
Abstract
Universal drug susceptibility testing (DST) is an important requirement of the End TB Strategy. The Sensititre broth micro-dilution assay (BMD) tests multiple drugs quantitatively. We defined interpretive criteria for this assay and analysed genotypic-phenotypic relationships. 385 Mycobacterium tuberculosis clinical isolates were processed for BMD and whole genome sequencing. The epidemiological cut-off value 99% (ECV99) amongst genotypically wild type (gWT) strains defined susceptibility. Minimum inhibitory concentration distributions of the resistance-associated variants (RAVs) for each drug were analysed. Susceptibility (µg/mL) criteria were determined as follows: rifampicin (≤0.125), isoniazid (≤0.25), ethambutol (≤2.0), moxifloxacin (≤0.5), levofloxacin (≤1.0), amikacin (≤2.0), kanamycin (≤8.0), capreomycin (≤4.0), clofazimine (≤0.25) and linezolid (≤2.0). Most drugs showed clear separation between gWT and RAV. Isoniazid showed a tri-modal pattern with 14/17 strains at ECV99 harbouring a fabG1 c. -15C > T RAV. Ethambutol RAVs at embB codons 306, 405 and 497 were responsible for resistance and showed differential distributions. Moxifloxacin RAVs (gyrA codon 90) were a dilution or two higher than the ECV99 while gyrB RAVs were uncommon and showed drug specific resistance propensity. Interpretive criteria established were robust facilitating progress towards universal DST and individualised precision medicine. This study demonstrates the value of quantitative DST to accurately interpret mutation data.
Similar content being viewed by others


Introduction
Tuberculosis (TB) is a major contributor to morbidity and mortality globally and efforts to address this public health threat has shown positive signs of decline in recent years1. However, multi-drug resistant TB (MDR-TB) defined as TB with resistance to the core first line drugs, rifampicin and isoniazid, is forecasted to increase into the future2. This form of TB is more complex to manage and microbiological testing is required to determine the resistance profile and guide appropriate treatment regimens. Universal drug susceptibility testing (DST) is highlighted as an important component of the World Health Organizations (WHO) End TB strategy3. The increased availability of molecular tests that effectively detect resistance associated variants (RAVs) in the 81 bp hotspot of the rpoB gene which almost universally confers resistance to rifampicin has been very successful4. However, for other drugs, genetic targets and markers are less well defined and testing is not ideal for resistance determination.
Phenotypic drug susceptibility testing (pDST) in contrast provides a wide array of drugs that could be tested, however, such methods are often technically challenging. Testing at a single concentration recommended by the WHO, referred to as a critical concentration (CC)5 is the current approach. These CCs were established on less robust criteria and consensus. The use of epidemiological cut-off values (ECVs) are widely used in general microbiology and are preferred. Studies have highlighted the weaknesses of the CC based approach compared to the ECV based approach for Mycobacterium tuberculosis(Mtb)6 and the ECV criteria are now adopted by WHO7.
The limited arsenal of mycobacterial drugs available is driving an emerging need for pDST that could provide levels of resistance allowing continued use of certain drugs in cases of “borderline” or “low-level” resistance with dose adjustment8. In addition, drug regimens for TB have historically been established on available knowledge at the time, and lacked pharmacokinetic/pharmacodynamic (PK/PD) assessments that could reliably provide the optimal dosage for effect. Unfortunately, as combination therapy is mandatory for TB, clinical outcome data cannot easily be used to confirm the interpretive criteria.
The Sensititre MYCOTB is a commercial broth microdilution (BMD) assay for Mycobacterium tuberculosis MIC testing introduced by TREK Diagnostic Systems (forming part of the Thermo Fischer Scientific group). Multiple drugs are tested at a pre-defined concentration in a 96-well plate. Results are available 14–21 days from inoculation. This method is increasingly being used, and does have the potential to address gaps in achieving universal DST for Mtb. Unfortunately; a robust approach to determine break points for this method has not been established. Results are often interpreted against break points established for other methods including agar proportion and MGIT9,10,11. This is inappropriate and not suitable for clinical use. In addition, no interpretive criteria are provided in the package insert12. Furthermore, many of the Mtb ECV studies have not included this method6,13,14,15.
We undertook to determine the ECVs for the customized panel of anti-mycobacterial drugs, and evaluated these against known genotypic resistance determinants as a secondary validation.
Results
A total of 385 strains were analysable having MIC determined on BMD for all drugs and whole genome sequencing (WGS) data for the genetic targets of interest. The ECVs at 95%, 97.5% and 99.0% were determined for each drug and shown in Table 1, while the graphs used to derive these ECVs are shown in Supplementary Material (Figs. S1–S10). The ECVs at all three values were the same except for isoniazid, kanamycin and clofazimine.
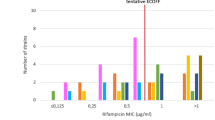
The distribution of MICs among strains with RAVs is shown in Fig. 1. Applying the ECV99 to strains with RAVs; the overlap was minimal for most drugs except isoniazid and clofazimine. The misclassification for these two drugs would be 7% and 11% respectively, while for all other drugs misclassification was 3% or lower (Table S1). For clofazimine, the number of resistant strains were low (n = 9) and one of these was misclassified (MIC = 0.12 µg/mL).

Overall distribution of MICs among RAVs for each drug. MIC: minimum inhibitory concentration. RAV: Resistance associated variant. Shaded area represents the testing range for each drug. The solid line in each distribution is the ECV99 that was derived by the ECOFFinder. The gWT distribution plot including raw count and fitted for each drug is shown in Supplementary Figs. 1–10. *Isoniazid RAVs: katG, inhA, fabG1, ahpC, ahpC promoter. Among the injectable drugs, 52 strains had a RAV (NC_000962.3: g.1473246A > G) and 1 strain had a RAV (NC_000962.3: g.1473247C > T). The MIC was >16 for amikacin and kanamycin while for capreomycin the distribution is shown in Fig. 3.
For rifampicin the MIC distribution of the RAVs were clearly separated with 95% (305/320) having an MIC > 4 µg/mL. Importantly, the ECV99 at 0.125 µg/mL correctly classified the majority of strains including the “disputed” rpoB RAVs (99%; 316/320). Drug resistance associated with these RAVs are often missed by the MGIT 960 (Becton Dickinson, USA), a broth based phenotypic method when tested at a CC of 1 µg/mL. Some have suggested that the CC of 1 µg/mL is too high, and have recommended this to be revised16.
There were 14/17 misclassified RAVs that had an MIC at the ECV99 for isoniazid (0.25 µg/mL) and harboured a RAV in the promoter for the fabG1-inhA-hemZ polycistronic operon (fabG1 promoter) fabG1 c. –15 known to confer low-level resistance (Fig. 1). In contrast all RAVs in katG were clearly separated having a modal MIC of 4–8 µg/mL, while those with both a katG and fabG1 promoter RAV had a modal MIC of 16 µg/mL or higher. There were 15 strains with an Ile194Thr RAV in the inhA gene and all were clearly in the resistant category, ranging from 0.5 µg/mL to 8 µg/mL.
Ethambutol (embB) resistance was due to a variety of RAVs in combination. The most dominant RAVs occurred at codons 306, 405 and 497 (Fig. 2), and some of their distributions were close to the ECV99 (2 µg/mL) i.e. one two fold dilution above the ECV99. These were observed at the following codons: 306 (9%;15/164), 497 (18%;2/11) and 405 (0%;0/17). In contrast, RAVs at codon 406 were commonly observed at MIC of 4 µg/mL (63%; 5/8).

Distribution of MICs and RAVs at specific positions for selected first line drugs. MIC: minimum inhibitory concentration. RAV: Resistance associated variant. Shaded area represents the drug resistant testing range for each drug. The solid line in each distribution is the ECV99 that was derived by the ECOFFinder. The gWT distribution plot including raw count and fitted for each drug is shown in Supplementary Figs. 1–10. Nucleotide and codons in bold represent the targets at specific positions included in WHO endorsed Genotype LPA first line assay.
Two fluoroquinolones were analysed, levofloxacin and moxifloxacin, both showed minimal misclassification applying the ECV99 of 1 µg/mL and 0.5 µg/mL respectively (Fig. 3). The modal MIC for RAVs at gyrA codon 94 for levofloxacin was >4 µg/mL, while for moxifloxacin it was 4 µg/mL. For RAVs at other codons the modal MIC was 4 µg/mL and 2 µg/mL respectively. For moxifloxacin, 5% (3/58) of RAVs at gyrA codon 94 had an MIC of one twofold dilution above the ECV99 with two of the three having a gyrA p.Asp94Ala RAV known to confer lower MIC. In contrast, at other hotspot codons, 33% (14/43) of RAVs had MICs of 2 µg/mL, being a dilution above the ECV99. When assessing the gyrB RAVs that were uncommon, there appeared to be a differential susceptibility pattern between the two drugs. RAVs at codon 501 were consistently resistant to moxifloxacin, while at codon 461 moxifloxacin was consistently susceptible, while levofloxacin was resistant.

Distribution of MICs and RAVs at specific positions for selected second line drugs. MIC: minimum inhibitory concentration. RAV: Resistance associated variant. Shaded area represents the drug resistant testing range for each drug. The solid line in each distribution is the ECV99 that was derived by the ECOFFinder. The gWT distribution plot including raw count and fitted for each drug is shown in Supplementary Figs. 1–10.
Amongst the second line injectable drugs, the rrs c. 1401 A > G RAV were common and conferred cross-resistance to all three drugs clearly separating from the gWT distribution (Figs. 1 and 3). For capreomycin, only 10% (5/52) of strains had an MIC > 16 while for the other two drugs it was 100% (52/52). Among RAVs which are known to confer low-level resistance to kanamycin, the eis promoter c. −14C > T RAV was identified in six strains, of which 2/6 had an MIC at the ECV99.
Linezolid strains with resistance were few (n = 3) and the ECV99 was appropriate, correctly classifying the strains (Fig. 1). Similarly, for clofazimine, the number of strains with resistance was also limited. Eight of nine were correctly classified, with one outlier having an MIC of 0.12 µg/mL. It should be noted that the Rv0678 gene encodes for an efflux pump and has previously been identified to demonstrate higher MICs in those with drug exposure17.
Discussion
General microbiology has for a long time used MIC testing as the standard method for pDST, however, this has been lacking for mycobacteriology. Although the Sensititre MYCOTB assay, a commercial BMD method has been available for many years, a robust study to define interpretative criteria for it has been lacking, limiting its broader utilization. This study has addressed this need applying CLSI standards to derive appropriate interpretive criteria, albeit using a customised panel of drugs. An additional validation was included, comparing the MIC distributions of gWT to known high confidence RAVs. This study, not only formally established criteria for both first and second line drugs for this method, but has also highlighted the important value of quantitative DST for Mtb. The RAV distributions for several gene-drug combinations have MICs close to the ECV99 necessitating quantitative DST. Additionally, some specific RAVs conferred either low or high level resistance, raising the potential value for using this method combined with PK/PD measurements to optimise therapies and improve patient outcomes.
A case in point has been ethambutol which is widely used in first line, and now in the WHO endorsed short course MDR-regimen. Reliability of testing this drug has been questioned when applying a single critical concentration6,13 and, therefore, pDST is usually not recommended5. Empiric use of this drug in the presence of high resistance levels and disabling side effects such as optic neuritis are concerning18. Testing for resistance is thus important to ensure patients are benefitted and not harmed. We observed overlapping distributions which explains reproducibility issues when performing DST for this drug. Using the quantitative DST approach resistance and susceptibility are clearly distinguished when not close to the ECV99. In the case of RAVs at embB codon 306, which were most common and known to confer resistance, 9% had MICs at the ECV99. Nonetheless, 91% would be clearly separated aiding appropriate clinical management. It is also interesting to note that strains with RAV at embB codon 406 commonly had MICs around the ECV99. Thus clinical studies could be targeted addressing this selection of patients to assess the clinical relevance, or alternatively the potential value of a higher dose in these cases, where ethambutol may serve as a useful oral option.
For isoniazid resistance, the distributions appeared tri-modal, split between combined inhA & fabG1 promoter, katG alone and katG & inhA & fabG1 promoter combination RAVs. Although molecular testing does provide an indication of low or high level RAVs, it is clear that RAVs with the fabG1promoter have a broad MIC distribution ranging between 0.25 µg/mL to 16 µg/mL, and having the actual MIC is valuable. Of interest, was the fabG1 c. −15C > T RAV, which is detected by WHO endorsed commercial assays, generally having lower MICs with 90% (26/29) having an MIC ≤ 1 µg/mL compared with the katG p. Ser315Thr RAV where only 1% (1/86) had an MIC ≤ 1 µg/mL. This has important value in decisions related to the use of high dose isoniazid for treating drug resistant TB as applied in the WHO recommendations19. Of note, was the 7% of the fabG1 promoter RAVs that fell within the gWT distribution and could potentially be treated with standard dose therapies avoiding dose related peripheral neuropathy. When restricting to the key RAVs included in commercial molecular assays, the sensitivity for molecular detection is 91.6% (230/251), which is consistent with a recent review of RAV data4,20. An important RAV that is not included in molecular assays is the inhA p. Ile194Thr which accounted for 6% (15/251) of resistance in this study, which, if incorporated could improve molecular test performance significantly (~97.6%).
The fluoroquinolones, levofloxacin and moxifloxacin both had ECV99 that separated the gWT and resistant populations making them appropriate for use. An interesting observation in this study was the relatively high proportion of strains with moxifloxacin MICs at gyrA codon 90 RAVs having MICs of 1–2 µg/mL while those with gyrA codon 94 RAVs had MICs of ≥4 µg/mL. The use of high dose moxifloxacin has been suggested to potentially treat strains with an MIC of 1–2 µg/mL in some patients21 and does offer hope to retain an important core second line drug especially when treatment options are limited. A second interesting observation was the differential susceptibility pattern among gyrB RAVs, though numbers were small and more data would be required to confirm this pattern. This does however raise the need to consider individual drug testing in selected patient groups and the added value of a 96 well plate BMD method, where, simultaneous multi-drug testing is simpler compared with agar or MGIT 960, thus facilitating universal quantitative DST.
Linezolid resistance is uncommon and only three genotypically resistant strains were observed in this study. The criteria established accurately separated susceptible and resistant strains. This drug has not been widely used, as it is expensive and does have a high adverse event profile22. This possibly explains the limited number of resistance isolates we found. Furthermore, acquired drug resistance to Linezolid has been shown to be prevented when using current recommended doses of 300 mg or 600 mg correlating with a susceptibility cut off value of 2 ug/mL23. As this drug has moved to category “A” forming the backbone of drug resistant TB regimens, greater vigilance for emergent resistance testing is needed, and the BMD is an appropriate method providing MIC data. This method would also be advantageous as the separation between genotypic resistant and gWT was distinct and monitoring gradual increases in MIC overtime would be important.
Clofazimine is a repurposed drug used for treatment of DR-TB and is now included as a category B drug by WHO to always be included where possible. The ECV99 ascertained was 0.25 µg/mL, which is consistent with what we previously published17, correctly classifying 8 of 9 RAVs in this study. The Rv0678 gene encodes for an efflux pump and thus variances in MIC are expected dependent on drug exposure. We have previously shown this to be the case for bedaquiline17 and may also explain the occurrence of the mutant with a clofazimine MIC in the WT range in this study. Although not described here, bedaquiline testing can be performed on the same plate and performs well, achieving universal DST with a single assay.
There have been several previous studies evaluating the standard format MYCOTB plate, but often these have been applied as comparisons with other methods while applying the criteria of the comparator for the BMD9,10,11. The resultant discordances particularly with strains close to the cut-off used, led Lee and colleagues to propose allowing a 1-dilution variance between methods24. This would be overcome if method specific criterion were applied following standardised approaches such as that provided by regulatory authorities and as used in this study. Another area of future research is applying the MICs derived from this assay in clinical cohorts with PK/PD sampling. It is encouraging to note that a clinical trial is planned25 aiming to address this gap, however, it is a single country study and multi-country data are still required. Additionally, triangulating the PK/PD findings with the ECV based methods to ascertain clinical breakpoints is what is ultimately required.
Our findings have provided a first step in standardised reporting criteria for the BMD method but needs to be seen in context of the specific limitations. This is a single country study and may not fully represent strains in other parts of the world. However, from reviews of MIC data using other methods conducted by WHO, the distributions have been similar26. Nonetheless, multi-country studies are required and these are currently underway. The strains that appeared to be outliers were not re-tested and although it would have been preferable, it does provide a more realistic routine scenario. In conclusion, the current study despite the noted limitations has provided robust validated criteria that will facilitate wider use of BMD as a method for quantitative pDST for Mtb. This will hopefully facilitate progress towards individualised precision medicine for TB and DR-TB. The value of quantitative pDST has also been demonstrated in this study to more accurately interpret mutation data and predict the potential range of MICs.
Methods
Mycobacterium tuberculosis strains which are broadly representative of strains circulating in South Africa were used. These strains were previously used for the determination of bedaquiline interpretive criteria on multiple methods including the commercial BMD plate17. In this study, we analysed the results of the other available anti-mycobacterial drugs on the plate which included: rifampicin (rif), isoniazid (inh), ethambutol (emb), ofloxacin (ofx), moxifloxacin (mxf), levofloxacin (levo), amikacin (ami), kanamycin (kana), capreomycin (cap), clofazimine (cfz) and linezolid (lzd), all key drugs for both first and second line treatment of Mtb. Of the 391 strains selected for testing, six had >3 drug inconsistencies between phenotypic and genotypic results and were excluded due to possible technical errors. Of the 385 strains with valid results, 68 (18%) were rifampicin susceptible, 317 (82%) were rifampicin resistant (RR) or MDR. Of the RR/MDR strains, 109 (34%) were pre-XDR/XDR, being MDR-TB with either fluoroquinolone or second line injectable resistance, or both, respectively. The strains represented the common lineages and were diverse (Fig. 4). The BMD plates were prepared and shipped frozen from the manufacturer in accordance with Food and Drug Agency (FDA) requirements.

Phylogenetic clustering of strains included in the study and associated lineages (N = 385).
The isolates were tested on BMD and had whole genome sequencing (WGS) performed as previously described17,27. In brief, WGS was performed using the MiSeq (Illumina, UK). Library preparation was performed using the Nextera-XT library preparation kit (Illumina, UK) and sequencing performed using the 2 × 300 bp MiSeq cartridge v.3 (Illumina, UK) with a target of 30×–50× paired coverage (~80–100X coverage). CLC Genomics Workbench 8.5.1(Qiagen, Venlo, The Netherlands) was used to detect RAVs using Reference mapping against the annotated reference genome H37Rv (NC00962.3) and the quality-based variant analysis tools where SNPs were filtered and qualified. Association of mutations as resistance predictors were primarily identified using the TB Drug Resistance Mutation Database (TBDReaMDB)28. If a mutation was not listed, literature, including newer published databases such as TBProfiler and PhyResSE, was surveyed to identify the association29,30. Mycobacterium tuberculosis lineages were assigned using the Single Nucleotide Polymorphism (SNP) barcode described by Coll et al.31. Individual SNP files for isolates were generated using CLC Genomics workbench v 8.5.1 (Qiagen, Venlo, The Nederlands). The SNPs for each genome were concatenated to an alignment and phylogeny inferred based on a comparison of SNP alignments of strains. SNP alignments were analysed using IQ-TREE (default settings) to generate a maximum-likelihood phylogenetic tree32. The output was visualized and lineage annotated with ITOL v.4.4.133.
The epidemiological cut off values (ECV) for all drugs were determined using the ECOFF finder34. For the ECV determination, we used strains that were genotypically wild type (gWT) considering well-known resistance associated variants (RAVs). A-priori an ECV of 99% (ECV99) was selected which is recommended by regulatory bodies (EUCAST and CLSI) and adopted by WHO recently. The ECV99 is the MIC value identifying the upper limit (99%) of the wild type population. The derived ECVs were evaluated for each drug and compared to the WGS data for the presence of mutations with known resistance associated variants (RAVs) and related MICs to validate the appropriateness of the ECV99 selected. The genetic targets for the following drug and gene combinations were analysed: rifampicin (rpoB); isoniazid (katG, inhA, ahpC, ahpC promoter and fabG1 promoter); ethambutol (embB); levofloxacin/moxifloxacin (gyrA and gyrB); amikacin/kanamycin/capreomycin (rrs), kanamycin (eis), linezolid (rplC and rrL), clofazimine (Rv0678). For clarity of interpretation, RAVs for a specific drug were analysed against strains with only a single gene having RAVs, e.g. strains with gyrA RAVs were analysed having no gyrB RAVs. However, for isoniazid, more than one RAV is frequent and we present distributions for both single and dual RAVs.
Data availability
The data presented in this manuscript are available in the European Nucleotide Archive under accession number: PRJEB25997.
References
WHO. Global TB Report, http://apps.who.int/iris/bitstream/handle/10665/274453/9789241565646-eng.pdf?ua=1 (2018).
Sharma, A. et al. Estimating the future burden of multidrug-resistant and extensively drug-resistant tuberculosis in India, the Philippines, Russia, and South Africa: a mathematical modelling study. Lancet Infect. Dis. 17, 707–715, https://doi.org/10.1016/S1473-3099(17)30247-5 (2017).
WHO. WHO END TB Strategy, http://www.who.int/tb/End_TB_brochure.pdf?ua=1 (2015).
Miotto, P., Zhang, Y., Cirillo, D. M. & Yam, W. C. Drug resistance mechanisms and drug susceptibility testing for tuberculosis. Respirol. 23, 1098–1113, https://doi.org/10.1111/resp.13393 (2018).
WHO. Technical manual for drug susceptibility testing of medicines used in the treatment of tuberculosis, https://www.who.int/tb/publications/2018/WHO_technical_drug_susceptibility_testing/en(2018).
Angeby, K., Jureen, P., Kahlmeter, G., Hoffner, S. E. & Schon, T. Challenging a dogma: antimicrobial susceptibility testing breakpoints for Mycobacterium tuberculosis. Bull. World Health Organ. 90, 693–698 (2012).
WHO. Technical report on critical concentrations for TB drug susceptibility testing of medicines used in the treatment of drug-resistant TB (2018).
Heysell, S. K. et al. Quantitative Drug-Susceptibility in Patients Treated for Multidrug-Resistant Tuberculosis in Bangladesh: Implications for Regimen Choice. PLOS ONE 10, e0116795, https://doi.org/10.1371/journal.pone.0116795 (2015).
Heysell, S. K. et al. Sensititre MycoTB plate compared to Bactec MGIT 960 for first- and second-line antituberculosis drug susceptibility testing in Tanzania: a call to operationalize MICs. Antimicrob. Agents Chemother. 59, 7104–7108, https://doi.org/10.1128/AAC.01117-15 (2015).
Hall, L. et al. Evaluation of the Sensititre MycoTB plate for susceptibility testing of the Mycobacterium tuberculosis complex against first- and second-line agents. J. Clin. Microbiol. 50, 3732–3734, https://doi.org/10.1128/JCM.02048-12 (2012).
Abuali, M. M., Katariwala, R. & LaBombardi, V. J. A comparison of the Sensititre(R) MYCOTB panel and the agar proportion method for the susceptibility testing of Mycobacterium tuberculosis. Eur. J. Clin. Microbiol. Infect. Dis. 31, 835–839, https://doi.org/10.1007/s10096-011-1382-z (2012).
Scientific, T. Sensititre™ Mycobacterium tuberculosis MYCOTB AST Plate, https://www.thermofisher.com/order/catalog/product/MYCOTB (2012).
Schon, T. et al. Evaluation of wild-type MIC distributions as a tool for determination of clinical breakpoints for Mycobacterium tuberculosis. J. Antimicrob. Chemother. 64, 786–793 (2009).
Schon, T. et al. Wild-type distributions of seven oral second-line drugs against Mycobacterium tuberculosis. Int. J. Tuberc. Lung Dis. 15, 502–509 (2011).
Jureen, P. et al. Wild-type MIC distributions for aminoglycoside and cyclic polypeptide antibiotics used for treatment of Mycobacterium tuberculosis infections. J. Clin. Microbiol. 48, 1853–1858 (2010).
Rigouts, L. et al. Rifampin resistance missed in automated liquid culture system for Mycobacterium tuberculosis isolates with specific rpoB mutations. J. Clin. Microbiol. 51, 2641–2645, https://doi.org/10.1128/JCM.02741-12 (2013).
Ismail, N. A. et al. Defining Bedaquiline Susceptibility, Resistance, Cross-Resistance and Associated Genetic Determinants: A Retrospective Cohort Study. EBioMedicine 28, 136–142, https://doi.org/10.1016/j.ebiom.2018.01.005 (2018).
Wang, M. Y. & Sadun, A. A. Drug-related mitochondrial optic neuropathies. J. Neuroophthalmol. 33, 172–178 (2013).
WHO. WHO consolidated guidelines on drug-resistant tuberculosis treatment. (2019).
WHO. The use of next-generation sequencing technologies for the detection of mutations associated with drug resistance in Mycobacterium tuberculosis complex: technical guide, http://apps.who.int/iris/handle/10665/274443 (2018).
Zvada, S. P. et al. Moxifloxacin population pharmacokinetics and model-based comparison of efficacy between moxifloxacin and ofloxacin in African patients. Antimicrobial agents chemotherapy 58, 503–510, https://doi.org/10.1128/AAC.01478-13 (2014).
Maartens, G. & Benson, C. A. Linezolid for Treating Tuberculosis: A Delicate Balancing Act. EBioMedicine 2, 1568–1569, https://doi.org/10.1016/j.ebiom.2015.10.014 (2015).
Srivastava, S. et al. Linezolid Dose That Maximizes Sterilizing Effect While Minimizing Toxicity and Resistance Emergence for Tuberculosis. Antimicrobial Agents Chemotherapy 61, e00751–00717, https://doi.org/10.1128/AAC.00751-17 (2017).
Lee, J. et al. Sensititre MYCOTB MIC plate for testing Mycobacterium tuberculosis susceptibility to first- and second-line drugs. Antimicrob. Agents Chemother. 58, 11–18, https://doi.org/10.1128/AAC.01209-13 (2014).
Davies Forsman, L. et al. Plasma concentrations of second-line antituberculosis drugs in relation to minimum inhibitory concentrations in multidrug-resistant tuberculosis patients in China: a study protocol of a prospective observational cohort study. BMJ Open. 8, e023899, https://doi.org/10.1136/bmjopen-2018-023899 (2018).
WHO. Technical report on critical concentrations for drug susceptibility testing of medicines used in the treatment of drug-resistant tuberculosis, http://apps.who.int/iris/handle/10665/260470 (2018).
Kaniga, K. et al. A Multilaboratory, Multicountry Study To Determine Bedaquiline MIC Quality Control Ranges for Phenotypic Drug Susceptibility Testing. J. Clin. microbiology 54, 2956–2962, https://doi.org/10.1128/JCM.01123-16 (2016).
Koser, C. U. et al. Whole-genome sequencing for rapid susceptibility testing of M. tuberculosis. N. Engl. J. Med. 369, 290–292 (2013).
Coll, F. et al. Rapid determination of anti-tuberculosis drug resistance from whole-genome sequences. Genome Med. 7, 51 (2015).
Feuerriegel, S. et al. PhyResSE: a Web Tool Delineating Mycobacterium tuberculosis Antibiotic Resistance and Lineage from Whole-Genome Sequencing Data. J. Clin. Microbiol. 53, 1908–1914 (2015).
Coll, F. et al. A robust SNP barcode for typing Mycobacterium tuberculosis complex strains. Nat. Commun. 5, 4812, https://doi.org/10.1038/ncomms5812 (2014).
Nguyen, L. T., Schmidt, H. A., von Haeseler, A. & Minh, B. Q. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274, https://doi.org/10.1093/molbev/msu300 (2015).
Letunic, I. & Bork, P. Interactive Tree Of Life (iTOL) v4: recent updates and new developments. Nucleic Acids Res, https://doi.org/10.1093/nar/gkz239 (2019).
Turnidge, J. & Paterson, D. L. Setting and revising antibacterial susceptibility breakpoints. Clin Microbiol Rev, 20, 391–408, table of contents (2007).
Acknowledgements
We acknowledge the diligence of staff at the Centre for Tuberculosis at the National Institute for Communicable Diseases who carried out the primary testing. Furthermore, we thank the clinical staff at the MDR initiation sites and also the MDR Advisory group for supporting the surveillance program. Janssen Pharmaceuticals provided funding for consumables used in the study and had no role in data collection, data analysis, or data interpretation of the report. All study procedures, data collection and analyses were independently conducted by the WHO Supranational TB Reference Laboratory at the Centre for Tuberculosis (CTB), the National Institute for Communicable Diseases (NICD) in Johannesburg, South Africa.
Author information
Authors and Affiliations
Contributions
N.A.I., K.K. and S.V.O. were involved in the conception and design of the study. N.A.I., S.V., L.J., N.G., L.B. and F.I. were involved in study implementation. N.A.I., S.V.O. and L.B. did the data analysis. N.A.I., F.I. and S.V.O. interpreted the data and provided important intellectual input. N.A.I., S.V.O. and F.I. wrote the first draft. All authors assure the accuracy and completeness of the data reported. The corresponding author had full access to all the data in the study and had final responsibility for the decision to submit for publication.
Corresponding author
Ethics declarations
Competing interests
K.K. is an employee of Janssen Pharmaceuticals. All other authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ismail, N.A., Ismail, F., Joseph, L. et al. Epidemiological cut-offs for Sensititre susceptibility testing of Mycobacterium tuberculosis: interpretive criteria cross validated with whole genome sequencing. Sci Rep 10, 1013 (2020). https://doi.org/10.1038/s41598-020-57992-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-57992-x
- Springer Nature Limited
This article is cited by
-
Evaluation of the broth microdilution plate methodology for susceptibility testing of Mycobacterium tuberculosis in Peru
BMC Infectious Diseases (2022)