbone. The plasmid was transformed into E. coli strain SY327λpir, isolated and verified by analytical digestion, PCR using the control primers 1074 and 1092 to amplify the whole insertion, and by Sanger sequencing (Table S1).
The plasmid pKD11 was constructed by subcloning the gentamicin cassette from the NcoI-HF/SacII digested plasmid pBBR-1MCS-5; a fragment of 828 bp was processed with Mung Bean nuclease and subsequently cloned into the PvuII-HF/NaeI digested pKD4 backbone. The plasmid was transformed into E. coli strain SY327λpir, isolated and verified by analytical digestion, PCR using control primers 1074 and 1092 to amplify the whole insertion, and by Sanger sequencing (Table S1).
Construction of pMC1 and pMC2
The plasmid pMC1 was constructed by amplifying the intI integrase gene of PAI I536 (including the IHF binding site) with Q5 High-Fidelity DNA polymerase using primers MC115 and MC14 and chromosomal DNA of E. coli strain 536 as template. The plasmid pMC2 was constructed by amplifying intI (excluding the IHF binding site) with Q5 High-Fidelity DNA polymerase using primer pair MC124/MC14 and chromosomal DNA of E. coli strain 536 as template. The PCR products were cloned into EcoRV-HF digested low copy plasmid pWKS30. Verification of pMC1 and pMC2 was done by PCR using the primer pair MC111/MC112 and by Sanger sequencing.
Construction of pMC3
The plasmid pMC3 was constructed by amplifying the intI::yfp-cat construct with Q5 High-Fidelity DNA polymerase using primer pair MC11/MC14 and chromosomal DNA of E. coli strain 536 PAI I intI::yfp-cat37 as template. The PCR product was cloned into EcoRV-HF digested low copy plasmid pWKS30. Verification of pMC3 was done by PCR using the primer pairs MC111/MBP206 and MB5/MC112 to control 5´- and 3´-junctions of insert and plasmid, respectively, as well as by Sanger sequencing.
Construction of pMC5 and pMC7
The plasmid pMC5 was constructed by amplifying the int-yfp operon with Q5 High-Fidelity DNA polymerase using primers MC125 and MC14 and chromosomal DNA of E. coli strain 536 intI-yfp as template. The PCR product was cloned into EcoRV-HF digested low copy plasmid pWKS30. The plasmid pMC7 was constructed by amplifying the intI-yfp operon with Q5 High-Fidelity DNA Polymerase using primer pair MC124/MC14 and chromosomal DNA of E. coli strain 536 intI-yfp as template. The PCR product was cloned into EcoRV-HF digested low copy plasmid pWKS30. Verification of both plasmids was done with the primer pairs MC111/MBP206 and 1720/MC112 to control 5′- and 3′-junctions of insert and plasmid, respectively, as well as by Sanger sequencing.
Construction of pUC19Pint and pUC19Pint ∆IHFbs
The plasmid pUC19PintI was constructed by amplifying the intI promoter region with Q5 High-Fidelity DNA polymerase using primers MC125 and MC14 and chromosomal DNA of E. coli strain 536 intI::yfp-catas template37. The PCR product was cloned into the SmaI digested high-copy vector pUC19. The plasmid pUC19PintI∆IHFbs was constructed by amplifying the intI promoter region with Q5 High-Fidelity DNA polymerase using primer pair MC124/MC14 and chromosomal DNA of E. coli strain 536 as template37. The PCR product was cloned into the SmaI digested high-copy vector pUC19. Verification of both plasmids was done with primer pairs 385/MBP206 and 384/MBP5 to control 5′- and 3′- junctions of insert and plasmid, and by Sanger sequencing.
Computational mapping of IHF binding sites
The computational mapping of IHF binding sites was performed as previously described63. The IHF consensus binding sequence was created based on the 124 sequences available in the TF binding sites experimental dataset on RegulonDB64. The 13-nt long consensus sequence was generated with MEME65 and then submitted to FIMO66 for scanning the occurrence of motifs with p < 0.0001 in E. coli strain 536 genome. The list of IHF binding site candidates was further examined for motifs found in the vicinity of intI-VI.
Electric mobility shift assays (EMSA)
scIHF was purified from plasmid pETscIHF2 as described previously40. The protein concentration was determined using the micro BCA protein assay kit (Thermo Fisher Scientific) and the quality of the protein preparation was controlled using SDS-PAGE (Fig. S2). A digested plasmid offers DNA fragments that do have binding sites and other fragments that are not expected to have binding sites in stoichiometric amounts, i.e. this experimental setup has the advantage of internal positive, as well as negative controls. The binding reaction was performed at 37 °C for 5 min by premixing a constant amount of digested pUC19PintI and pUC19PintI ∆IHFbs vectors (250 ng) with the indicated amount of purified protein (0.5 to 0.2 µg) in 20 µl final volume of STE buffer (100 mM NaCl, 10 mM Tris-Cl, pH8, 1 mM EDTA). Afterwards, the binding reactions were loaded on a 1.5% TAE agarose gel and run at 130 V for 3 h. The DNA was then visualized by EtBr staining.
Additionally, PCR products of the intI promoter region were generated with Q5 High-Fidelity DNA polymerase using primers MC125/MBP206 and MC124/MBP206, and the plasmids pUC19PintI and pUC19PintI ∆IHFbs as templates, respectively. EMSAs were carried out using the DIG Gel Shift Kit, 2nd Generation (Sigma-Aldrich, Taufkirchen, Germany) according to the manufacturer’s instructions. 25 ng of DIG-labeled probes were mixed with decreasing concentrations of purified scIHF (80 to 20 µg), and the reaction mixture was incubated at room temperature for 30 min. Protein-DNA complexes were then applied to an 8% (w/v) polyacrylamide gel, run for 2 h at 100 V in 0.5x TBE buffer using the Owl Dual-Gel Vertical Electrophoresis Systems (Thermo Fisher Scientific) and then electrically transferred to a nylon membrane. The DIG-label were then detected by chemiluminescence using the ChemiDoc MP System (BioRad, Munich,Germany).
Media
Lysogeny broth (LB; containing 10 g/L tryptone, 5 g/L yeast extract, 5 g/L NaCl) was used as standard growth medium. For the solid medium preparation, agar was added to a final concentration of 1.5% (w/v). When required, the medium was supplemented with the appropriate concentration of the selected antibiotic. Phosphate buffered saline (PBS; containing 8 g/L NaCl, 0.2 g/L KCl, 1.42 g/L Na2HPO4, 0.27 g/L KH2PO4, pH 7.4) was used for the washing step and for resuspending bacterial cells prior flow cytometric measurements.
Flow cytometry and cell sorting
Flow cytometric measurements were done using a Gallios flow cytometer (Beckman Coulter Life Sciences, Krefeld, Germany) equipped with a blue air-cooled argon laser (488 nm) and fluorescence filter sets for fluorescence FL1 (BP 515–545 nm) and FL3 (LP > 650 nm). For sorting of bacterial cells we used the BD FACSAria III model cell sorter (Becton-Dickinson Biosciences, Heidelberg, Germany). The instrument settings have been made utilizing the BD FACSDiva software. The sample preparation and the programs of the instruments have been described previously37. Briefly, the samples for the measurement of the stability of the islands in E. coli 536 wild type and different isogenic variants were prepared from independent overnight cultures grown under standard laboratory conditions and inactivated with an equal volume of 70% (v/v) isopropanol. For the measurements of the integrase promoter activity in its native position or when cloned into the different newly created vectors bacterial samples transitioning from logarithmic to stationary phase were used and prepared for the measurements as described above and elsewhere37.
Standard fluorescence signal detection and OD595 measurements
The samples were grown in LB at 37 °C under shaking standard laboratory conditions until the transition from logarithmic to stationary growth phase, and then prepared for the measurement. Prior each measurement 1 mL from every independent culture was washed once with PBS and re-suspended into an equal volume of PBS. The average fluorescence intensity as well as the optical density was measured with the TECAN infinite M200 plate reader (TECAN, Männedorf, Switzerland). Optical density was measured at 595 nm and the fluorescence signal measured using an excitation wavelength of 514 nm and detecting the emission signal at 550 nm with a detector gain of 100. Optical density signal was corrected for blank and fluorescence signal was corrected for background fluorescence signal (E. coli K-12 MG1655 for all strains). Afterwards YFP expression (Fluo/OD595) was calculated by normalizing corrected fluorescence signal (Fluo) to corrected optical density (OD595).
References
Stamm, W. E. & Norrby, S. R. Urinary Tract Infections: Disease Panorama and Challenges. J Infect Dis 183, S1–S4, https://doi.org/10.1086/318850 (2001).
Abbo, L. M. & Hooton, T. M. Antimicrobial stewardship and urinary tract infections. Antibiotics 3, 174–192, https://doi.org/10.3390/antibiotics3020174 (2014).
Foxman, B. Urinary Tract Infection Syndromes Occurrence, Recurrence, Bacteriology, Risk Factors, and Disease Burden. Infect Dis Clin NA 28, 1–13, https://doi.org/10.1016/j.idc.2013.09.003 (2014).
Hooton, T. M. Uncomplicated Urinary Tract Infection. New Engl J Med 366, 1028–1037, https://doi.org/10.1056/NEJMcp1104429 (2012).
Brzuszkiewicz, E. et al. How to Become a Uropathogen: comparative genomic analysis of extraintestinal pathogenic Escherichia coli strains. Proc Natl Acad Sci USA 103, 12879–12884, https://doi.org/10.1073/pnas.0603038103 (2006).
Dobrindt, U. et al. Genetic structure and distribution of four pathogenicity islands (PAI I(536) to PAI IV(536)) of uropathogenic Escherichia coli strain 536. Infect Immun 70, 6365–6372, https://doi.org/10.1128/iai.70.11.6365-6372.2002 (2002).
Dobrindt, U. & Hacker, J. Whole genome plasticity in pathogenic bacteria. Curr Opin Microbiol 4, 550–557, https://doi.org/10.1016/s1369-5274(00)00250-2 (2002).
Middendorf, B. et al. Instability of Pathogenicity Islands in Uropathogenic Escherichia coli 536. J Bacteriol 186, 3086–3096, https://doi.org/10.1128/JB.186.10.3086 (2004).
Dobrindt, U., Chowdary, M. G., Krumbholz, G. & Hacker, J. Genome dynamics and its impact on evolution of Escherichia coli. Med Microbiol Immunol 199, 145–154, https://doi.org/10.1007/s00430-010-0161-2 (2010).
Hacker, J., Blum-Oehler, G., Mühldorfer, I. & Tschäpe, H. Pathogenicity islands of virulent bacteria: Structure, function and impact on microbial evolution. Mol Microbiol 23, 1089–1097, https://doi.org/10.1046/j.1365-2958.1997.3101672.x (1997).
Boyd, E. F., Almagro-Moreno, S. & Parent, M. A. Genomic islands are dynamic, ancient integrative elements in bacterial evolution. Trends Microbiol 17, 47–53, https://doi.org/10.1016/j.tim.2008.11.003 (2009).
Napolitano, M. G., Almagro-Moreno, S. & Boyd, E. F. Dichotomy in the evolution of pathogenicity island and bacteriophage encoded integrases from pathogenic Escherichia coli strains. Infect Genet Evol 11, 423–436, https://doi.org/10.1016/j.meegid.2010.12.003 (2011).
Schmidt, H. & Hensel, M. Pathogenicity islands in bacterial pathogenesis. Clin Microbiol Rev 17, 14–56, https://doi.org/10.1128/CMR.17.1.14 (2004).
Ahmed, N., Dobrindt, U., Hacker, J. & Hasnain, S. E. Genomic fluidity and pathogenic bacteria: applications in diagnostics, epidemiology and intervention. Nature Rev Microbiol 6, 387–394, https://doi.org/10.1038/nrmicro1889 (2008).
Dobrindt, U., Hochhut, B., Hentschel, U. & Hacker, J. Genomic islands in pathogenic and environmental microorganisms. Nat Rev Microbiol 2, 414–424, https://doi.org/10.1038/nrmicro884 (2004).
Frost, L. S., Leplae, R., Summers, A. O. & Toussaint, A. Mobile genetic elements: The agents of open source evolution. Nat Rev Microbiol 3, 722–732, https://doi.org/10.1038/nrmicro1235 (2005).
Hochhut, B. et al. Role of pathogenicity island-associated integrases in the genome plasticity of uropathogenic Escherichia coli strain 536. Mol Microbiol 61, 584–595, https://doi.org/10.1111/j.1365-2958.2006.05255.x (2006).
Esposito, D. & Scocca, J. J. The integrase family of tyrosine recombinases: Evolution of a conserved active site domain. Nucl Acids Res 25, 3605–3614, https://doi.org/10.1093/nar/25.18.3605 (1997).
Argos, P. et al. The integrase family of site-specific recombinases: regional similarities and global diversity. EMBO J 5, 433–440, https://doi.org/10.1099/jmm.0.030015-0 (1986).
Campbell, A. M. Chromosomal insertion sites for phages and plasmids. J Bacteriol 174, 7495–7499, https://doi.org/10.1128/jb.174.23.7495-7499.1992 (1992).
Ravatn, R., Studer, S., Zehnder, A. J. B. & Van Der Meer, J. R. Int-B13, an unusual site-specific recombinase of the bacteriophage P4 integrase family, is responsible for chromosomal insertion of the 105- kilobase clc element of Pseudomonas sp. strain B13. J Bacteriol 180, 5505–5514 (1998).
Darmon, E. & Leach, D. R. F. Bacterial Genome Instability. Microbiol Mol Biol Rev 78, 1–39, https://doi.org/10.1128/MMBR.00035-13 (2014).
Nash, H. A. Integration and Excision of Bacteriophage λ: The Mechanism of Conservative Site Specific Recombination. Ann Rev Genet 15, 143–167, https://doi.org/10.1007/BF02247811 (1981).
Seah, N. E. et al. Nucleoprotein architectures regulating the directionality of viral integration and excision. Proc Natl Acad Sci USA 111, 12372–12377, https://doi.org/10.1073/pnas.1413019111 (2014).
Pierson, L. S. & Kahn, M. L. Integration of satellite bacteriophage P4 in Escherichia coli. DNA sequences of the phage and host regions involved in site-specific recombination. J Mol Biol 196, 487–496, https://doi.org/10.1016/0022-2836(87)90026-X (1987).
Grindley, N. D. F., Whiteson, K. L. & Rice, P. A. Mechanisms of Site-Specific Recombination. Ann Rev Biochem 75, 567–605, https://doi.org/10.1146/annurev.biochem.73.011303.073908 (2006).
Piazzolla, D. et al. Expression of phage P4 integrase is regulated negatively by both Int and Vis. J Gen Virol 87, 2423–2431, https://doi.org/10.1099/vir.0.81875-0 (2006).
Yu, A. & Haggård-Ljungquist, E. Characterization of the binding sites of two proteins involved in the bacteriophage P2 site-specific recombination system. J Bacteriol 175, 1239–1249 (1993).
Rice, P. A., Yang, S. W., Mizuuchi, K. & Nash, H. A. Crystal structure of an IHF-DNA complex: A protein-induced DNA U-turn. Cell 87, 1295–1306, https://doi.org/10.1016/S0092-8674(00)81824-3 (1996).
Zulianello, L., de la Gorgue de Rosny, E., van Ulsen, P., van de Putte, P. & Goosen, N. The HimA and HimD subunits of integration host factor can specifically bind to DNA as homodimers. EMBO J 13, 1534–1540, https://doi.org/10.1002/j.1460-2075.1994.tb06415.x (1994).
Thompson, J. F. & Landy, A. Empirical estimation of protein-induced DNA bending angles: Applications to λ site-specific recombination complexes. Nucl Acids Res 16, 9687–9705, https://doi.org/10.1093/nar/16.20.9687 (1988).
Craig, N. L. & Nash, H. A. E. coli integration host factor binds to specific sites in DNA. Cell 39, 707–716, https://doi.org/10.1016/0092-8674(84)90478-1 (1984).
Freundlich, M., Ramani, N., Mathew, E., Sirko, A. & Tsui, P. The role of integration host factor in gene expression in Escherichia coli. Mol Microbiol 6, 2557–2563, https://doi.org/10.1111/j.1365-2958.1992.tb01432.x (1992).
Stonehouse, E., Kovacikova, G., Taylor, R. K. & Skorupski, K. Integration host factor positively regulates virulence gene expression in Vibrio cholerae. J Bacteriol 190, 4736–4748, https://doi.org/10.1128/JB.00089-08 (2008).
Fogg, P. C. M., Rigden, D. J., Saunders, J. R., McCarthy, A. J. & Allison, H. E. Characterization of the relationship between integrase, excisionase and antirepressor activities associated with a superinfecting Shiga toxin encoding bacteriophage. Nucl Acids Res 39, 2116–2129, https://doi.org/10.1093/nar/gkq923 (2011).
Esposito, D. & Gerard, G. F. The Escherichia coli Fis protein stimulates bacteriophage λ integrative recombination in vitro. J Bacteriol 185, 3076–3080, https://doi.org/10.1128/JB.185.10.3076-3080.2003 (2003).
Chittò, M., Berger, M., Klotz, L. & Dobrindt, U. Sub-Inhibitory Concentrations of SOS-Response Inducing Antibiotics Stimulate Integrase Expression and Excision of Pathogenicity Islands in Uropathogenic Escherichia coli Strain 536. Int J Med Microbiol, 151361–151361, https://doi.org/10.1016/j.ijmm.2019.151361 (2019).
Berger, M. et al. Genes on a wire: The nucleoid-associated protein HU insulates transcription units in Escherichia coli. Sci Rep 6, 1–12, https://doi.org/10.1038/srep31512 (2016).
Moitoso de Vargas, L. & Landy, A. A switch in the formation of alternative DNA loops modulates λ site-specific recombination. Proc Natl Acad Sci USA 88, 588–592, https://doi.org/10.1073/pnas.88.2.588 (1991).
Bao, Q., Christ, N. & Dröge, P. Single-chain integration host factors as probes for high-precision nucleoprotein complex formation. Gene 343, 99–106, https://doi.org/10.1016/j.gene.2004.08.030 (2004).
Corona, T. et al. Activation of site-specific DNA integration in human cells by a single chain integration host factor. Nucl Acids Res 31, 5140–5148, https://doi.org/10.1093/nar/gkg711 (2003).
Blum, G. et al. Excision of Large DNA Regions Termed Pathogenicity Islands from tRNA- Specific Loci in the Chromosome of an Escherichia coli Wild-Type Pathogen. Infect Immun 62, 606–614, doi:0019-9567/94/$04.00+0 (1994).
Messerer, M., Fischer, W. & Schubert, S. Investigation of horizontal gene transfer of pathogenicity islands in Escherichia coli using next-generation sequencing. PLoS ONE 12, 1–17, https://doi.org/10.1371/journal.pone.0179880 (2017).
Subashchandrabose, S. & Mobley, H. L. T. Virulence and Fitness Determinant of Uropathogenic Escherichia coli. Microbiol Spectr 3, 1–32, https://doi.org/10.1128/microbiolspec.UTI-0015-2012.Virulence (2015).
Hiszczyńska-Sawicka, E. & Kur, J. Effect of Escherichia coli IHF mutations on plasmid p15A copy number. Plasmid 38, 174–179, https://doi.org/10.1006/plas.1997.1307 (1997).
Prieto, A. I. et al. Genomic analysis of DNA binding and gene regulation by homologous nucleoid-associated proteins IHF and HU in Escherichia coli K12. Nucl Acids Res 40, 3524–3537, https://doi.org/10.1093/nar/gkr1236 (2012).
Engelhorn, M. & Geiselmann, J. Maximal transcriptional activation by the IHF protein of Escherichia coli depends on optimal dna bending by the activator. Mol Microbiol 30, 431–441, https://doi.org/10.1046/j.1365-2958.1998.01078.x (1998).
Macchi, R. et al. Recruitment of sigma54-RNA polymerase to the Pu promoter of Pseudomonas putida through integration host factor-mediated positioning switch of alpha subunit carboxyl-terminal domain on an UP-like element. J Biol Chem 278, 27695–27702, https://doi.org/10.1074/jbc.M303031200 (2003).
Parekh, B. S. & Hatfield, G. W. Transcriptional activation by protein-induced DNA bending: Evidence for a DNA structural transmission model. Proc Natl Acad Sci USA 93, 1173–1177, https://doi.org/10.1073/pnas.93.3.1173 (1996).
Mumm, J. P., Landy, A. & Gelles, J. Viewing single λ site-specific recombination events from start to finish. EMBO J 25, 4586–4595, https://doi.org/10.1038/sj.emboj.7601325 (2006).
Panis, G., Duverger, Y., Champ, S. & Ansaldi, M. Protein binding sites involved in the assembly of the KplE1 prophage intasome. Virology 404, 41–50, https://doi.org/10.1016/j.virol.2010.04.027 (2010).
Panis, G. et al. Tight regulation of the intS Gene of the KplE1 prophage: A new paradigm for integrase gene regulation. PLoS Genet 6, 1–15, https://doi.org/10.1371/journal.pgen.1001149 (2010).
Hwang, E. S. & Scocca, J. J. Interaction of integration host factor from Escherichia coli with the integration region of the Haemophilus influenzae bacteriophage HP1. J Bacteriol 172, 4852–4860, https://doi.org/10.1128/jb.172.9.4852-4860.1990 (1990).
Calì, S. et al. Bacteriophage P4 Vis protein is needed for prophage excision. Virology 322, 82–92, https://doi.org/10.1016/j.virol.2004.01.016 (2004).
Arfin, S. M. et al. Global gene expression profiling in Escherichia coli K12: The effects of integration host factor. J Biol Chem 275, 29672–29684, https://doi.org/10.1074/jbc.M002247200 (2000).
Mangan, M. W. et al. The integration host factor (IHF) integrates stationary-phase and virulence gene expression in Salmonella enterica serovar Typhimurium. Mol Microbiol 59, 1831–1847, https://doi.org/10.1111/j.1365-2958.2006.05062.x (2006).
McLeod, S. M. & Johnson, R. C. Control of transcription by nucleoid proteins. Curr Opin Microbiol 4, 152–159, https://doi.org/10.1016/S1369-5274(00)00181-8 (2001).
Baños, R. C. et al. Differential regulation of horizontally acquired and core genome genes by the bacterial modulator H-NS. PLoS Genet 5, e1000513, https://doi.org/10.1371/journal.pgen.1000513 (2009).
Navarre, W. W. The Impact of Gene Silencing on Horizontal Gene Transfer and Bacterial Evolution. Adv Microb Physiol 69, 157–186, https://doi.org/10.1016/bs.ampbs.2016.07.004 (2016).
Queiroz, M. H., Madrid, C., Paytubi, S., Balsalobre, C. & Juárez, A. Integration host factor alleviates H-NS silencing of the Salmonella enterica serovar Typhimurium master regulator of SPI1, hilA. Microbiology 157, 2504–2514, https://doi.org/10.1099/mic.0.049197-0 (2011).
Datsenko, K. A. & Wanner, B. L. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci USA 97, 6640–6645, https://doi.org/10.1073/pnas.120163297 (2000).
Wang, R. F. & Kushner, S. R. Construction of versatile low-copy-number vectors for cloning, sequencing and gene expression in Escherichia coli. Gene 100, 195–199, https://doi.org/10.1016/0378-1119(91)90366-j (1991).
Berger, P. et al. The primary transcriptome of the Escherichia coli O104:H4 pAA plasmid and novel insights into its virulence gene expression and regulation. Sci Rep 6, 1–10, https://doi.org/10.1038/srep35307 (2016).
Santos-Zavaleta, A. et al. RegulonDB v 10.5: Tackling challenges to unify classic and high throughput knowledge of gene regulation in E. coli K-12. Nucl Acids Res 47, D212–D220, https://doi.org/10.1093/nar/gky1077 (2019).
Bailey, T. L. & Elkan, C. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proceedings /… International Conference on Intelligent Systems for Molecular Biology; ISMB. International Conference on Intelligent Systems for Molecular Biology 2, 28–36 (1994).
Grant, C. E., Bailey, T. L. & Noble, W. S. FIMO: Scanning for occurrences of a given motif. Bioinformatics 27, 1017–1018, https://doi.org/10.1093/bioinformatics/btr064 (2011).
Acknowledgements
Our work was supported by the Interdisciplinary Center for Clinical Research, Medical Faculty Münster (grant number Dob2/013/12) and the ERA-NET PathoGenoMics III (Federal Ministry of Education and Research grant number 0315904A). The work of P.B. was funded by the German Research Foundation (project number 276606594). Data reported in this study appear in the PhD thesis of M.C. Support by the Münster Graduate School of Evolution (MGSE) to M.C. is gratefully acknowledged.
Author information
Authors and Affiliations
Contributions
M.B. and U.D. conceived the idea, designed the project, supervised the experiments. M.C. performed the experiments and analyzed the data. P.B. determined IHF binding sites in the upstream region of integrase genes. M.C., M.B., P.B. and U.D. drafted the manuscript. All authors (M.C., M.B., P.B., L.K., P.D. and U.D.) critically revised and approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chittò, M., Berger, M., Berger, P. et al. IHF stabilizes pathogenicity island I of uropathogenic Escherichia coli strain 536 by attenuating integrase I promoter activity. Sci Rep 10, 9397 (2020). https://doi.org/10.1038/s41598-020-66215-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-66215-2
- Springer Nature Limited