
Abstract
The upper cervical spinal cord is measured in a large longitudinal amyotrophic lateral sclerosis (ALS) cohort to evaluate its role as a biomarker. Specifically, the cervical spinal cord´s cross-sectional area (CSA) in plane of the segments C1–C3 was measured semi-automatically with T1-weighted 3T MRI sequences in 158 ALS patients and 86 controls. Six-month longitudinal follow-up MRI scans were analyzed in 103 patients. Compared to controls, in ALS there was a significant mean spinal cord atrophy (63.8 mm² vs. 60.8 mm², p = 0.001) which showed a trend towards worsening over time (mean spinal cord CSA decrease from 61.4 mm² to 60.6 mm² after 6 months, p = 0.06). Findings were most pronounced in the caudal segments of the upper cervical spinal cord and in limb-onset ALS. Baseline CSA was related to the revised ALS functional rating scale, disease duration, precentral gyrus thickness and total brain gray matter volume. In conclusion, spinal cord atrophy as assessed in brain MRIs in ALS patients mirrors the extent of overall neurodegeneration and parallels disease severity.
Similar content being viewed by others
Introduction
Spinal cord involvement is a well-known and prominent feature of amyotrophic lateral sclerosis (ALS), that reflects both anterior horn cell (“amyotrophy”) and pyramidal tract degeneration/sclerosis of the lateral columns (“lateral sclerosis”) – which has been highlighted since Charcot’s pathology studies in 18651. Autopsy studies correspondingly reveal mild to marked loss of motor neurons in around 80% of the ALS patients which can affect the anterior horn of the entire spinal cord from the level of C2 to the level of its caudal sacral segments2. Anterior horn cell loss is furthermore closely associated with large myelinated fiber degeneration of the pyramidal tract2,3,4.
In-vivo studies applying magnetic resonance imaging (MRI) correspondingly display a reduction of the spinal cord cross-sectional area (CSA) in ALS, i.e. showing spinal cord atrophy together with pyramidal tract integrity loss in diffusion tensor imaging (DTI)5,6,7.
Controversies remain, however, on the relationship of in-vivo MRI spinal cord atrophy to clinical features and biomarkers in ALS. While some studies reported an association between CSA reduction and worse motor performance or longer disease duration8,9, others failed to find such a clinical correlation10,11. Conflicting or negative results might stem from small sample sizes in that most studies included less than 30 patients8,9,12. Additionally, only a few ALS studies thus far took advantage of the capability of spinal cord in-vivo MRI to monitor CSA evolution over time5,12,13.
In consideration of these uncertainties, we conducted a retrospective analysis of the upper cervical spinal cord within brain MRIs that were acquired during a large cross-sectional (N = 158) and longitudinal (N = 103) multicenter study to understand how in-vivo spinal cord atrophy relates to clinical features, other biomarkers, and, how it evolves over time in ALS.
Material and Methods
Sample
This is a retrospective study of 3T brain MRI scans of ALS patients acquired between 04/2011 and 01/2014 and controls scanned between 09/2013 and 03/2014. Data analysis took place between 01/2017 and 12/2018. The cohort comprised N = 158 ALS patients recruited at the neuromuscular outpatient clinics of the Departments of Neurology at Otto-von-Guericke University, Magdeburg; Hannover Medical School, Hannover; and University Medical Center, Rostock, Germany. ALS diagnosis was based on the El Escorial criteria and its revisions14,15,16. Classification of clinical phenotypes along the ALS disease spectrum was in accordance with operational definitions and comprised classic, lower motor neuron dominant (LMND) and upper motor neuron dominant (UMND) ALS17.
In short, all patients with LMND ALS had clinical and electrophysiological evidence of sporadic progressive pure LMN involvement in one or more regions without clinical signs of UMN dysfunction. LMN involvement must be the predominant finding for at least 12 months after the symptom onset. Other LMN diseases, such as multifocal motor neuropathy, spinal muscular atrophy, monomelic amyotrophy, Kennedy disease, and post-polio syndrome were excluded by extensive clinical and laboratory examinations18,19.
In UMND ALS patients, LMN signs were restricted to only one neuraxis level (bulbar, cervical, or lumbosacral), and electromyography (EMG) abnormalities were limited to sparse fibrillation potentials/positive sharp waves or minor enlargement of motor unit potentials in one or at most two muscles20,21 for at least 12 months after symptoms onset.
We also included patients with primary lateral sclerosis (PLS), a syndrome of pure upper motor neuron involvement that has been considered part of the motor neuron disease spectrum22,23. The diagnostic criteria for PLS required only UMN signs on examination at the time of baseline MRI24. Based on those criteria, at this time PLS diagnosis has been suspected in N = 9 patients, but N = 4 of them still not fulfilled the demand of a period of at least 4 years in which only UMN signs remained on examination24. At the time of data analysis, which took place some years after baseline MRI (please see above), all of those N = 4 patients with initially suspected PLS fulfilled the diagnostic criteria for PLS in that there were only UMN signs for a period of at least 4 years. We thus considered all N = 9 patients to suffer from PLS. The following were rigorously excluded from the PLS category: patients with clinical or EMG signs of LMN involvement; those with a disease that could mimic motor neuron disease; a family history of spastic paraparesis/tetraparesis; mutations of genes related to hereditary spastic paraplegia (SPG3A, SPG4. SPG7, SPG10, and SPG1125); and those with symptom onset before an age of 40 years.
N = 4 ALS patients fulfilled the diagnostic criteria for behavioral variant frontotemporal dementia26.
The study was approved by the Ethik-Kommission der Otto-von-Guericke-Universität in Magdeburg, the Ethik-Kommission der Medizinischen Hochschule Hannover and the Ethikkommission an der Medizinischen Fakultät der Universität Rostock (Approval No. 150/09, No. 07/17 and No. 16/17), and all subjects gave written informed consent. All methods were performed in accordance with the relevant guidelines and regulations.
Cross-sectional data ALS sample
As part of an ongoing multicenter study, all patients underwent 3T MRI of the brain (N = 141 in Magdeburg, N = 17 in Rostock, please see below), which also included the upper portion of the cervical spinal cord. Overall motor function was assessed by applying the revised ALS functional rating scale (ALSFRS-R) at baseline. Baseline disease duration was defined as the timespan between symptom onset and the first 3T MRI.
Ventilation status was available in N = 138 patients (87%), and of those, 16 (10%) were using non-invasive ventilation (NIV).
Criteria for NIV comprised dyspnea, a forced vital capacity of < 75% as predicted and/or daytime hypercapnia (pCO2 ≥ 45 mmHg)27,28.
In a subset of N = 63 ALS cases (40%), genetic testing was performed for superoxide dismutase 1 protein (SOD1) and chromosome 9 open reading frame 72 protein (C9orf72) gene mutations.
There were no differences between the ALS patients who had genetic data available and those who did not with regard to demographics (age, sex, height, weight) and clinical variables (baseline ALSFRS-R, baseline disease duration, onset site, clinical phenotype; data not shown).
Longitudinal data ALS sample
Of the 158 ALS patients, N = 103 cases (65%) underwent a second 3T MRI of the brain and upper cervical spinal cord and a second assessment of their motor function, i.e. ALSFRS-R, after 6 months. The baseline ALSFRS-R score of those who had longitudinal imaging was significantly better than that of the patients who only had a single scan (39 vs. 36 points, t(84) = −2.6, p = 0.01; independent-samples t-test). This is not unexpected because the less severe patients at baseline were more likely to undergo repeated imaging at 6-month follow-up. There were no further differences between patients with or without follow-up MRI with regard to their demographics (age, sex, height, weight) or clinical variables (baseline disease duration, onset site, clinical phenotype; data not shown).
Controls
For group comparisons, a community-based cohort of N = 86 participants recruited through public advertisements in Magdeburg (N = 69) and Rostock (N = 17), underwent a baseline 3T MRI of the brain and the upper cervical spinal cord using the same MRI protocol. None of the control subjects suffered from neuromuscular disorders, i.e. peripheral neuropathies, muscle or motor neuron diseases, nor from any brain injury, epilepsy or major psychiatric disease, nor did they display any specific abnormalities on the neurological exam29,30.
3T MRI of the brain and the upper cervical spinal cord
At both imaging sites (Magdeburg and Rostock) cerebral 3D-MPRAGE (magnetization-prepared rapid gradient-echo) images were acquired on a MAGNETOM Verio 3T MRI scanner (Siemens, Erlangen, Germany) using a 32-channel head coil applying exactly the same MRI protocol: TE (echo time) = 4.82 ms, TR (repetition time) = 2500 ms, TI (inversion time) = 1100 ms, flip angle = 7°, voxel size = 1 × 1 × 1 mm3, matrix = 256 × 256 × 192. MRI data acquisition time was 52 minutes.
Determination of the cervical spinal cord area
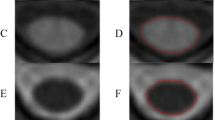
All scans were visually inspected to exclude spondylotic myelopathy. The cervical spinal cord CSA was determined with the freely available software SpineSeg, developed by Bergo et al.31. The contour of the spinal cord in the sagittal and coronal plane was tracked using a spline approximation and the volume was subsequently resampled into slices perpendicular to this guiding curve. The spinal cord was then semi-automatically segmented in each of these resampled axial slices using a tree pruning approach32 starting from the upper boundary of the dens axis to the middle of the second intervertebral disk, covering the upper cervical spinal cord segments C1, C2 and C3 (Fig. 1). Scans in which the delineation at C3 was not possible, either because of a decreased contrast resulting from the intensity attenuation towards the inferior edge of the field of view or because C3 was not covered, were excluded from the study. For baseline MRI, the scans of N = 2 controls had to be excluded; for follow-up MRI, the scans of N = 5 ALS patients had to be excluded. The mean number of slices per subject did not differ between patients and controls (t(240) = −0.8, p = 0.4).

Measurement of the cervical spinal cord cross-sectional area (CSA) applying SpineSeg. (a) demonstrates the cerebral 3T MRI, covering the upper segments of the cervical spinal cord, in a sagittal view. A guiding curve (pink) was placed along the spinal cord in the coronal and sagittal plane; the image is consequently resampled into perpendicular slices. Within the slices from the dens axis until the second intervertebral disk (b), the cervical spinal CSA segmentation was derived using a semi-automated tree pruning approach. Transversal view of the spinal cord is demonstrated before (c) and after (d) segmentation, respectively. (d) Seed in red, segmentation in yellow.
For group comparison, the mean of each subject’s CSA of all available spinal cord slices was calculated. All measures were performed twice by one researcher (TW) blinded to the clinical diagnosis. Corresponding intra-class correlation coefficient (ICC) was excellent (0.99). In 33 randomly chosen ALS patients and 26 controls, CSA measurements were repeated by a second researcher (FS), with an excellent inter-rater reliability (ICC = 0.98).
For analysis of distribution along the examined spinal cord length, five intermediate positions between dens axis and the center of the first intervertebral disk and two between first and second intervertebral disk were calculated. The slices closest to these chosen positions were used for comparison, creating an approximately equidistant sampling with a median of 5 [4–6] mm spacing.
Cortical Thickness and brain volumetric measures
To relate the cervical spinal cord CSA to structural measures of cerebral motor and extra-motor involvement, for each patient cortical thickness of the bilateral precentral gyrus was obtained from the native-space MPRAGE scans using the automated FreeSurfer 6.0 parcellation33. In both ALS and controls the right precentral gyrus was significantly thinner than the left (for ALS: 2.33 mm vs 2.43 mm, Z(11232) = 8.8, p < 0.001; for controls: 2.4 mm vs 2.49, Z(3127) = −6.6, p < 0.001; Wilcoxon signed-rank test). For statistical analysis the right and left motor cortex were thus considered separately. Total cerebral gray matter volume (GMV) normalized for head size was further estimated using the SIENAX algorithm from the SIENA-package of FMRIB Software Library (FSL) v5.0.
Statistics
Statistical analysis was conducted using IBM SPSS Statistics (Version 22.0).
Normal distribution of the data was assessed with a Shapiro-Wilk Test.
Group comparisons of the baseline mean cervical spinal cord CSA between (i) ALS vs. controls, (ii) limb- vs. bulbar-onset ALS, (iii) selected clinical phenotypes (e.g. classic ALS) vs. the remainder, (iv) sporadic vs. familial ALS and (v) non-ventilated patients vs. those requiring assisted ventilation were conducted using an analysis of variance (ANOVA). Additional group comparisons between ALS vs. controls along the length of the cervical spinal cord were performed, also applying an ANOVA. Baseline cervical spinal cord CSA depended on baseline age (see Results), and thus all models were adjusted for baseline age.
The relationship between mean spinal cord CSA and the patients’ and controls’ demographics (age, sex, height, weight); the patients’ clinical features (baseline ALSFRS-R and its sub-scores for bulbar fine motor, gross motor, respiratory function, baseline disease duration) and imaging data (motor cortex thickness, GMV) were assessed applying bivariate correlations and a χ² test.
Differences between baseline and follow-up cervical mean spinal cord CSA were calculated for ALS and for limb- and bulbar-onset ALS separately, applying a repeated measure ANOVA. Further, another repeated measure ANOVA was conducted to calculate any differences between baseline and follow-up ALSFRS-R. For ALS, additionally, differences between baseline and follow-up local CSA along the spinal cord were computed, also using a repeated measure ANOVA. All models were adjusted for baseline age.
P-values ≤ 0.05 were deemed statistically significant.
Results
Sample
Demographics of the whole cohort and the clinical data of the ALS patients are given in Table 1. There were no group differences between ALS and controls with respect to age, sex, height and weight.
Cross-sectional data
For the whole cohort, baseline mean spinal cord CSA was not normally distributed (D[242] = 1.0, p = 0.03). When considering ALS patients and controls separately, mean CSA was normally distributed in ALS (D[158] = 1.0, p = 0.5), but not in controls (D[84] = 1.0, p = 0.003) (Fig. 2a). Considering the whole cohort, mean CSA was related to baseline age (r = −0.2, p = 0.003) in that older age was associated with a smaller spinal cord CSA (Fig. 3a). Mean CSA was not related to sex, height and weight. These results remained unchanged when considering ALS and controls separately.

Quantile function of baseline mean cervical spinal cord cross-sectional area in ALS and controls. (a) CSA in ALS was significantly smaller than in controls. (b) CSA was further significantly smaller in limb- compared to bulbar-onset ALS. *p ≤ 0.05, **p ≤ 0.001 (all statistical models are adjusted for baseline age).

Relationship between baseline mean cervical spinal cord cross-sectional area (CSA) and various variables in ALS. CSA relates to baseline (a) age, (b) total revised ALS functional rating scale (ALSFRS-R), (c) disease duration, (d) cortical thickness of left precentral gyrus, (e) total adjusted gray matter (GM) volume. Some patients labelled PLS did not fulfill the demand of a period of at least 4 years with UMN signs only at the time of 3T MRI, but would go on to be diagnosed with PLS after the study’s conclusion. FS, Freesurfer; FSL, FMRIB Software Library; LMND, lower motor neuron dominant ALS; UMND, upper motor neuron dominant ALS; PLS, primary lateral sclerosis. P-values ≤ 0.05 were deemed statistically significant.
In ALS compared to controls, mean CSA was significantly smaller (60.8 mm² vs. 63.8 mm², F(1,240) = 10.7, p = 0.001; Fig. 2a).
The results of the CSA analysis along the length of the upper cervical spinal cord are displayed in Fig. 4a,b. At the dens axis level there were no significant group differences between ALS and controls, while all of the more caudal points down to the first intervertebral disk revealed a group difference with p < 0.05. The points of the first intervertebral disk until the second intervertebral disk revealed more significant differences with p < 0.001.

Cross-sectional area along the studied upper cervical spinal cord length in ALS and controls. For analysis of CSA distribution along the spinal cord, five intermediate positions between dens axis and the center of the first intervertebral disk (IVD) and two between first and second IVD were calculated, as indicated in (a). The cross-sectional area in these positions is compared between ALS and controls in (b), displaying means for ALS and controls as well as the interquartile range (IQR) for both groups. Significantly smaller CSA values were found in ALS in all positions below the dens axis. The longitudinal change in CSA along the spinal cord in ALS in shown in (c), indicating that significant CSA changes are only found in the more caudal areas between IVD 1 and IVD 2. *p ≤ 0.05, **p ≤ 0.001 (all statistical models are adjusted for baseline age).
Furthermore, ALS patients with limb-compared to bulbar-onset had a significantly smaller baseline mean spinal cord CSA (60.2 mm² vs. 62.5 mm², F(1,153) = 5.4, p = 0.02; Fig. 2b). Limb- and bulbar-onset ALS patients did not differ significantly with respect to age, disease duration or disease severity (according to baseline ALSFRS-R); group differences were thus not driven by those variables.
There were, however, no group differences for the mean spinal cord CSA when considering the ALS phenotypes, sporadic against familial ALS or non-ventilated against ventilated patients. Detailed results of all group comparisons are given in Table 2.
In the ALS sample, mean CSA was related to clinical variables, i.e. positively correlated with baseline ALSFRS-R total score (r = 0.2, p = 0.04); its gross motor function subscore (r = 0.2, p = 0.03); and negatively correlated with disease duration (r = −0.3, p < 0.001) (Fig. 3b,c). It was also positively correlated with precentral gyrus thickness (left r = 0.3, p < 0.001; right r = 0.2, p = 0.04) and GMV (r = 0.2, p = 0.02) (Fig. 3d,e). In summary, a larger mean spinal cord CSA was related to shorter disease duration, higher functional scoring, greater motor cortex thickness and GMV.
Longitudinal data
Longitudinal analysis revealed a trend towards a significant decline of the cervical spinal cord mean CSA from baseline to follow-up in ALS (61.4 vs. 60.6 mm², F(1,101) = 3.7, p = 0.06) (Fig. 5a). As displayed in Fig. 4c, CSA decline was most pronounced in the more caudal parts of the upper cervical spinal cord towards the second intervertebral disk. When considering limb- vs. bulbar-onset patients separately, only those with limb-onset displayed a significant mean CSA decline (60.6 vs. 59.6 mm2, F(1,75) = 4.8, p = 0.03), while bulbar-onset patients did not (63.9 vs. 63.6 mm2, F(1,23) = 0.1, p = 0.7) (Fig. 5a). Likewise, there was a significant decline of the ALSFRS-R from baseline to follow-up in ALS, with a larger effect size than the decline of the spinal cord mean CSA (39.1 vs. 35.8, F(1,99) = 15.9, p < 0.001) (Fig. 5b). There was nevertheless a small effect-size correlation between ΔCSA and ΔALSFRS-R in ALS (bivariate correlation, r = 0.2, p = 0.03).

Baseline and follow-up mean cervical spinal cord cross-sectional area and ALSFRS-R in ALS. Markers are spread out to display measurement quantiles, as given in the top axis. *p ≤ 0.05 (all statistical models are adjusted for baseline age).
Discussion
This large, multicenter study of the brain confirmed atrophy of the adjacent upper cervical spinal cord cross-sectional area and a trend towards its ongoing decline over 6 months in ALS, especially in the upper cervical spinal cord’s caudal segments and in limb-onset disease. Mean cervical spinal cord area related further to several clinical variables and measures, such as motor function, disease duration, motor cortex thickness and whole brain gray matter volume. In-vivo MRI upper cervical spinal cord atrophy assessed in a brain MRI can thus be considered a reliable biomarker that reflects disease severity. This highlights that measuring the upper spinal cord as part of the (routine) brain MRI scan is a valid marker in research studies.
Compared to most previous studies, our ALS cohort was nearly fivefold larger, allowing for subgroup comparisons along the motor neuron disease spectrum. The most severe cord atrophy was found in PLS in the present study, although this did not reach statistical significance, almost certainly because of insufficient power due to the small PLS subgroup size. We propose that upper spinal cord atrophy across the motor neuron disease spectrum is largely driven by corticospinal tract degeneration - a hypothesis that would explain the PLS finding in that this sub-group would not be expected to have significant anterior horn involvement. This is furthermore reinforced by the fact that the upper spinal cord is dominated by the white matter fraction (around 85% of the CSA), while it declines along lower cervical spinal cord levels (around 80%)34. However, more severe cord atrophy in PLS patients could additionally be explained by a longer disease duration than in ALS patients.
Upper cervical spinal cord atrophy could thus be considered to serve as a biomarker of upper motor neuron involvement, which by now is mainly assessed clinically depending on the experience of the physician. This could be one major role of cross-sectional and longitudinal spinal cord imaging despite its effect size of change is smaller than that of the longitudinal alteration of the clinical state of the ALS patients, i.e. of scoring their motor function applying the ALSFRS-R. Indeed, the ALSFRS-R is a composite score measuring the function of 4 different levels, comprising the patients’ bulbar state; its decline does thus not reflect the loss of any specific motor ability. The ALSFRS-R is further a measure that predominantly indicates the involvement of the lower motor neuron. The correlation between baseline spinal cord CSA and ALSFRS-R as well as the relationship between the decline of the two variables, however, reflects the common direction of imaging and clinical variables. Clinical trials should thus carefully take into account the, presumably, additive value of imaging and clinical measures in ALS.
Spinal cord atrophy and its ongoing decline was more pronounced in limb- compared to bulbar-onset patients confirming the results of one large autopsy study in ALS2. These findings are also consistent with a recent case-series that failed to identify significant cervical spinal cord atrophy in bulbar predominant ALS11. In addition, in bulbar-onset ALS spinal cord CSA did not decline within a period of 6 months. This resonates with the clinical observation that in bulbar predominant ALS spinal cord involvement takes place at later disease stages2,35, which has relevance when considering spinal cord atrophy as a monitor variable in clinical trials. One has, however, to consider some recruitment bias in that patients with severe bulbar involvement that cannot lie down for a longer period did not undergo MRI, especially longitudinal scanning. In that instance, it has to be considered remarkable that our cohort included 10% ALS patients with NIV, which are suffering from the same restrictions and are thus otherwise rarely included into MRI studies. The exclusion of patients with severe bulbar involvement might indeed have shifted our cohort towards (bulbar-onset) patients with a less rapidly progressive overall disease course and thus, less severe spinal cord involvement.
In the present study, reduction of mean upper cervical spinal cord CSA was around 5% in ALS compared to controls. Atrophy was greater in all other thus far conducted upper cervical spinal cord studies ranging between 10% to 17%5,8,10,11,13. All of these studies focused their measurements at the slice in the center of the intervertebral disc between C2 and C3 and few adjacent slices. In contrast, this study covered the whole length of the upper cervical spinal cord segments C1, C2 and C3 to the middle of the second intervertebral disk, to establish the atrophy along this section. The non-uniform manifestation of CSA reduction along the evaluated length results in lower mean atrophy estimates, despite the locally higher CSA reduction found in more caudal segments. Moreover, our ALS sample was at an earlier disease stage, reflected by a shorter median baseline disease duration of 16 months and relatively preserved motor function (median baseline ALSFRS of 39 points), compared to all previous cohorts. Accordingly, the ALS study reporting the greatest cord atrophy (17%) had the longest disease duration (44 months) and a lower ALSFRS-R (35 points)11. However, differences in measurement methods, MRI sequences (T1, T2) and field strengths (1.5 T vs. 3T) may have also played a role. Furthermore, the large white matter fraction of the upper cervical spinal cord (see above) should give rise to worse atrophy in samples with greater proportions of PLS and UMND ALS. As only one previous study reported ALS phenotypes11, it remains speculation as to whether this might have contributed to differences between studies.
A major strength of our study was the availability of longitudinal MRI datasets. The spinal cord CSA atrophy rate in our ALS sample was −1.7% over 6 months, which is somewhat smaller than that of previous studies showing rates of −2.5% (over 9 months) or −4.4% (over 8 months)5,13. Importantly, our longitudinal sample was also approximately four- to five times larger than that of past studies, suggesting the present atrophy rate is, arguably, a more robust estimate of reality. Different timespans, disease stages and various proportions of limb-onset ALS may have contributed to the differences as well. Overall, the results indicate that the upper spinal cord CSA might potentially be helpful to track ongoing disease even over a six-month interval but only provided the cohort is substantial (well in excess of 100 patients).
Turning to technical considerations, cross-sectional area is the most often used measure to assess spinal cord atrophy with excellent reliability31. Other measures such as the anterior-posterior diameter36 or spinal cord eccentricity8,13 are not useful biomarkers being insensitive to cord atrophy in ALS.
A range of methods has been employed to measure cervical cord CSA in ALS studies. In our study, the CSA was assessed with a semi-automated method (SpineSeg)31 which was highly reliable (both intra- and inter-rater)8,13,31. It took 10 to 15 minutes to process a subject, which is comparable to other semi-automated methods, e.g. Losseff’s approach37. Given the high reliability of semi-automated spinal cord CSA measures, new fully automated approaches could now be benchmarked against this approach.
Similar to several previous studies, we focused on the assessment of the upper part of the cervical spinal cord8,13. The main advantages of this approach are that it does not require additional MRI scans of the spinal cord, which are time consuming (potentially important given the difficulty many ALS patients have to lie flat for long periods); demand specific coils and dedicated sequences/protocols; need cardiac and respiratory gating; and are less standardized within multicenter studies. The upper cervical spinal cord can instead easily be included in an MRI brain acquisition and is, furthermore, useful in suspected limb-onset disease to exclude compressive myelopathy. One has, however, to take into account that – in a brain MRI - the upper part of the cervical spinal cord (i.e. especially C2 and C3 levels) is often at the limit of the field of view and its measure can be very variable between the acquisitions. The spinal cord area might thus often be difficult to be analyzed with a sufficient precision. Moreover, one has to keep in mind that the exploration of the lower parts of the cervical cord, although more technically challenging, is very relevant in ALS patients since lower motor neuron involvement is usually most pronounced at the C5 to Th1 level2.
Conclusion
Our study demonstrated mean upper cervical spinal cord atrophy that was – on a trend-level - detectable within a six-month window, and, that was related to clinical measures and markers of lower and upper motor neuron pathology in ALS. Atrophy was most pronounced in the caudal upper cervical spinal cord segments and in limb- compared to bulbar-onset disease, but was found in all clinical phenotypes along the ALS disease spectrum. The results emphasize the potential role of in-vivo MRI upper cervical spinal cord cross-sectional area to serve as a valid quantifiable marker for ALS, perhaps also for the longitudinal monitoring of ongoing disease. Future studies should focus on the differences between gray and white matter atrophy not only in the different ALS phenotypes, but also by taking into account brain and spinal DTI parameters and their correlation with the cervical spinal cord area.
Data availability
All custom scripts and all relevant data of this study are available from the corresponding author upon reasonable request.
References
Goetz, C. G. Amyotrophic lateral sclerosis: early contributions of Jean-Martin Charcot. Muscle Nerve 23, 336–343; 10.1002/(sici)1097-4598(200003)23: 3<336::aid-mus4>3.0.co;2-l (2000).
Brettschneider, J. et al. TDP-43 pathology and neuronal loss in amyotrophic lateral sclerosis spinal cord. Acta Neuropathol 128, 423–437, https://doi.org/10.1007/s00401-014-1299-6 (2014).
Brettschneider, J. et al. Stages of pTDP-43 pathology in amyotrophic lateral sclerosis. Ann Neurol 74, 20–38, https://doi.org/10.1002/ana.23937 (2013).
Geser, F. et al. Motor neuron disease clinically limited to the lower motor neuron is a diffuse TDP-43 proteinopathy. Acta Neuropathol 121, 509–517, https://doi.org/10.1007/s00401-011-0797-z (2011).
Agosta, F. et al. A longitudinal diffusion tensor MRI study of the cervical cord and brain in amyotrophic lateral sclerosis patients. J Neurol Neurosurg Psychiatry 80, 53–55, https://doi.org/10.1136/jnnp.2008.154252 (2009).
Cohen-Adad, J. et al. Involvement of spinal sensory pathway in ALS and specificity of cord atrophy to lower motor neuron degeneration. Amyotrophic lateral sclerosis & frontotemporal degeneration 14, 30–38, https://doi.org/10.3109/17482968.2012.701308 (2013).
Cohen-Adad, J. et al. 7-T MRI of the spinal cord can detect lateral corticospinal tract abnormality in amyotrophic lateral sclerosis. Muscle Nerve 47, 760–762, https://doi.org/10.1002/mus.23720 (2013).
Branco, L. M. T. et al. Spinal cord atrophy correlates with disease duration and severity in amyotrophic lateral sclerosis. Amyotroph Lat. Scl. Fr. 15, 93–97, https://doi.org/10.3109/21678421.2013.852589 (2014).
Paquin, M.-Ê. et al. Spinal Cord Gray Matter Atrophy in Amyotrophic Lateral Sclerosis. AJNR. American journal of neuroradiology 39, 184–192, https://doi.org/10.3174/ajnr.A5427 (2018).
Valsasina, P. et al. Diffusion anisotropy of the cervical cord is strictly associated with disability in amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry 78, 480–484, https://doi.org/10.1136/jnnp.2006.100032 (2007).
Olney, N. T. et al. Measurement of spinal cord atrophy using phase sensitive inversion recovery (PSIR) imaging in motor neuron disease. PLoS One 13, e0208255, https://doi.org/10.1371/journal.pone.0208255 (2018).
El Mendili, M. M. et al. Multi-parametric spinal cord MRI as potential progression marker in amyotrophic lateral sclerosis. PLoS One 9, e95516, https://doi.org/10.1371/journal.pone.0095516 (2014).
Albuquerque, M. et al. Longitudinal evaluation of cerebral and spinal cord damage in Amyotrophic Lateral Sclerosis. Neuroimage Clin 14, 269–276, https://doi.org/10.1016/j.nicl.2017.01.024 (2017).
Brooks, B. R., Miller, R. G., Swash, M. & Munsat, T. L. El Escorial revisited. Revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotrophic Lateral Sc 1, 293–299, https://doi.org/10.1080/146608200300079536 (2000).
Brooks, B. R. El Escorial World Federation of Neurology criteria for the diagnosis of amyotrophic lateral sclerosis. J. Neurol Sci 124, 96–107, 10.1016/0022-510×(94)90191-0(1994).
Carvalho, M. et al. Electrodiagnostic criteria for diagnosis of ALS. Clin Neurophysiol 119, 497–503 (2008).
Schreiber, S. et al. Peripheral nerve ultrasound in amyotrophic lateral sclerosis phenotypes. Muscle Nerve 51, 669–675, https://doi.org/10.1002/mus.24431 (2015).
van den RM, B.-V. et al. The spectrum of lower motor neuron syndromes. J Neurol 250, 1279–1292 (2003).
van den RM, B.-V. et al. Sporadic lower motor neuron disease with adult onset. Classification of subtypes. Brain 126, 1036–1047 (2003).
Soraru, G. et al. Natural history of upper motor neuron-dominant ALS. Amyotroph. Lateral. Scler. 11, 424–429 (2010).
Gordon, P. H., Cheng, B., Katz, I. B., Mitsumoto, H. & Rowland, L. P. Clinical features that distinguish PLS, upper motor neuron-dominant ALS, and typical ALS. Neurology 72, 1948–1952 (2009).
Al-Chalabi, A. et al. Amyotrophic lateral sclerosis: moving towards a new classification system. The Lancet Neurology 15, 1182–1194, https://doi.org/10.1016/S1474-4422(16)30199-5 (2016).
Finegan, E., Chipika, R. H., Shing, S. L. H., Hardiman, O. & Bede, P. Primary lateral sclerosis: a distinct entity or part of the ALS spectrum? Amyotrophic lateral sclerosis & frontotemporal degeneration 20, 133–145, https://doi.org/10.1080/21678421.2018.1550518 (2019).
Gordon, P. H. et al. The natural history of primary lateral sclerosis. Neurology 66, 647–653 (2006).
Tartaglia, M. C. et al. Differentiation between primary lateral sclerosis and amyotrophic lateral sclerosis. Examination of symptoms and signs at disease onset and during follow-up. Arch. Neurol. 64, 232–236 (2007).
Rascovsky, K. et al. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain 134, 2456–2477 (2011).
Prell, T. et al. Assessment of pulmonary function in amyotrophic lateral sclerosis: when can polygraphy help evaluate the need for non-invasive ventilation? J Neurol Neurosurg Psychiatry 87, 1022–1026, https://doi.org/10.1136/jnnp-2015-312185 (2016).
Andersen, P. M. et al. EFNS guidelines on the clinical management of amyotrophic lateral sclerosis (MALS)–revised report of an EFNS task force. Eur J. Neurol. 19, 360–375, https://doi.org/10.1111/j.1468-1331.2011.03501.x (2012).
Machts, J. et al. Basal ganglia pathology in ALS is associated with neuropsychological deficits. Neurology 85, 1301–1309, https://doi.org/10.1212/WNL.0000000000002017 (2015).
Schreiber, S. et al. Significance of CSF NfL and tau in ALS. J Neurol 265, 2633–2645, https://doi.org/10.1007/s00415-018-9043-0 (2018).
Bergo, F. P. G., França, M. C., Chevis, C. F. & Cendes, F. SpineSeg: A segmentation and measurement tool for evaluation of spinal cord atrophy. 7th Iberian Conference on Information Systems and Technologies (CISTI 2012) pp. 1–4 (2012).
Bergo, F. P. G., Falcão, A. X., Miranda, P. A. V. & Rocha, L. M. Automatic Image Segmentation by Tree Pruning. Journal of Mathematical Imaging and Vision 29, 141–162, https://doi.org/10.1007/s10851-007-0035-4 (2007).
Fischl, B. et al. Whole Brain Segmentation. Neuron 33, 341–355, https://doi.org/10.1016/S0896-6273(02)00569-X (2002).
Kameyama, T., Hashizume, Y. & Sobue, G. Morphologic features of the normal human cadaveric spinal cord. Spine 21, 1285–1290 (1996).
Turner, M. R. et al. The diagnostic pathway and prognosis in bulbar-onset amyotrophic lateral sclerosis. J. Neurol. Sci. 294, 81–85, https://doi.org/10.1016/j.jns.2010.03.028 (2010).
Sperfeld, A.-D. et al. MR-pathologic comparison of the upper spinal cord in different motor neuron diseases. Eur. Neurol. 53, 74–77, https://doi.org/10.1159/000084650 (2005).
Losseff, N. A. et al. Spinal cord atrophy and disability in multiple sclerosis. A new reproducible and sensitive MRI method with potential to monitor disease progression. Brain 119(Pt 3), 701–708, https://doi.org/10.1093/brain/119.3.701 (1996).
Acknowledgements
We thank Anne-Katrin Baum, Department of Neurology, Otto-von-Guericke University, Magdeburg, Germany, for technical assistance, and Christa Sobetzko, Department of Neurology, Otto-von-Guericke University, Magdeburg, Germany, for data collection. “This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors”.
Author information
Authors and Affiliations
Contributions
T.W.: data analysis, data interpretation, study concept and design, drafting the manuscript. F.S.: data analysis, data interpretation, critical revision of the manuscript for important intellectual content. C.G.: acquisition of data. N.H., J.K., J.M., S.V., J.P., R.D., S.P., P.N. and H.J.H.: acquisition of data, critical revision of the manuscript for important intellectual content. P.N.: critical revision of the manuscript for important intellectual content. S.V.: acquisition of data, data interpretation, study concept and design, critical revision of the manuscript for important intellectual content, study supervision. S.S.: data analysis, data interpretation, study concept and design, critical revision of the manuscript for important intellectual content, study supervision. T.W. and S.S. had full access to all of the data of the study, and take responsibility for the integrity of the data and the accuracy of the data analysis.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wimmer, T., Schreiber, F., Hensiek, N. et al. The upper cervical spinal cord in ALS assessed by cross-sectional and longitudinal 3T MRI. Sci Rep 10, 1783 (2020). https://doi.org/10.1038/s41598-020-58687-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-58687-z
- Springer Nature Limited
This article is cited by
-
Association between brain and upper cervical spinal cord atrophy assessed by MRI and disease aggressiveness in amyotrophic lateral sclerosis
Neuroradiology (2023)
-
Analysis of brain and spinal MRI measures in a common domain to investigate directional neurodegeneration in motor neuron disease
Journal of Neurology (2023)