
Abstract
Altered pulmonary angiogenesis contributes to disrupted alveolarization, which is the main characteristic of bronchopulmonary dysplasia (BPD). Transforming growth factor β (TGFβ) plays an important role during lung vascular development, and recent studies have demonstrated that endoglin is engaged in the modulation of TGFβ downstream signalling. Although there are two different isoforms of endoglin, L- and S-endoglin, little is known about the effect of S-endoglin in developing lungs. We analysed the expression of both L- and S-endoglin in the lung vasculature and its contribution to TGFβ-activin-like kinase (ALK)-Smad signalling with respect to BPD development. Hyperoxia impaired pulmonary angiogenesis accompanied by alveolar simplification in neonatal mouse lungs. S-endoglin, phosphorylated Smad2/3 and connective tissue growth factor levels were significantly increased in hyperoxia-exposed mice, while L-endoglin, phosphor-Smad1/5 and platelet-endothelial cell adhesion molecule-1 levels were significantly decreased. Hyperoxia suppressed the tubular growth of human pulmonary microvascular endothelial cells (ECs), and the selective inhibition of ALK5 signalling restored tubular growth. These results indicate that hyperoxia alters the balance in two isoforms of endoglin towards increased S-endoglin and that S-endoglin attenuates TGFβ-ALK1-Smad1/5 signalling but stimulates TGFβ-ALK5-Smad2/3 signalling in pulmonary ECs, which may lead to impaired pulmonary angiogenesis in developing lungs.
Similar content being viewed by others


Introduction
The main pathologic finding of bronchopulmonary dysplasia (BPD) in the postsurfactant era is an alveolar arrest resulting in alveolar simplification (the development of fewer but larger alveoli with decreased septation) and decreased density of capillary beds1,2,3,4,5,6. Recent studies have revealed that impaired angiogenesis may lead to decreased numbers of alveoli; therefore, enhancement of normal angiogenesis could promote alveolarization in preterm infants1,3,5,7,8,9,10. Indeed, evidence suggests that pulmonary vessels actively promote alveolar development in the distal saccules and that dysmorphic changes in the pulmonary vasculature contribute to alveolar hypoplasia in animal models of BPD1,3,4,5,6,7,11,12. Postmortem studies on infants who died after prolonged mechanical ventilation have also quantified abnormal pulmonary microvascular growth13,14.
Transforming growth factor β (TGFβ) plays a pivotal role during lung development and angiogenesis through the regulation of endothelial cell (EC) growth, differentiation, migration, senescence, and extracellular matrix (ECM) production15,16,17,18,19. TGFβ-mediated signalling is initiated by binding to a TGFβ specific membrane receptor complex in ECs that contains 3 types of receptors: type I and type II serine/threonine kinase receptors and a co-receptor or TGFβ type III receptor named endoglin, also known as CD105. These receptors propagate the signal to the downstream nuclear effectors, Smads, by phosphorylation16,18,20,21,22. Phosphorylated Smads bind to the common Smad4 protein, translocate into the nucleus and regulate the transcription of their target genes. Depending on the TGFβ type I receptor involved, the signal is propagated to two different Smad protein subfamilies with specificities for activin-like kinase 1 (ALK1) phosphorylating Smad1/5 or ALK5 phosphorylating Smad2/321,22,23,24,25,26. It has also been reported that vascular endothelial growth factor (VEGF) expression increases through enhancement of the TGFβ-ALK1-Smad1/5 pathway, while connective tissue growth factor (CTGF) expression increases through enhancement of the TGFβ-ALK5-Smad2/3 pathway27,28,29,30,31.
Several independent findings have demonstrated that endoglin is engaged in TGFβ receptor complex formation and the modulation of downstream signalling32,33,34,35. The expression of two alternatively spliced isoforms, long (L-) endoglin and short (S-) endoglin, has been demonstrated in human and mouse lung tissues in vivo33,34,36,37. L-endoglin is the predominant isoform, and its pre-mRNA can be alternatively spliced by intron retention, producing the less abundant form, S-endoglin36,37. Evidence indicates that L-endoglin, the predominant isoform in ECs, promotes EC proliferation via TGFβ-ALK1 signalling, whereas S-endoglin acts as an antagonist of L-endoglin via activation of the TGFβ-ALK5 pathway32,33,36,37.
To date, most studies published on endoglin have focused on L-endoglin. It has been found that L-endoglin enhances ALK1-Smad1/5 signalling in ECs, leading to proliferation and migration, which are the main characteristics of the activation phase of angiogenesis15,18,20,22,38,39,40,41,42,43. However, a role of S-endoglin during endothelial senescence was recently described36,37. The S-endoglin:L-endoglin ratio increases during the senescence of ECs in vitro as well as during ageing in vascularized tissues, and the switch from L-endoglin to S-endoglin affects TGFβ-mediated cell signalling towards promoting the ALK5-Smad2/3 pathway instead of the ALK1-Smad1/5 pathway23,37,43,44,45.
Although the role of S-endoglin in TGFβ signalling was recently addressed in mature systemic ECs, little is known about the expression and role of S-endoglin in immature pulmonary ECs and their impacts on BPD-associated dysangiogenesis. In the present study, we analysed the expression of both L-endoglin and S-endoglin in the neonatal lung vasculature and its contribution to TGFβ-ALK-Smad signalling with respect to BPD development.
Results
Hyperoxia impairs alveolar and pulmonary vascular development in neonatal mice
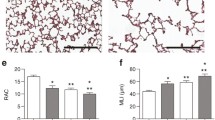
The unexposed lungs of control mice showed normal alveolarization, indicated by small and complex distal air spaces. However, the hyperoxia group showed impaired alveolarization, with fewer, larger and simpler alveoli on P14 (Fig. 1A). The hyperoxia group had a significantly smaller alveolar surface area (SA) and a significantly longer mean chord length than the control group on P14 (Fig. 1C,D). Pulmonary vessel staining with platelet-endothelial cell adhesion molecule (PECAM)-1 was significantly lower in the hyperoxia group than in the control group during the entire experimental period (Fig. 1B,E,F). Quantification of these parameters indicated that exposure of newborn mice to hyperoxia during the early alveolar period resulted in decreased sprouting angiogenesis and impaired alveolarization.

Effect of hyperoxia on alveolar and pulmonary vascular development. (A) Representative light microscopic images of mouse lungs. Apparently thinned septal walls and markedly large and simple airspaces were observed in the hyperoxia group compared with the control group. H&E staining: original magnification 200×. Bars, 100 µm. (B) Representative light microscopic images of PECAM-1 immunohistochemistry of mouse lungs. Markedly fewer blood vessels and decreased PECAM-1 staining were observed in the hyperoxia group compared with the control group. PECAM-1 staining was visualized with a DAB reaction (brown colour). Light H&E staining was used as a counterstain: original magnification 200×. Bars, 100 µm. (C) The alveolar SA was smaller in the hyperoxia group than in the control group; it increased as the lungs developed in the control group but not in the hyperoxia group. (D) The mean cord length, indicating the average alveolar size, was greater in the hyperoxia group than in the control group; it decreased as the lungs developed in the control group, but not in the hyperoxia group. (E) The number of blood vessels per HPF was significantly lower in the hyperoxia group than in the control group. (F) Immunohistochemical staining for PECAM-1 was significantly decreased in the hyperoxia group compared with the control group. (C–F) White bars denote the control group, and black bars denote the hyperoxia group. The data are expressed as the mean ± SEM (n = 5–7 in each group). Significant differences (p < 0.05) compared with the control group are marked by *.
Hyperoxia attenuates the expression of L-endoglin but promotes the expression of S-endoglin
The level of L-endoglin mRNA was significantly decreased at P9 and P14 in the hyperoxia group, whereas S-endoglin mRNA expression was significantly increased over the entire experimental period in the hyperoxia group compared to the control group (Fig. 2A,B). Although VEGF mRNA expression was reduced in the hyperoxia group, the difference was not significant (Fig. 2C). CTGF mRNA expression was higher in the hyperoxia group than in the control group (Fig. 2D). As CTGF seems to be a key mediator of the profibrotic effect of TGFβ36,46, we assessed whether hyperoxia could modify CTGF expression via L- or S-endoglin. Hyperoxia in newborn mice attenuated the mRNA expression of L-endoglin but promoted the mRNA expression of S-endoglin. These results indicate that hyperoxia enhances switching from L-endoglin to S-endoglin, leading to attenuation of the active proliferation of lung and pulmonary vessels as well as promotion of pulmonary fibrosis.

Expression of L-endoglin, S-endoglin, VEGF and CTGF mRNAs in hyperoxia. (A) Compared with the control group, the mRNA levels of L-endoglin were significantly decreased at P9 and P14 in the hyperoxia group. (B) Those of S-endoglin were significantly increased in the hyperoxia group over the entire experimental period. (C) VEGF showed no statistically significant difference between the control and hyperoxia groups. (D) The mRNA levels of CTGF were significantly increased at P2, P5 and P14 in the hyperoxia group. White bars denote the control group, and black bars denote the hyperoxia group. The data are expressed as the mean ± SEM (n = 4–6 in each group). Groups of data were compared with the corresponding time point by the Mann-Whitney U test. Significant differences (p < 0.05) compared with the control group are marked by *.
Hyperoxia alters the production of L- and S-endoglin and phosphorylated Smad proteins in neonatal mice
The protein levels of L-endoglin and phosphorylated Smad1/5 were significantly decreased in the hyperoxia group compared to the control group, while those of S-endoglin and phosphorylated Smad2/3 were significantly increased (Fig. 3A–G), demonstrating that these proteins were oppositely regulated by hyperoxia exposure. The calculated S-/L-endoglin ratio was significantly higher in the hyperoxia group than in the control group (Fig. 3C). Endoglin is an important accessory receptor for TGFβ-mediated signalling, and it is well known that TGFβ type I receptors (ALK1 and ALK5) have very high specificities for Smad phosphorylation (ALK1 phosphorylates Smad1/5, while ALK5 phosphorylates Smad2/3)23,37,43,44,45. Given all these findings, we postulate that hyperoxia exposure increases the production of S-endoglin, resulting in upregulation of Smad2/3 phosphorylation through ALK5.

Hyperoxia-induced alterations in L-/S-endoglin and Smad proteins in mice. (A,B) The protein level of L-endoglin was significantly decreased at P5, P9 and P14 in the hyperoxia group. In contrast, S-endoglin was significantly increased at P9 and P14 in the hyperoxia group. (C) The calculated S-endoglin/L-endoglin ratio was significantly increased at P5, P9 and P14 in the hyperoxia group. (D–F) The protein levels of phosphorylated Smad1/5 were significantly decreased in the hyperoxia group, whereas the protein levels of phosphorylated Smad2 and phosphorylated Smad3 were significantly increased in the hyperoxia group compared to the control group at P9 and P14. (G) Representative images of western blots in the control (C) and hyperoxia (H) groups. The data are expressed as the mean ± SEM (n = 4–6 in each group). White bars denote the control group, and black bars denote the hyperoxia group. Groups of data were compared with the corresponding time point by the Mann-Whitney U test. Significant differences (p < 0.05) compared with the control group are marked by *.
Hyperoxia impairs the tubular growth of ECs, while blockade of ALK5 restores normal tubular growth
We observed a pronounced decrease in tubular growth and branching in human pulmonary microvascular endothelial cells (HPMECs) exposed to hyperoxia compared to control cells (Fig. 4A,B). To examine the effect of ALK5 blockade on hyperoxia-induced tubular growth impairment, we added the synthetic ALK5 inhibitor SB431542. As shown in Fig. 4A,B,D, ALK5 inhibition restored normal tubular growth and extinguished the effect of hyperoxia on HPMECs. The relative fluorescence effects were more obvious for PECAM-1 than for γ-tubulin (Fig. 4C,D). From the above results, we can hypothesize that the downstream action of ALK5 is involved in hyperoxia-related pulmonary dysangiogenesis.

Tubular growth of human pulmonary microvascular endothelial cells (HPMECs). (A) Representative light microscopic images of the tubular growth of HPMECs. Magnification 40×. Bars, 500 µm. (B) Tube formation assays showed decreased branch points of HPMECs exposed to hyperoxia. The synthetic ALK5 inhibitor SB431542 restored normal tubular growth. (C) Immunofluorescent triple staining with antibodies against PECAM-1, DAPI and ɣ-tubulin. Magnification 200×. Bars, 100 µm. (D) Relative fluorescence of PECAM-1 was decreased in hyperoxia-exposed HPMECs. SB431542 restored its fluorescence similar to that observed in control HPMECs. There was no difference in the relative fluorescence of ɣ-tubulin. The data are expressed as the mean ± SEM (n = 4–6 in each group). Groups of data were compared with the corresponding time point by the Mann-Whitney U test. Significant differences (p < 0.05) compared with the control group are marked by *.
Hyperoxia alters the production of L- and S-endoglin and phosphorylated Smad proteins in ECs
In hyperoxia-exposed HPMECs compared to control HPMECs, the protein levels of L-endoglin and phosphorylated Smad1/5 were significantly decreased, while the protein levels of S-endoglin and phosphorylated Smad2/3 were significantly increased, which were restored by treatment with the SB431542 ALK5 inhibitor (Fig. 5A–G). The calculated S-/L-endoglin ratio was significantly higher in the hyperoxia group than in the control group, but balance was restored by ALK inhibition (Fig. 5C). Taken together, these results indicate that hyperoxia exposure causes the increased production of S-endoglin in HPMECs, resulting in the upregulation of Smad2/3 phosphorylation, and that these actions are mediated by ALK5. This result also reflects the possibility that immature human pulmonary ECs can synthesize considerable amounts of S-endoglin together with L-endoglin. The absence of an anti-S-endoglin antibody renders it difficult to confirm this reaction as an endogenous protein.

Hyperoxia-induced alterations in L-/S-endoglin and Smad proteins in HPMECs. (A,B) The protein level of L-endoglin was significantly decreased in the hyperoxia group. In contrast, S-endoglin was significantly increased in the hyperoxia group at T24. These protein levels were restored by treatment with the SB431542 ALK5 inhibitor. (C) The calculated S-endoglin/L-endoglin ratio was significantly increased at T4 and T24 in the hyperoxia group. The balance was restored by SB431542. (D–F) The protein levels of phosphorylated Smad1/5 were significantly decreased in the hyperoxia group, whereas the protein levels of phosphorylated Smad2 and phosphorylated Smad3 were significantly increased in the hyperoxia group compared to the control group at T24. These protein levels were restored by treatment with SB431542. (G) Representative images of western blots in the control, hyperoxia and hyperoxia + SB431542 groups. The data are expressed as the mean ± SEM (n = 4–6 in each group). White bars denote T0, grey bars indicate T4, and black bars indicate T24 in each group. Groups of data were compared with the corresponding time point by the Mann-Whitney U test. Significant differences (p < 0.05) compared with the control group are marked by *.
Ectopic expression of L-or S-endoglin alone does not modify the production of phosphorylated Smads or VEGF, CTGF or PECAM-1 proteins
The ectopic expression of L- and S-endoglin in HPMECs was assessed by immunofluorescence and western blot analysis. Both L- and S-endoglin were homogeneously distributed on the cell membrane (Fig. 6A). L- and S-endoglin were also detected by western blot analysis (Fig. 6B,C). As we expected, no endoglin expression was observed in mock cells. TGFβ1-induced Smad1/5 phosphorylation was noted only in L-endoglin-transfected cells, whereas Smad2/3 phosphorylation was noted only in S-endoglin-transfected cells (Fig. 6D). VEGF and PECAM-1 expression was markedly augmented in L-endoglin- but not S-endoglin-transfected HPMECs. In contrast, CTGF expression was markedly increased in S-endoglin- but not L-endoglin-transfected HPMECs (Fig. 6E,F). However, hyperoxia did not modify Smad phosphorylation or VEGF, CTGF or PECAM-1 production in either L-endoglin- or S-endoglin-transfected cells (Fig. 6E,F). These results indicate that L- and S-endoglin regulate pulmonary angiogenesis differentially and that these contrasting effects are mediated by ALK1 or ALK5 phosphorylating their specific downstream Smad proteins.

Production of Smad proteins and PECAM-1 in L-/S-endoglin transfected HPMECs. Representative images of (A) immunofluorescence and (B) western blots of endoglin in mock, L-endoglin (L-ENG)- and S-endoglin (S-ENG)-transfected HPMECs. (C–F) Mock, L-ENG- and S-ENG-transfected cells were serum starved for 12 h before 4 h of TGFβ1 treatment. Experimental 90% O2 was administered during TGFβ1 treatment. (D–F) TGFβ1-induced Smad1/5 phosphorylation was noted only in L-ENG-transfected cells, whereas Smad2/3 phosphorylation was noted only in S-ENG-transfected cells. (D) Hyperoxia did not modify Smad phosphorylation or VEGF, CTGF or PECAM-1 production in either L-ENG- or S-ENG-transfected cells. The data are expressed as the mean ± SEM (n = 4–6 in each group). Groups of data were compared by the Mann-Whitney U test. Non-significant differences are expressed as n.s.
Discussion
Here, we demonstrated, for the first time, the existence of two endoglin isoforms and an altered balance between these isoforms in hyperoxia-induced BPD models. We found that S-endoglin expression was increased by high oxygen exposure in a murine BPD model and in human pulmonary ECs. This upregulation of S-endoglin led to decreased angiogenesis and alveolar simplification in neonatal murine lungs and inhibited tube formation in a 3D culture of human pulmonary microvascular ECs.
Endoglin (also known as CD105 or TGFβ type III receptor) is not a true receptor for TGFβ, but it regulates the phosphorylation levels of the TGFβ type II receptors, ALK1 and ALK547. Recent studies have reported that downregulation of TGFβ-endoglin-ALK1-Smad1/5 signalling and upregulation of TGFβ-endoglin-ALK5-Smad2/3 signalling play important roles in the developmental arrest of not only pulmonary angiogenesis but also alveolarization25,42,44,48,49. However, it is unclear how endoglin determines this pathway. To date, most studies published on endoglin have focused on L-endoglin, although L-endoglin and S-endoglin are co-expressed and distinguishable in vivo32,36,37,39,40,41,42,43,48,50,51,52. This finding explains the contradictory results in the quantitative expression of endoglin in experimental BPD models or postmortem BPD patients in which the total quantity of endoglin has been evaluated9,11,18,25,32,40,42,45,53.
Two alternatively spliced isoforms of endoglin have been reported as follows: L- and S-endoglin32,36,37,39,40,41,42,43,48,50,51,52. S-endoglin is characterized by the retention of intron 14 in the mature mRNA34,36,37,39,40,41,42,43. Because the stop codon is included in the non-spliced intron 14, S-endoglin contains only 14 amino acids in the cytoplasmic domain, while L-endoglin contains 47 amino acids in its intracellular domain34,39. These structural differences may account for the distinct functions of L- and S-endoglin34,37. Experimental data have demonstrated that the S-endoglin:L-endoglin ratio is increased during senescence and oxidative stress in ECs in vitro as well as during ageing in vascularized tissues37,39,40,54. The switch between L-endoglin and S-endoglin affects TGFβ-mediated cell signalling towards promoting the ALK5-Smad2/3 pathway instead of the ALK1-Smad1/5 pathway. This phenomenon is thought to be due to the preferential affinity of the short cytoplasmic domain for ALK534,36,37,43.
Several studies have demonstrated that L-endoglin-ALK1-Smad1/5 signalling enhances the migration, proliferation, and tube formation of ECs, while the S-endoglin-ALK5-Smad2/3 pathway inhibits the activity of ECs. In addition, the expression of S-endoglin is increased in senescent ECs, and increased S-endoglin levels are related to endothelial ageing, damage or dysfunction32,37,54,55,56. Unfortunately, little is known about the role of endoglin isoforms in developing lungs and immature pulmonary ECs during the alveolarization period. Therefore, we hypothesized that endoglin may be the key molecule determining the TGFβ signalling pathway and that elevated S-endoglin could be linked to the pathogenesis of BPD.
Indeed, we observed that oxygen exposure increased S-endoglin levels and the S-/L-endoglin ratio in both mouse and pulmonary ECs. According to our results, S-endoglin behaves as an antiangiogenic molecule in contrast to proangiogenic L-endoglin. Elevated Smad2/3 phosphorylation, a marker of ALK5 activity, was also observed with paucity of vessels and angiogenesis accompanied by impaired alveolarization in mice and by tubular growth impairment in pulmonary ECs. Furthermore, the inhibition of ALK5 increased L-endoglin levels and Smad1/5 phosphorylation similar to control cells, with the restoration of normal tubular growth in pulmonary ECs. These data lend support that the unbalanced hyper-expression of S-endoglin alters the balance between TGFβ-ALK1-Smad1/5 and TGFβ-ALK5-Smad2/3 signalling in pulmonary ECs towards ALK5 activation with Smad2/3 phosphorylation and suggest that this process may underlie the impaired pulmonary angiogenesis resulting in disrupted alveolarization in the developing lung. To assess the impact of the two isoforms of endoglin in pulmonary ECs, transfected cells overexpressing human L-endoglin or S-endoglin as well as mock transfectants were investigated. Although this experimental method does not reflect the in vivo situation, the results indicate that L- and S-endoglin regulate pulmonary angiogenesis differentially, and these contrasting effects are mediated by ALK1 or ALK5 phosphorylating their specific downstream Smad proteins. We could not analyse the functional effects of S-endoglin silencing. The only difference between S-endoglin and L-endoglin mRNA transcripts is an intronic sequence of 135 bp that is retained in S-endoglin37. For this reason, there is no consensus small interfering RNA motif for this small sequence in the current study.
In the context of pulmonary vascular remodelling, the abnormally enhanced S-endoglin expression during exposure to hyperoxia together with ALK5 upregulation we observed are noteworthy, since S-endoglin and ALK5 activities are related to a decreased vasodilatory response and decreased endogenous nitric oxide synthase expression in lungs23,24,36,37. This finding means that S-endoglin overexpression may contribute to the development of pulmonary hypertension associated with BPD. In addition, because hyperoxia-exposed ECs did not form neoangiogenic webs on the Matrigel assay and displayed a high S-/L-endoglin ratio in the present study (Fig. 5), we can infer that unbalanced S-endoglin expression may be linked to precocious lung and pulmonary vascular maturation at the expense of developmental potential in preterm infants.
To our knowledge, the present study is the first report to demonstrate the dysregulated expression of two endoglin isoforms in the neonatal lungs and pulmonary microvascular ECs in the context of BPD pathogenesis. Our observations provide supportive proof for the crucial role of endoglin in the TGFβ signalling pathway in the developing lung and suggest a new therapeutic target for prevention or treatment of BPD. Although signalling by S-endoglin may play an important role in hyperoxia-induced BPD models, the association of S-endoglin with other factors contributing to BPD, such as inflammation, glucocorticoids, and mechanical ventilator-induced lung injuries, remains to be determined. In addition, much more needs to be learned about the mechanisms that regulate the production of S-endoglin, including the alternative splicing of endoglin mRNA in premature infants, and the effects of altered S-/L-endoglin balance on the function, growth and maturation of other vascular organs, especially the kidney, pulmonary and systemic vasculatures themselves.
Methods
Animals and hyperoxia exposure
All animal studies were performed at Inha University Hospital Biomedical Research Institute. The experimental protocol and procedures were approved by the Institutional Animal Care and Use Committee of Inha University (INHA 170330-488-1). All experiments were performed in accordance with relevant guidelines and regulations. Three-day-old male C57BL/6 J mice were used to examine the saccular and early alveolar stages of lung development in the mouse. Because sex-specific differences are well known in hyperoxic lung injury, we selected only male pups to exclude the bias effect from the sex difference57,58. The sex of mice was determined based on the morphological features of external genitalia59. In total, 54 mouse pups from 10 pregnant mice were used in the experiment. Animals were randomly divided into two groups as follows: control and hyperoxia groups.
The mice in the hyperoxia group were kept with a dam in a cage within a Plexiglas hyperoxic chamber with a continuous flow of oxygen to reach an oxygen concentration of 70% for 14 days60. The mice in the control group were continuously maintained in room air for 14 days. In mice, postnatal day (P) 14 is considered to represent the infant period of humans and the timing of active alveolarization and microvascular maturation61. Nursing dams were switched on alternate days between hyperoxic and normoxic cages to prevent oxygen toxicity. The oxygen concentration inside the Plexiglas hyperoxic chamber was continuously monitored using an oxygen sensor (Coy Laboratory Products, Grass Lake, MI, USA). At each time point (P2, P5, P9 and P14), mice were sacrificed. After thoracotomy, 10 to 40 μl (4 μl/g of body weight for each pup) of phosphate buffered saline (PBS) was infused through the trachea. The right lungs of the pups were immediately stored at −70 °C for biochemical analysis, and the left lungs were fixed in 4% paraformaldehyde solution, embedded in paraffin and stored at 4 °C for histological analysis.
Lung morphometry
Haematoxylin and eosin (H&E)-stained sections were photographed using a digital camera system (Leica DFC 280; Leica, Wentzler, Germany) with a microscope (Olympus BX51; Olympus, Tokyo, Japan) at 100 × magnification. Images were analysed using the morphometric method as described previously25. From the distal lung sections, 4 random non-overlapping high-powered fields (HPFs) per pup were used for morphometric evaluations. The mean chord length (Lm) provided an estimate of the distance from one airspace wall to an adjacent airspace wall25. The alveolar SA was calculated using the following equation: SA = 4 × VDT × lung volume / Lm25. Tissue volume densities (VDT) were determined using a 10 × 10 grid (the grid element side length was approximately 29 μm)25. Lung volume was determined by measuring the displacement of water by lungs after fixation25. A total of 20 measurements from 5 random areas on 4 random HPFs were performed for each pup25.
Immunohistochemistry of mouse lungs
For the immunohistochemical evaluation of PECAM-1 (CD31), the deparaffinized lung sections were incubated with a PECAM-1 antibody (Millipore, MA, USA) and a biotin-labelled donkey anti-mouse secondary antibody (Jackson ImmunoResearch Laboratories, West Grove, PA). Immune complexes were visualized using 3,3-diaminobenzidine tetrachloride (DakoCytomation, Denmark). Sections were lightly counterstained with haematoxylin, cleared and mounted with mounting medium. Immunostained sections were examined using a camera system with a microscope at 200 × magnification. Three mouse pups were examined per group at each time point. Four random non-overlapping distal lung sections per pup were used and 5 random non-overlapping fields per section were captured for quantitative imaging (Leica DFC 280; Leica, Wentzler, Germany)25.
Real-time RT-PCR
The quality and purity of the isolated total RNA from lung tissues were determined by spectrophotometry at 260 and 280 nm. Single-stranded cDNA was synthesized from 1 μg of RNA using a cDNA synthesis kit (Bio-Rad, Hemel Hempstead, UK) according to the manufacturer’s instructions. The transcripts were amplified, and real-time PCRs were run with SYBR Green Mastermix (Bio-Rad, Hercules, CA, USA). The data were analysed using Opticon Monitor software (Ver 3.1, Bio-Rad). β-Actin was used as the housekeeping gene, and the relative gene expression of L-endoglin, S-endoglin, VEGF and CTGF was calculated. For experiments with mouse lungs, the cDNA was used as a template for real- time PCR performed with specific primers. The mouse primers were as follows: L-endoglin (FW, 5′-GACCTGTCTGGTAAAGGCCTTGTCCTG-3′; RV, 5′-CTGGGGCCACGTGTGTGAGAATAG-3′); S-endoglin (FW, 5′-GACCTGTCTGGTAAAGGCCTTGTCCTG-3′; RV, 5′-CTGAGGGGCGTGGGTGA AGG-3′); VEGF (FW, 5′- AGAAAGCCCTGAAGTGGTG-3′; RV, 5′- ACTCCAGGGCTTCATCATTG-3′); and CTGF (FW, 5′-ACCCAACTATGATTAGAGCC-3′; RV, 5′-TTGCCCTTCTTAATGTTCTC-3′)43,46,62.
Cell culture and hyperoxia exposure
HPMECs (PromoCell, Heidelberg, Germany) isolated from a 60-year-old male Caucasian donor were used at passages 3‒4 and cultured in EC growth medium MV containing 5% foetal calf serum (FCS), 0.4% EC growth supplement (bovine hypothalamic extract), 10 ng/ml recombinant human epidermal growth factor, 1 μg/ml hydrocortisone and 90 μg/ml heparin (PromoCell, Heidelberg, Germany). HPMECs were grown in a humidified incubator containing 5% CO2 at 37 °C as recommended by the manufacturer (PromoCell, Heidelberg, Germany). For hyperoxia culture, cells were grown in a multi-gas incubator (APM-30D, ASTEC, Fukuoka, Japan) with regulated delivery of 95% O2 and 5% CO2 gas.
Angiogenic tubular formation assay
HPMECs grown to approximately 80% confluence in 6-well plates were silenced. After 24 h of silencing, the cells were detached with TrypLE Express (Invitrogen, CA, USA), and 1 × 105 cells were added to 250 μl of serum-free Matrigel (PromoCell, Heidelberg, Germany), gently mixed, polarized for 30 min at 37 °C, deposited into a 24-well culture plate (Bio-Rad, Redmond, WA, USA) and treated with 100 pM recombinant human TGFβ1 (R&D Systems, Minneapolis, MN, USA). Cells were then covered with 50 μl of complete medium containing 10% FCS and grown at 37 °C under 5% CO2 or 90% O2 for the indicated time periods (0, 4, 12, 24, and 48 h). To inhibit the ALK5 pathway, the cells were treated with 10 μM SB431542, an inhibitor of Smad2 phosphorylation (R&D Systems, Minneapolis, MN, USA). Angiogenesis was evaluated by counting the number of tube and network formations. Only tubular structures connecting two cell clusters were considered for branch measurements. Representative images were taken using a BX51 microscope (Olympus, Tokyo, Japan) with an attached camera (Leica DFC 280; Leica, Wentzler, Germany) at 40 × magnification. At each time point, cells were processed with Dispase (Bio-Rad, Hercules, CA, USA) for biochemical analysis.
Immunofluorescence analysis and confocal microscopy
Cells were grown in chambered cover glasses with or without Matrigel for 24 h. The cells were fixed with 2% paraformaldehyde for 20 min at room temperature and permeabilized with 0.5% Triton X-100 in PBS for 10 min at room temperature. The cells were rinsed three times with 100 mM glycine in PBS for 15 min and blocked with TNB [0.1 M Tris-HCl, 0.15 M NaCl, 0.5% blocking reagent (Boehringer Mannheim GmbH, Germany)] plus 10% FBS for 2 h at 37 °C. The cells were then incubated with the indicated antibodies diluted in TNB overnight at room temperature, and after washing with 0.1% NP-40 in PBS, the cells were incubated with goat anti-mouse secondary antibodies (Invitrogen, CA, USA) for 2 h at 37 °C. A goat anti-mouse secondary antibody marked with the red fluorochrome Alexa Fluor 594 (Invitrogen, CA, USA) was used to detect PECAM-1 or ɤ-tubulin. A goat anti-rabbit secondary antibody marked with the green fluorochrome Alexa Fluor 488 (Invitrogen, CA, USA) was used to detect ɤ-tubulin or human influenza haemagglutinin (HA). For nuclear counterstaining, blue-fluorescent DAPI staining was performed (Vector Laboratories, CA, USA). For multiple staining, samples were incubated sequentially with the indicated primary and secondary antibodies. The preparations were analysed with a Leica TCSSP5 (Leica Microsystems, Wentzler, Germany) confocal microscope. The analyses were captured at 200 × magnification. A total of 60 measurements from five-well slides were performed per group.
Transfection of L- or S-endoglin in HPMECs
The cDNAs encoding full-length human L- and S-endoglin polypeptides were cloned by application of the complete open reading frame (C-ORF) strategy and bioinformatics. L- and S-endoglin containing an N-terminal HA-tag were amplified by RT-PCR using the universal primers CMV 5′- AGAGAGTAGGTACAACGAAGGAGG- 3′ and 5′-ACGTTCTCGAAGGCATCTCTGGAA-3′, and the L- and S-endoglin expression vectors (pcDNA3.1 + /N-HA_L-ENG and pcDNA3.1 + /N-HA_S-ENG) were constructed by cloning the RT-PCR product into the BamHI site of pcDNA3.1 + (Invitrogen, San Diego, CA, USA). Cells were transfected with the expression vectors for 2 h using SuperFect® transfection reagent (Qiagen, Valencia, CA, USA) as indicated by the manufacturer. The cells were grown in FBS-free medium for 12 h and treated with TGFβ1 (100 pM) for 4 h at 37 °C under 5% CO2 or 90% O2. Then, the cells were lysed for western blot analysis. Detection of co-precipitated endoglin protein isoforms in the western blots was performed with anti-HA antibodies.
Western blot analysis
Lung tissues and cells were homogenized in lysis buffer with protease inhibitor cocktail (Roche, IN, USA)25. After determining the protein concentration by DC protein assay kits (Bio-Rad, Hercules, CA, USA), total soluble proteins (20 μg) were separated on 4–12% gradient gels (Invitrogen, CA, USA)25. The proteins were transferred to a nitrocellulose membrane (Amersham, Little Chalfont, UK). Nonspecific binding was blocked with 5% skim milk (BD, CA, USA) at room temperature for 1 h25. Then, the protein blots were probed with specific primary antibodies (1:1000 dilutions) against phospho-Smad1/5 (Cell Signaling, Frankfurt, Germany), phospho-Smad2 (Millipore, MA, USA), phospho-Smad3 (Millipore, MA, USA), PECAM-1 (Millipore, MA, USA), and L- and S-endoglin (R&D Systems, Minneapolis, MN, USA) overnight at 4 °C. After incubating with host-specific secondary antibodies (goat anti-rabbit IgG-HRP) for 1 h at room temperature, the immunoreaction was detected with a chemiluminescent substrate kit (Thermo Scientific, Rockford, IL, USA) and quantitated by densitometric scanning of the X-ray film with SLB MyImager (UVP Inc, Upland, CA, USA)25. The blots were normalized for protein loading by washing in stripping solution and reprobing with a monoclonal antibody against β-actin (Cell Signaling, Frankfurt, Germany)25.
Statistical analysis
Statistical analyses were performed using SPSS version 21.0. The data are expressed as the mean ± standard error of the mean (SEM) of four or more independent experiments. The Mann-Whitney U test was used for comparisons between the hyperoxia and control groups. P < 0.05 was considered statistically significant.
Data availability
The datasets generated and analysed in the present study are available upon request from the corresponding author.
References
Thébaud, B. Angiogenesis in Lung Development, Injury and Repair: Implications for Chronic Lung Disease of Prematurity. Neonatology 91, 291–297, https://doi.org/10.1159/000101344 (2007).
Kalikkot Thekkeveedu, R., Guaman, M. C. & Shivanna, B. Bronchopulmonary dysplasia: A review of pathogenesis and pathophysiology. Respir. Med. 132, 170–177, https://doi.org/10.1016/j.rmed.2017.10.014 (2017).
Thébaud, B. & Abman, S. H. Bronchopulmonary Dysplasia. Am. J. Respir. Crit. Care Med. 175, 978–985, https://doi.org/10.1164/rccm.200611-1660PP (2007).
Coalson, J. J. Pathology of Bronchopulmonary Dysplasia. Semin. Perinatol. 30, 179–184, https://doi.org/10.1053/j.semperi.2006.05.004 (2006).
Silva, D. M. G., Nardiello, C., Pozarska, A. & Morty, R. E. Recent advances in the mechanisms of lung alveolarization and the pathogenesis of bronchopulmonary dysplasia. Am. J. Physiol. Lung. Cell. Mol. Physiol. 309, L1239–L1272, https://doi.org/10.1152/ajplung.00268.2015 (2015).
Jobe, A. H. The new BPD: an arrest of lung development. Pediatr. Res. 46, 641–643 (1999).
Madurga, A., Mizikova, I., Ruiz-Camp, J. & Morty, R. E. Recent advances in late lung development and the pathogenesis of bronchopulmonary dysplasia. Am. J. Pphysiol. Lung Cell. Mol. Physiol. 305, L893–L905, https://doi.org/10.1152/ajplung.00267.2013 (2013).
Pasha, A. B., Chen, X.-Q. & Zhou, G. Bronchopulmonary dysplasia: Pathogenesis and treatment (Review). Exp. Ther. Med. 4315–4321, https://doi.org/10.3892/etm.2018.6780 (2018).
De Paepe, M. E., Greco, D. & Mao, Q. Angiogenesis-related gene expression profiling in ventilated preterm human lungs. Exp. Lung Res. 36, 399–410, https://doi.org/10.3109/01902141003714031 (2010).
Collins, J. J. P., Tibboel, D., de Kleer, I. M., Reiss, I. K. M. & Rottier, R. J. The Future of Bronchopulmonary Dysplasia: Emerging Pathophysiological Concepts and Potential New Avenues of Treatment. Front. Med. 4, 1–17, https://doi.org/10.3389/fmed.2017.00061 (2017).
Yang, W. C., Chen, C. Y., Chou, H. C., Hsieh, W. S. & Tsao, P. N. Angiogenic Factors in Cord Blood of Preterm Infants Predicts Subsequently Developing Bronchopulmonary Dysplasia. Pediatr. Neonatol. 56, 382–385, https://doi.org/10.1016/j.pedneo.2015.02.001 (2015).
Bourbon, J., Boucherat, O., Chailley-Heu, B. & Delacourt, C. Control mechanisms of lung alveolar development and their disorders in bronchopulmonary dysplasia. Pediatr. Res. 57, 38R–46R, https://doi.org/10.1203/01.PDR.0000159630.35883.BE (2005).
De Paepe, M. E. et al. Growth of pulmonary microvasculature in ventilated preterm infants. Am. J. Respir. Crit. Care Med. 173, 204–211, https://doi.org/10.1164/rccm.200506-927OC (2006).
Tomashefiski, J. F. J., Oppermann, H. C., Vawter, G. F. & Reid, L. M. Bronchopulmonary dysplasia: a morphometric study with emphasis on the pulmonary vasculature. Pediatr Pathol 2, 469–487 (1984).
Goumans, M.-J., Liu, Z., ten Dijke, P. & Dijke, P. TGF-beta signaling in vascular biology and dysfunction. Cell Res. 19, 116–27, https://doi.org/10.1038/cr.2008.326 (2009).
Walshe, T. E. TGF-β and microvessel homeostasis. Microvasc. Res. 80, 166–173, https://doi.org/10.1016/j.mvr.2010.03.003 (2010).
Vander Ark, A., Cao, J. & Li, X. TGF-β receptors: In and beyond TGF-β signaling. Cell. Signal. 52, 112–120, https://doi.org/10.1016/j.cellsig.2018.09.002 (2018).
Browne, S. et al. TGF-β1/CD105 signaling controls vascular network formation within growth factor sequestering hyaluronic acid hydrogels. PLoS One 13, 1–17, https://doi.org/10.1371/journal.pone.0194679 (2018).
Upton, P. D. & Morrell, N. W. The transforming growth factor-β-bone morphogenetic protein type signalling pathway in pulmonary vascular homeostasis and disease. Exp. Physiol. 98, 1262–1266, https://doi.org/10.1113/expphysiol.2012.069104 (2013).
Kamato, D. et al. Transforming growth factor β-mediated site-specific Smad linker region phosphorylation in vascular endothelial cells. J. Pharm. Pharmacol. 66, 1722–1733, https://doi.org/10.1111/jphp.12298 (2014).
Kamato, D. et al. Transforming growth factor-β signalling: Role and consequences of Smad linker region phosphorylation. Cell. Signal. 25, 2017–2024, https://doi.org/10.1016/j.cellsig.2013.06.001 (2013).
Zhang, H., Du, L., Zhong, Y., Flanders, K. C. & Roberts, J. D. Transforming growth factor-β stimulates Smad1/5 signaling in pulmonary artery smooth muscle cells and fibroblasts of the newborn mouse through ALK1. Am. J. Physiol. Lung Cell. Mol. Physiol. 313, L615–L627, https://doi.org/10.1152/ajplung.00079.2017 (2017).
Thomas, M. et al. Activin-like kinase 5 (ALK5) mediates abnormal proliferation of vascular smooth muscle cells from patients with familial pulmonary arterial hypertension and is involved in the progression of experimental pulmonary arterial hypertension induced by monocrot. Am. J. Pathol. 174, 380–389, https://doi.org/10.2353/ajpath.2009.080565 (2009).
Wu, S. et al. ALK-5 mediates endogenous and TGFβ1-induced expression of connective tissue growth factor in embryonic lung. Am. J. Respir. Cell Mol. Biol. 36, 552–561, https://doi.org/10.1165/rcmb.2006-0320OC (2007).
Jin, M. et al. Alteration of TGF-β-ALK-Smad signaling in hyperoxia-induced bronchopulmonary dysplasia model of newborn rats. Exp. Lung Res. 42, 354–364, https://doi.org/10.1080/01902148.2016.1226448 (2016).
Obreo, J. et al. Endoglin expression regulates basal and TGF- β1-induced extracellular matrix synthesis in cultured L6E9 myoblasts. Cell. Physiol. Biochem. 14, 301–310, https://doi.org/10.1159/000080340 (2004).
Boström, K., Zebbondj, A. F., Yao, Y., Lin, T. S. & Torres, A. Matrix GLA protein stimulates VEGF expression through increased transforming growth factor-β1 activity in endothelial cells. J. Biol. Chem. 279, 52904–52913, https://doi.org/10.1074/jbc.M406868200 (2004).
Wang, S. & Hirschberg, R. Diabetes-relevant regulation of cultured blood outgrowth endothelial cells. Microvasc. Res. 78, 174–179, https://doi.org/10.1016/j.mvr.2009.06.002 (2009).
Shieh, J. M., Tsai, Y. J., Chi, J. C. Y. & Wu, W. Bin. TGFβ mediates collagen production in human CRSsNP nasal mucosa-derived fibroblasts through Smad2/3-dependent pathway and CTGF induction and secretion. J. Cell. Physiol. 234, 10489–10499, https://doi.org/10.1002/jcp.27718 (2019).
Cheong, M. L., Lai, T. H. & Wu, W. Bin. Connective. tissue growth factor mediates transforming growth factor β-induced collagen expression in human endometrial stromal cells. PLoS One 14, 1–15, https://doi.org/10.1371/journal.pone.0210765 (2019).
Wu, Y. et al. Rapamycin upregulates connective tissue growth factor expression in hepatic progenitor cells through TGF-β-Smad2 dependent signaling. Front. Pharmacol. 9, 1–17, https://doi.org/10.3389/fphar.2018.00877 (2018).
Meurer, S. K., Tihaa, L., Borkham-Kamphorst, E. & Weiskirchen, R. Expression and functional analysis of endoglin in isolated liver cells and its involvement in fibrogenic Smad signalling. Cell. Signal. 23, 683–699, https://doi.org/10.1016/j.cellsig.2010.12.002 (2011).
Pérez-Gómez, E. et al. Characterization of murine S-endoglin isoform and its effects on tumor development. Oncogene 24, 4450–4461, https://doi.org/10.1038/sj.onc.1208644 (2005).
Bellón, T. et al. Identification and expression of two forms of the human transforming growth factor-beta-binding protein endoglin with distinct cytoplasmic regions. Eur. J. Immunol. 23, 2340–2345, https://doi.org/10.1002/eji.1830230943 (1993).
Gougos, A. & Letarte, M. Primary structure of endoglin, an RGD-containing glycoprotein of human endothelial cells. J. Biol. Chem. 265, 8361–8364 (1990).
Velasco, S. et al. L- and S-endoglin differentially modulate TGFbeta1 signaling mediated by ALK1 and ALK5 in L6E9 myoblasts. J. Cell Sci. 121, 913–9, https://doi.org/10.1242/jcs.023283 (2008).
Blanco, F. J. et al. S-endoglin expression is induced in senescent endothelial cells and contributes to vascular pathology. Circ. Res. 103, 1383–1392, https://doi.org/10.1161/CIRCRESAHA.108.176552 (2008).
Gore, B. et al. Key role of the endothelial TGF-β/ALK1/endoglin signaling pathway in humans and rodents pulmonary hypertension. PLoS One 9, e100310, https://doi.org/10.1371/journal.pone.0100310 (2014).
Jang, Y.-S. & Choi, I.-H. Contrasting roles of different endoglin forms in atherosclerosis. Immune. Netw. 14, 237–40, https://doi.org/10.4110/in.2014.14.5.237 (2014).
Tian, H. et al. Endoglin Mediates Vascular Maturation by Promoting Vascular Smooth Muscle Cell Migration and Spreading. Arterioscler. Thromb. Vasc. Biol. 37, 1115–1126, https://doi.org/10.1161/ATVBAHA.116.308859 (2017).
Lastres, P. et al. Endoglin modulates cellular responses to TGF-β1. J. Cell Biol. 133, 1109–1121, https://doi.org/10.1083/jcb.133.5.1109 (1996).
Lebrin, F. et al. Endoglin promotes endothelial cell proliferation and TGF-β/ALK1 signal transduction. EMBO J. 23, 4018–4028, https://doi.org/10.1038/sj.emboj.7600386 (2004).
Aristorena, M. et al. Expression of endoglin isoforms in the myeloid lineage and their role during aging and macrophage polarization. J. Cell Sci. 127, 2723–2735, https://doi.org/10.1242/jcs.143644 (2014).
Mancini, M. L. et al. Endoglin plays distinct roles in vascular smooth muscle cell recruitment and regulation of arteriovenous identity during angiogenesis. Dev. Dyn. 238, 2479–2493, https://doi.org/10.1002/dvdy.22066 (2009).
De Paepe, M. E., Patel, C., Tsai, A., Gundavarapu, S. & Mao, Q. Endoglin (CD105) up-regulation in pulmonary microvasculature of ventilated preterm infants. Am. J. Respir. Crit. Care Med. 178, 180–187, https://doi.org/10.1164/rccm.200608-1240OC (2008).
Chen, S. et al. CTGF disrupts alveolarization and induces pulmonary hypertension in neonatal mice: implication in the pathogenesis of severe bronchopulmonary dysplasia. Am. J. Physiol. Cell. Mol. Physiol. 300, L330–L340, https://doi.org/10.1152/ajplung.00270.2010 (2011).
Guerrero-Esteo, M., Sanchez-Elsner, T., Letamendia, A. & Bernabeu, C. Extracellular and cytoplasmic domains of endoglin interact with the transforming growth factor-beta receptors I and II. J. Biol. Chem. 277, 29197–29209, https://doi.org/10.1074/jbc.M111991200 (2002).
Ten Dijke, P., Goumans, M. J. & Pardali, E. Endoglin in angiogenesis and vascular diseases. Angiogenesis 11, 79–89, https://doi.org/10.1007/s10456-008-9101-9 (2008).
Tian, F. et al. Endothelial cells are activated during hypoxia via endoglin/ALK-1/SMAD1/5 signaling in vivo and in vitro. Biochem. Biophys. Res. Commun. 392, 283–288, https://doi.org/10.1016/j.bbrc.2009.12.170 (2010).
Jerkic, M. et al. Endoglin regulates cyclooxygenase-2 expression and activity. Circ. Res. 99, 248–256, https://doi.org/10.1161/01.RES.0000236755.98627.69 (2006).
Higa, R. et al. CD105 maintains the thermogenic program of beige adipocytes by regulating Smad2 signaling. Mol. Cell. Endocrinol. 474, 184–193, https://doi.org/10.1016/j.mce.2018.03.008 (2018).
Scherner, O., Meurer, S. K., Tihaa, L., Gressner, A. M. & Weiskirchen, R. Endoglin differentially modulates antagonistic transforming growth factor-β1 and BMP-7 signaling. J. Biol. Chem. 282, 13934–13943, https://doi.org/10.1074/jbc.M611062200 (2007).
Long, L. et al. Altered bone morphogenetic protein and transforming growth factor-β signaling in rat models of pulmonary hypertension. Potential for activin receptor-like kinase-5 inhibition in prevention and progression of disease. Circulation 119, 566–576, https://doi.org/10.1161/CIRCULATIONAHA.108.821504 (2009).
Blanco, F. J. & Bernabeu, C. Alternative splicing factor or splicing factor-2 plays a key role in intron retention of the endoglin gene during endothelial senescence. Aging Cell 10, 896–907, https://doi.org/10.1111/j.1474-9726.2011.00727.x (2011).
Meurer, S. K., Alsamman, M., Scholten, D. & Weiskirchen, R. Endoglin in liver fibrogenesis: Bridging basic science and clinical practice. World J. Biol. Chem. 5, 180–203, https://doi.org/10.4331/wjbc.v5.i2.180 (2014).
Wu, S. et al. High tidal volume ventilation activates Smad2 and upregulates expression of connective tissue growth factor in newborn rat lung. Pediatr Res 63, 245–250, https://doi.org/10.1203/PDR.0b013e318163a8cc (2008).
Lingappan, K. et al. Sex-specific differences in hyperoxic lung injury in mice: Implications for acute and chronic lung disease in humans. Toxicol Appl Pharmacol 272, 281–290, https://doi.org/10.1016/j.taap.2013.06.007 (2013).
Lingappan, K., Jiang, W., Wang, L. & Moorthy, B. Sex-specific differences in neonatal hyperoxic lung injury. Am. J. Physiol Lung. Cell. Mol. Physiol. 311, L481–L493, https://doi.org/10.1152/ajplung.00047.2016 (2016).
Murdaugh, L. B., Mendoza-Romero, H. N., Fish, E. W. & Parnell S. E. A novel method for determining sex in late term gestational mice based on the external genitalia. PLoS One 13, e0194767, 10.137/journal.pone.0194767 (2018).
Gortner, L. et al. Bronchopulmonary dysplasia in a double-hit mouse model induced by intrauterine hypoxia and postnatal hyperoxia: Closer to clinical features? Ann. Anat. 195, 351–358, https://doi.org/10.1016/j.aanat.2013.02.010 (2013).
Pinkerton, K. E. & Joad, J. P. The mammalian respiratory system and critical windows of exposure for children’s health. Environ. Health Perspect. 108, 457–462, https://doi.org/10.1289/ehp.00108s3457 (2000).
Qi, W. et al. The differential regulation of Smad7 in kidney tubule cells by connective tissue growth factor and transforming growth factor-beta1. Nephrology 12, 267–274, https://doi.org/10.1111/j.1440-1797.2007.00788.x (2007).
Acknowledgements
This work was supported by a National Research Foundation of Korea (NRF) grant funded by the Korea Government Ministry of Science and ICT (2016R1C1B2008221) and the Inha University Research Fund (Grant no. 55445-01).
Author information
Authors and Affiliations
Contributions
J.L. and Y.H.J. planned the study. Y.L., J.L. and S.K.N. conducted the experiments and performed statistical analyses. Y.L. and J.L. analysed the results and wrote the manuscript. All authors reviewed and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lee, Y., Lee, J., Nam, S.K. et al. S-endoglin expression is induced in hyperoxia and contributes to altered pulmonary angiogenesis in bronchopulmonary dysplasia development. Sci Rep 10, 3043 (2020). https://doi.org/10.1038/s41598-020-59928-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-59928-x
- Springer Nature Limited
This article is cited by
-
Association of cord blood Ang-1 and sCD105 levels with bronchopulmonary dysplasia in preterm infants
BMC Pediatrics (2024)
-
The role of rhIGF-1/BP3 in the prevention of pulmonary hypertension in bronchopulmonary dysplasia and its underlying mechanism
BMC Pulmonary Medicine (2023)
-
CircRNA, lncRNA, and mRNA profiles of umbilical cord blood exosomes from preterm newborns showing bronchopulmonary dysplasia
European Journal of Pediatrics (2022)
-
Alternative splicing in endothelial cells: novel therapeutic opportunities in cancer angiogenesis
Journal of Experimental & Clinical Cancer Research (2020)