
Abstract
Mycotoxins, such as aflatoxin B1 (AFB1), pose a serious threat as biological weapons due to their high toxicity, environmental stability, easy accessibility and lack of effective therapeutics. This study investigated if blood purification therapy with CytoSorb (CS) porous polymer beads could improve survival after a lethal aflatoxin dose (LD90). The effective treatment window and potential therapeutic mechanisms were also investigated. Sprague Dawley rats received a lethal dose of AFB1 (0.5–1.0 mg/kg) intravenously and hemoperfusion with a CS or Control device was initiated immediately, or after 30, 90, or 240-minute delays and conducted for 4 hours. The CS device removes AFB1 from circulation and significantly improves survival when initiated within 90 minutes of toxin administration. Treated subjects exhibited improved liver morphology and health scores. Changes in the levels of cytokines, leukocytes and platelets indicate a moderately-severe inflammatory response to acute toxin exposure. Quantitative proteomic analysis showed significant changes in the level of a broad spectrum of plasma proteins including serine protease/endopeptidase inhibitors, coagulation factors, complement proteins, carbonic anhydrases, and redox enzymes that ostensibly contribute to the therapeutic effect. Together, these results suggest that hemoadsorption with CS could be a viable countermeasure against acute mycotoxin exposure.
Similar content being viewed by others

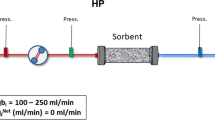
Introduction
Aflatoxins are toxic secondary fungal metabolites (mycotoxins) produced by fungus from the genus Aspergillus that cause severe acute reactions that can be lethal. Aspergillus species are important human pathogens and the toxic metabolites appear to act as virulence factors to suppress the immune system in invasive aspergillosis1. Aflatoxins cause damage to the liver resulting in hemorrhagic liver necrosis, steatosis, bile duct proliferation and subsequent organ failure and have been detected in pulmonary lesions of immune-compromised patients with systemic aspergillosis2. Aflatoxin B1 (AFB1), the most potent toxin of the 14 naturally occurring aflatoxin variants, is extremely cytotoxic, genotoxic, and carcinogenic3,4. In the liver, cytochrome P450-modified AFB1 forms DNA adducts that lead to impaired cellular function, carcinogenesis and/or cell death and organ failure5,6.
Acute aflatoxin poisoning from mold contaminated foods has been linked with numerous deaths in several instances7,8. Importantly, mycotoxins pose a serious threat as potential biowarfare agents due to their inherent stability and ease of manufacture. The toxins can be readily weaponized into aerosol form and dispersed over a wide area to elicit mass casualties through both inhalation and dermal exposure9. There have been several reported incidents of use of mycotoxins as bioweapons in Southeast Asia and the Gulf States10,11. Early symptoms of mycotoxin exposure in bio-warfare manifest rapidly in minutes to hours and can include burning, pain, wheezing, nausea, vomiting, tearing, weakness, bleeding and a host of other symptoms depending on the route of exposure making preparedness a critical element of any medical countermeasure. Extracorporeal removal methods, in conjunction with supportive care, have been employed to treat victims of acute intoxication with varying results12,13. Of note, a successful outcome was reported in a case of acute aflatoxicosis with fulminant hepatic failure and rhabdomyolysis case treated with hemodiafiltration14. Also, a case of amanita phalloides-induced liver failure was successfully treated with the Molecular Adsorbent Recirculating System (MARS)13. Nonetheless, the optimal approach is often unclear given the limited reports and the rapid distribution of many mycotoxins relative to medical presentation15.
Mycotoxin-induced inflammation and necrosis releases intracellular proteins, called damage associated molecular pattern proteins (DAMPs), that cause further tissue damage. A recent study demonstrated the rapid removal of AFB1 and T-2 toxin, as well as various DAMPs and cytokines from blood by CytoSorb® (CS; CytoSorbents Corporation, Monmouth Jct., NJ) porous polymer beads16, suggesting that hemoperfusion with CS could be useful in the treatment of acute mycotoxin exposure, such as might be predicted in a bioterrorist attack. In addition, intra-operative use of the device has been reported to reduce bleeding complications in patients who present for emergency cardiac surgery by the removal of the coagulation-active substances rivaroxaban and ticagrelor17. As such, it was of interest to evaluate if CS treatment could effectively mitigate the toxicity of an acutely lethal dose of AFB1 with systemic administration. Specifically, these studies were designed to determine if hemoperfusion with CS polymer beads could demonstrate in vivo AFB1 removal from circulation and ultimately improve survival of rats exposed to a lethal (LD90) dose of AFB1, to identify the effective treatment window through delayed hemoperfusion following AFB1 exposure, and lastly, to elucidate the impact of the hemoadsorption treatment on the plasma proteome.
Results
Aflatoxin in circulation
AFB1 concentration was measured in circulating plasma collected from the rats immediately after toxin injection and at various time points during and after hemoperfusion with Control and CS devices. Initial systemic AFB1 levels (T0) were not significantly different between Control and CS-treated groups in any of the treatment groups. Overall levels in the immediate, 30-minute, 90-minute, and 4-hour delayed treatment studies were 1464 ± 580 ng/mL, 1671 ± 743 ng/mL, 1579 ± 426 ng/mL, and 2703 ± 267 ng/mL, respectively.
The CS device directly removed AFB1 from the blood stream as the clearance rate was significantly faster in the CS-treated animals than the natural clearance seen in the Control group (Fig. 1). Five minutes after AFB1 administration, 63.5% of the initial circulating AFB1 remained in the blood of the Control group, whereas just 5.2% was detectable in that of the CS-treated group. During the later time points, aflatoxin levels post-CS device remained low (under 5% of initial levels). This contrasts with the aflatoxin levels in the Control group, which gradually decreased from 64% at 5 minutes to 17% at 30 minutes. Circulating aflatoxin concentrations were significantly lower in the CS-treated rats versus those of the Control group at 5, 10, 15, and 30 minutes post dosing (P < 0.03; Fig. 1). At 90 minutes, aflatoxin levels were almost identical between the two groups, dropping to 3.6% and 2.4% of initial in the Control and CS-treated groups, respectively (Fig. 1).

Circulating AFB1 Levels During Hemoperfusion. AFB1 levels measured by ELISA from plasma collected during hemoperfusion immediately after AFB1 IV administration (0.5 mg/kg). Hemoperfusion used either an empty (Control) or a CS device. Mean ± SD. n = 4 for both groups, *P < 0.05.
The 30-minute delay study had similar average initial AFB1 levels between Control and CS-treated groups (Supplementary Fig. 1A). At 1.5 hours post injection, AFB1 levels dropped 62-fold to 25.1 ± 6.4 ng/mL in the Control group and 149-fold to 11.9 ± 2.9 ng/mL in the CS-treated group, a statistically significant difference (P < 0.0001). By 4.5 hours post injection, AFB1 levels were essentially identical between the Control and CS-treated groups at 3.3 ng/mL (P = 0.97).
At the start of the 90-minute delayed hemoperfusion, AFB1 levels had decreased by >99% to 6.8 ± 1.4 ng/mL in the Control group and to 6.6 ± 2.4 ng/mL in the CS-treated group; (P = 0.89; Supplementary Fig. 1B). By the end of the hemoperfusion session at 5.5 hours, toxin levels had decreased further to 1.2 ± 0.4 ng/mL and 1.0 ± 0.4 ng/mL in the Control and CS-treated group, respectively (P = 0.52). After 24 hours, circulating toxin levels had decreased by 99.98% to 0.3 ± 0.1 ng/mL in both groups (P = 0.85).
With the 4-hour delay, blood AFB1 levels were <0.1% of initial dose levels at 1.9 ± 0.7 ng/mL and 1.4 ± 0.6 ng/mL in the Control and CS-treated group, respectively (P = 0.12; Supplementary Fig. 1C) and decreased to 0.7 ± 0.3 ng/mL in the Control group and 0.5 ± 0.3 ng/mL in the CS-treated group by the end of the hemoperfusion treatment (P = 0.17). By 24 hours, toxin levels declined to less than 0.01% of initial dose in all groups.
Survival
Preliminary dosing studies were used to adjust the IV dose of AFB1 to provide the targeted ~90% lethality within 3–4 days of dosing and ranged from 0.5–1.0 mg/kg among the immediate treatment, 30 and 90-min delayed treatment (0.5 mg/kg) and the 4-hour delayed treatment (1.0 mg/kg) studies.
Animals treated with the CS device immediately after toxin administration lived significantly longer than the Control animals (P = 0.025). Animals in the Control group survived an average of 3.6 days, which confirms that the AFB1 dose was appropriate for this study. Sixty percent of the CS-treated rats survived until study conclusion on day 7 with the others dying between day 2 and 6 (Fig. 2A), whereas only 12.5% of the Control animals survived to study conclusion with the other animals dying between day 1 and 6 (Fig. 2A).

Survival Plots. Effect of hemoperfusion on survival following a lethal AFB1 dose. Rats were dosed with AFB1 IV and subjected to hemoperfusion with a Control or a CS device for 4 hours either (A) immediately after toxin injection (0.5 mg/kg AFB1); n = 8 and 10 for Control and CS groups, respectively, *P < 0.025, (B) after a 30-min delay (0.5 mg/kg AFB1); n = 8 for both groups, *P < 0.003, (C) after a 90-min delay (0.5 mg/kg AFB1); n = 12 and 11 for Control and CS groups, respectively, *P < 0.04, or (D) after a 4-h delay (1 mg/kg AFB1); n = 8 for both groups.
The survival advantage remained when treatment was delayed by 30 minutes (Fig. 2B) and 90 minutes (Fig. 2C). During the 7-day observation period after the 30-min delayed treatment, none of the Control rats survived to day 7, while 75% of the CS-treated rats survived (Fig. 2B). CS treatment 90 minutes after toxin dosing increased survival from 66% with the Control-treated rats (Fig. 2C) to 100% of the CS-treated rats surviving to the end of the study period (P = 0.04).
When treatment was delayed 4 hours, 2 of 8 CS-treated rats survived to the end of the study period, compared to 0 of 8 Control rats (Fig. 2D), a nonsignificant improvement of 25% with CS treatment (P = 0.32).
Physical symptoms and weight loss
Animals treated immediately with CS displayed a marked reduction in physical symptoms of aflatoxicosis compared to Controls; rats were more active, had less chromodacryorrhea, and more normal posture and grooming habits (Supplementary Table S2). As expected, AFB1 exposure caused a loss of body weight in both the Control and CS-treated animals; however, the animals that survived the first few days began to regain the lost weight (Fig. 3A). Control group animals lost significantly more body weight than CS-treated rats by day 1 post-injection: 8.9 ± 2.7% vs. 6.2 ± 1.9% (P = 0.02). By day 2, Control rats lost 12.0 ± 1.9% body weight; significantly more weight loss than that experienced by CS-treated rats ̶ 10.0 ± 1.7% (P = 0.04). This trend continued through day 4, where the surviving Control rats had lost 13.8 ± 2.1% body weight compared to 11.2 ± 1.4% for CS-treated rats (P = 0.04). On the last day of survival, rats in the Control group had a significantly greater weight loss than CS-treated animals (P = 0.005).

Body Weights. AFB1 -induced body weight loss. Rats received 0.5 mg/kg AFB1 dose (IV) (immediate, 30-min, and 90-min delayed treatment) or 1 mg/kg dose (4-h delayed treatment) and after the indicated delay, were connected to a hemoperfusion circuit containing either a Control or a CS device. Mean ± SD, T0 n ≥ 8. Dashed line represents sole surviving control rat, *P < 0.05. Red line indicates threshold for euthanasia.
The Control animals in the 30-minute delayed treatment group exhibited clear signs of AFB1 poisoning, including lethargy, chromodacryorrhea, rough coat, and hypothermia. These symptoms were largely absent from the CS-treated rats (Supplementary Table S2). Control group animals lost significantly more body weight than CS-treated rats by day 1 post-injection: 9.1 ± 2.0% vs. 5.6 ± 1.6% P = 0.002). By day 2, Control rats lost 12.4 ± 1.0% body weight; significantly more weight loss than that experienced by CS-treated rats ̶ 8.3 ± 3.3% (P = 0.04) (Fig. 3B). By day 7, the surviving CS-treated rats had regained 20% of the weight lost during the study. Direct comparison of weight lost in CS-treated rats that received immediate treatment versus those that had a 30-minute delay of treatment showed no significant difference between the groups at any point during the study (P < 0.22).
Delaying treatment by 90 minutes also caused a reduction in chromodacryorrhea and improved grooming habits in CS-treated rats vs. Control rats (Supplementary Table S1). As expected, both Control and CS groups experienced significant weight loss (Fig. 3C). On day 1, Control rats lost 6.6% and CS rats lost 5.4% body weight, but there was no significant difference between the two groups (P = 0.71). On day 2, weight loss continued with both groups losing 10.4% body weight. The maximum average weight loss occurred on day 3 and was similar between Control and CS groups at 12.3% and 11.7%, respectively (P = 0.57). From day 4 through 7, both Control and CS-treated rats began to regain weight.
A 4-hour delay of treatment caused a reduction in toxin-mediated chromodacryorrhea and hypothermia but increased the incidence of lethargy in CS-treated vs. Control rats (Supplementary Table S2). Significant weight loss also occurred in this study, with Control and CS-treated groups losing similar amounts of weight. On day 1, Control rats lost 7.3% and CS-treated rats lost 5.7% body weight; however, there was no significant difference (P = 0.07). On day 2, rats continued to lose weight; Control rats lost 11.7% and CS-treated rats lost 11% (P = 0.58). Peak average weight loss occurred on day 4 and 5 for CS-treated rats (15.5%) and Control rats (15.7%), respectively. From day 4 through 7, the 2 surviving CS-treated rats were regaining weight (Fig. 3D).
Liver morphology
No gross lesions were noted upon necropsy in any of the rats dosed with AFB1. Histological analysis of liver sections collected at death revealed a distinct difference in the degree of AFB1-induced liver damage between the Control and CS-treated rats. Rats that died during the first 3 days had large hemorrhagic areas throughout the parenchyma with biliary hyperplasia and hepatocyte apoptosis and little to no liver regeneration (Fig. 4A,B). Rat livers were scored for key markers of toxin-induced damage; Control rat livers exhibited greater necrosis, inflammation, and hemorrhage than those of CS-treated rats (Fig. 5A and Supplementary Table S3). Biliary hyperplasia was higher in Control rat livers that survived through day 4; however, CS-treated animals that survived through day 7 exhibited greater hyperplasia than Control rats (Fig. 5A and Supplementary Table S3). Surviving CS-treated rats had significant regeneration of the liver, reduced inflammation, and residual tissue fibrosis (Fig. 5A and Supplementary Table S3). Rank order analysis of severity of tissue damage showed that, with one exception, livers from CS-treated rats had less toxin-associated lesions than those of the Controls.

Liver Pathology. Liver pathology caused by acute AFB1 intoxication. H&E stained liver sections from rats injected with AFB1 IV and treated with either a Control or CS device as indicated. Areas of severe hemorrhage and hepatocyte necrosis are visible in tissues at day 1. (A–D) On day 3–4, leukocyte infiltration is present. (G,H) Biliary hyperplasia is evident in the day 7 toxin-exposed animals (F,I,J). 100X mag.

Histological Scores. Scores of liver sections from AFB1 injected animals. Treatment with either a Control or CS device began immediately (A), after a 30-min delay (B), a 90-min delay (C), or a 4-h delay. (D) Dose: 0.5 mg/kg AFB1 for immediate, 30-min, and 90-min delayed treatments; 1 mg/kg for 4-h delayed treatment. Key markers of toxin-induced damage: necrosis, hemorrhage, inflammation, and hyperplasia were scored on increasing severity from 0 through 4, with 0 representing normal healthy tissue.
Delaying treatment for 30 minutes did not impact the therapeutic effects of CS treatment. Among animals that died on day 1, there was substantially more hemorrhaging and hepatocyte necrosis in livers of the Control group than in the CS-treated group (Figs. 4C,D, 5B and Supplementary Table S3). At day 7, the surviving CS-treated rats exhibited clear signs of hepatocyte regeneration in the parenchyma with modest inflammation; residual tissue fibrosis and biliary hyperplasia were also present (Fig. 5B and Supplementary Table S3).
Animals from the 90-minute delayed treatment study exhibited less overall liver damage, which agrees with the decreased mortality observed in the Control group. Control animals that died on day 3 had hemorrhaging and substantial leukocyte infiltration in the parenchyma (Fig. 5C and Supplementary Table S3). At day 7, liver regeneration had occurred in surviving Control and CS rats, with more inflammation present among Control animals. Again, moderate fibrosis and hyperplasia were evident around the portal triad (Figs. 4E,F, 5C and Supplementary Table S3).
The 4-hour delay of treatment animals experienced more severe liver damage than those of the 90-minute delay study, in congruence with the low survival outcome of both groups in this study. CS animals that died on day 2 had greater hemorrhage and necrosis than Control animals, but the difference was not significant. Control rats that died between day 3 and 4 had greater hemorrhage and necrosis, yet lower inflammation and hyperplasia than CS-treated animals; however, these differences were not significant (Figs. 4G,H, 5D and Supplementary Table S3). Animals that survived to day 5 to 7 had no detectable liver hemorrhage, minimal necrosis, substantial inflammation, and obvious biliary hyperplasia (Fig. 4I,J, 5D and Supplementary Table S3). The CS-treated animals appeared to have less severe lesions; however, differences were not significant.
Direct AFB1-induced hepatocyte apoptosis was assessed in rats that received 90-minute delayed treatment after a 1 mg/kg AFB1 dose using terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL). At 5.5 hours post-toxin dosing, there was substantial apoptosis visible in the parenchyma: 9.0 ± 2.4% and 9.3 ± 1.5% of liver cells in the Control and CS-treated rats were apoptotic, respectively, underscoring the rapid hepatotoxic effects of the AFB1 dose (Fig. 6).

Hepatocyte Apoptosis. AFB1-induced hepatocyte apoptosis in rats treated for 4 hours with either a Control or CS device 90-minutes after AFB1 dosing (1.0 mg/kg) then sacrificed. Liver sections were TUNEL stained to visualize apoptotic cells (stained brown) (A,B) or H&E-stained for gross morphology (C,D). 400X mag.
Circulating blood counts
Aflatoxicosis has been reported to cause lymphocytopenia and monocytopenia with increased neutrophil levels in certain species18, so changes in hematology were of interest in these studies. A complete blood count (CBC) was performed during the immediate and delayed treatment studies to analyze the direct AFB1 effects on the blood cell populations and any modulatory effects of CS treatment. Overall, AFB1 caused a 30–50% drop in white blood cell (WBC) numbers, which proceeded to rebound over the ensuing 24 hours, eventually reaching/surpassing baseline levels in all four studies. Except for immediate treatment, CS did not impact WBC counts. AFB1 had minimal effects on platelet and erythrocyte number, with the exception of the 30-min delayed treatment study, which showed a significant 46% decline in platelet numbers only in the Control group, and the 4-hour delayed treatment study, where erythrocyte number was significantly lower in the CS-treated group at 24 hours (Supplementary Table S4).
Cytokine response
AFB1 poisoning has been reported to cause extensive physiological effects from cellular damage, which can stimulate a widespread inflammatory response. As such, the impact of AFB1 administration on circulating cytokine levels as well as the influence of hemoperfusion on the cytokine response was assessed by immunoassay. Pro-inflammatory cytokines IL-6, IFN-γ, and TNF-α were elevated above baseline, peaking hours to days after AFB1 injection in both Control and CS-treated groups of the immediate, 30-minute and 90-minute delayed treatment studies (Fig. 7). IL-6 and IFN-γ were significantly elevated above baseline in the immediate, 30-minute and 90-minute delayed treatment studies, while TNF-α was significantly increased in the 30-minute delayed treatment study (Supplementary Tables S4–6). These changes were not significant in the case of the 4-hour delayed treatment study. IL-1α, IL-1β, IL-2, IL-4, IL-5, IL-10, IL-12, IL-13, and GM-CSF levels were also elevated over the course of the 7 days following AFB1 injection. IL-1α, IL-2, IL-4, IL-5, IL-10, IL-12 were significantly elevated in the immediate, 30-minute, and 90-minute studies, IL-1β and IL-13 were significantly elevated in the 30-minute and 90-minute studies, and GM-CSF was significantly increased above baseline in the 90-minute study (Supplementary Figs. S2–4 and Tables S4–6).

Cytokine Levels. Effect of AFB1 injection and subsequent hemoperfusion on levels of key circulating pro-inflammatory cytokines. AFB1 dose (mg/kg) was administered at time 0 with the hemoperfusion starting immediately (0.5 mg/kg), at 30 min (0.5 mg/kg), at 90 min (0.5 mg/kg), or at 4 hours (1.0 mg/kg). Mean ± SEM.
Modulation of protein levels
Since minimal AFB1 remains in circulation 90 minutes post-toxin dosing, identifying the role of direct adsorption or indirect removal of DAMPs and inflammatory proteins on the therapeutic mechanism responsible for the increased survival with CS treatment following AFB1 exposure (Fig. 2C) was of interest. Therefore, a comprehensive proteomic analysis of plasma samples from a 90-minute delayed treatment study following 1 mg/kg AFB1 dose was utilized to identify and quantify proteins involved in the toxin-mediated damage.
Over 300 unique proteins were unambiguously identified by liquid chromatography-mass spectrometry (LC-MS) proteomic analysis of the plasma samples. Examination of the samples 90 minutes following AFB1 administration, and prior to treatment, identified 34 proteins that were significantly altered in abundance (Supplementary Table S8) clustered around increases in complement proteins and serine protease inhibitors with decreased levels of hemoglobin subunits. Following hemoperfusion, at 5.5 hours post-toxin, 72 proteins were significantly altered from baseline in the Control animals, indicating further AFB1-induced changes (Supplementary Table S9). Serine protease inhibitors remained elevated with additional isoforms identified as well as significant increases in various coagulation factors. Hemoglobin subunits again were below baseline levels, as were a number of apolipoproteins. AFB1 induced significant changes in abundanc e of carbonic anhydrase 1 and 2 (CA1 and CA2) in the Control group after 1.5 hours with CA1 remaining significantly below baseline after 5.5 hours. AFB1 also impacted the abundance of two important redox enzymes, increasing superoxide dismutase 3 (SOD3) and decreasing peroxiredoxin-2 (PRDX2) levels at 1.5 hours. Examining the CS versus Control treatment effect (5.5 hours post-toxin), identified 109 proteins that were significantly different in abundance (Fig. 8 and Supplementary Table S10). Eight serine protease inhibitor isoforms were significantly decreased in the CS treated animals compared to Controls suggesting the importance of this group to the therapeutic effect. Levels of intrinsic coagulation factors and carboxypeptidase B2 (CPB2) are decreased in the CS treated animals compared to the Controls, while later stage common pathway factors remained elevated. Similarly, many complement proteins are decreased at 5.5 hours post-toxin in the CS compared to the Control animals where 8 different complement proteins were increased from baseline, suggesting another means of CS treatment may reduce toxin-mediated inflammation. CS treatment caused a significant increase in the abundance of both carbonic anhydrases when compared to the Control group at 5.5 hours post-toxin dosing. CS treatment resulted in increased PRDX2 and decreased SOD3 levels compared to those of the Control group. Additionally, treatment increased the level of prosaposin-isoform A (PSAP-1), glutathione peroxidase 1 (GPx-1), and glutathione peroxidase 3 (GPx-3).

Proteomic Analysis. Effect of CS treatment on AFB1 (1.0 mg/kg)-induced changes in circulating protein abundance at 5.5 hours (n = 8). Key proteins that were significantly different between Control and CS treatment (P < 0.05) are color coded by function: red, hemoglobin; black, pH and fluid homeostasis; orange, coagulation; purple, complement; green, protease inhibitor; blue, ROS detoxification; brown, heat shock. Mean difference (Control-CS) vs. statistical P-value at 5.5 h post-toxin dose following 4 hours of hemoperfusion.
Discussion
These studies demonstrate that treatment with the CS hemadsorption device removes AFB1 from the blood stream and significantly improves survival after exposure to an acutely lethal dose of AFB1 when treatment is initiated within 90 minutes of administration. The reduced scale device used in this study is based on the 300 mL CytoSorb® adsorber, a CE-Mark certified device approved for use as an extracorporeal cytokine adsorber. The indications for use have been expanded to include the adsorption of myoglobin19 and bilirubin20. The device has been shown to adsorb a number of small lipophilic molecules from blood in vitro including amitriptyline, diazepam, digoxin, quetiapine, and rivaroxaban21 as well as aflatoxin and T-2 toxin16, which prompted this investigation. Notably the device has been used alongside supportive care to treat intoxication by the serotonin and norepinephrine reuptake inhibitor, venlafaxine and the mushroom product psilocybin22,23, illustrating the translational use in treating acute toxin poisoning.
Treatment with the CS device remains just as effective in improving survival, liver morphology, and health scores when delayed by 30 or 90 minutes as when applied immediately after direct systemic administration of the toxin (Figs. 2–5; Supplementary Tables S2–3). The broad removal capabilities of the device suggest several possible, complementary mechanisms for the survival benefit observed with this acute mycotoxin poisoning model. Direct aflatoxin adsorption can readily explain at least part of the survival benefit observed when treatment is initiated within 30 minutes of AFB1 exposure as substantial levels of aflatoxin remain in circulation. However, the survival benefit extends beyond this time pointing to modulation of the plasma proteome as an additional therapeutic mechanism. CS has been shown to remove DAMPs16, including activated complement, indicating that the reduction of tissue damage associated and inflammatory mediators as a likely explanation for the survival benefit observed when treatment is initiated when nominal levels of toxin remain in circulation. A reduction in the primary and secondary release of DAMPs and inflammatory factors could be an additional mechanism to explain the treatment efficacy, beyond direct AFB1 adsorption. This is supported by the proteomic analysis that identified a broad spectrum of proteins from different biochemical pathways that were modulated by CS treatment. Five notable protein groups were influenced by the treatment: serine protease/endopeptidase inhibitors, coagulation factors, complement proteins, carbonic anhydrases, and redox enzymes (Fig. 8). The broad range of affected pathways is similar to proteomic surveys of acute-on-chronic liver failure, which causes sweeping changes in the plasma proteome24,25 and an in vitro model of mycotoxin-induced hepatocyte toxicity that found non-housekeeping pathway proteins to be affected most26.
AFB1 causes significant hemorrhage in the liver and intrinsic coagulation factors are fundamental to clot formation in response to vascular damage. However excessive clot formation can result in thrombosis, poor microvascular perfusion and systemic depletion of coagulation factors. A reduction in the level of several intrinsic coagulation factors and CPB2 could prevent excessive clotting and expedite existing clot fibrinolysis, possibly resulting in stabilization of hemostasis and protection of the endothelial layer. Furthermore, serine protease inhibitors help regulate proteolytic cascades that directly modulate coagulation, inflammation and immune responses, and tissue remodeling27 providing multiple potential beneficial mechanism(s) of action of their removal. The decrease in complement factors, which are vital components of the innate immune system that promote inflammation28, observed in the present study may reduce toxin-induced tissue necrosis and systemic inflammation as a potential therapeutic action of CS treatment.
Acutely toxic doses of AFB1 rapidly overwhelm endogenous redox enzyme-mediated detoxification, leading to widespread inflammation and release of DAMPS, amplifying the direct toxin effects29,30,31,32. Indeed, reactive aflatoxin metabolites can directly bind nucleic acids, disrupt protein synthesis and ultimately cause widespread cellular damage7,32,33. Consistent with prior reports34,35, elevated levels of TNF-α and IFN-γ were observed. Coupled with an excess of reactive oxygen species (ROS), TNF-α has been shown to cause hepatocyte apoptosis36,37,38. Although CS-treated rats had similarly elevated levels of TNF-α after AFB1 dosing as those of Control animals, there was markedly reduced liver damage (Fig. 5 and Supplementary Table S2), indicating an alternative to cytokine removal alone may explain the reduced tissue damage.
Carbonic anhydrases transport and remove dissolved carbon dioxide from the tissues and circulation; their depletion could result in decreased systemic pH as well as hyperammonemia, elevated lactate, metabolic acidosis and hypoglycemia. It is apparent that CS treatment abrogates the observed AFB1-mediated depletion of these enzymes, which could contribute to the overall therapeutic effect of the treatment. It is known that AFB1 detoxification causes elevated levels of ROS39,40,41. SOD directly converts superoxide into less-reactive oxygen species such as hydrogen peroxide and oxygen, whereas PRDX catalyzes the breakdown of hydrogen peroxide into water. While CS treatment moderately reduced the levels of SOD3, it caused a substantially larger increase in PRDX2. PRDX2 is a key component of the downstream ROS detoxification pathway39,40,41,42. Depending on the type of ROS present during CS treatment after AFB1 dosing, SOD3 may not be as critical as PRDX2 in preventing further tissue damage. Moreover, CS treatment also increased the level of GPx-1 and GPx-3, which directly catalyze the reduction of organic ROS and hydrogen peroxide by glutathione43. An increase in their protein abundance will likely result in decreased accumulation of ROS species, thereby protecting cells against further oxidative damage.
The changes observed in cytokines, WBC and platelet counts, are consistent with a moderately severe inflammatory response to acute toxin exposure. Furthermore, the rapid decline in WBCs after the single toxin dose parallels the effect seen in chronic AFB1 animal models14,18. However, unlike the consistently reduced WBC numbers observed with chronic toxin exposure, a single acute AFB1 dose causes a transient decline in WBCs followed by a rebound above baseline (Supplementary Table S3). This is supported by Marin 2002, who observed a biphasic response of decreasing and increasing WBC counts, with low and high toxin concentrations in piglets44. Immediate CS treatment may limit the subsequent toxin-induced increase of circulating WBCs; however, this effect is not observed when CS treatment is delayed by 30 or 90 minutes, limiting the generalizability of this observation.
Early hemoperfusion approaches to treat intoxication with modified charcoal have been reported to have undesirable effects on hemostasis and coagulation activity leading to their disuse45. In contrast, longer-term clinical studies (30- & 60-day) have established CS treatment to be safe and effective for a number of indications in critically ill patients46 easing general concerns about disturbances to physiologic homeostasis through untoward removal of beneficial substances, however close drug level monitoring is essential as some antibiotics and other drugs are known to be adsorbed47.
Overall, this study supports further investigation into the possibility of implementing CS treatment as a medical countermeasure against potential bio-warfare attacks with mycotoxins to lessen loss of life in military or civilian casualty situations if rapid deployment mechanisms are implemented. Consideration of such logistics is prudent, given the broad-spectrum nature of the device as a hemoadsorbent to address not only the initial toxin adsorption but the subsequent tissue damaging inflammatory response. This secondary mechanism is likely to have a greater influence in clinical practice as patients are typically treated for 24 hrs/day for several days with CytoSorb® and the rats were limited to a single 4-hour treatment in this study. The identification of a multitude of proteins from key physiologic pathways modulated by the treatment may potentially aide future studies evaluating treatment responses for a number of indications such as acute liver failure and invasive aspergillosis.
Material and Methods
Animal treatment
Eighty-one Sprague Dawley rats (350–450 g) cannulated in the jugular and femoral veins (Taconic Labs) were anesthetized with 1.5% isoflurane and injected with 0.5–1.0 mg/kg of AFB1 (1.4 mg/mL solution- 44:56 (v/v) DMF:saline solution) intravenously via the tail vein48,49,50. Hemoperfusion was initiated immediately or after a 30, 90, or 240-minute delay and conducted for 4 hours at a flow rate of 1.5 mL/min from the jugular vein through a device containing 3 mL CS polymer beads or an empty device (Control) and back into the rat through the femoral vein catheter. Humane endpoints for euthanasia were included (i.e., inability to eat and drink for 2 days, loss of >15% body weight or >6 °C drop in body temperature). Heparin was dosed at 200 IU/kg/hour during the hemoperfusion and proper heparinization levels monitored by ACT with a Vetscan i-STAT (Abaxis). Rats were observed for a 7-day period following AFB1 exposure with body weight and temperature recorded daily.
Analytical methods
Blood was collected at specific times prior to, during, and following the hemoperfusion session. Hematological analysis was conducted with a Hemavet analyzer (Drew Scientific). Circulating AFB1 and cytokines were measured by ELISA (Helica Biosystems, Santa Ana, CA) and Multi-plex immunoassay (Bio-Plex Th1/Th2 Rat Cytokine Assay Kit; Bio-Rad Laboratories), respectively.
Histology
At death, livers were preserved in 10% neutral buffered formalin (NBF). Liver tissue sections were stained with haematoxylin and eosin dyes (H&E) and scored for key hepatotoxicity markers: hemorrhage, necrosis, biliary hyperplasia, and inflammation.
Proteomic analysis
For the 90-min delayed treatment proteomic study: blood samples were collected 30 min (baseline) prior to AFB1 dose (1 mg/kg IV) and immediately before and after the 4-hour hemoperfusion. The plasma was frozen and shipped to MS Bioworks (Ann Arbor, MI) for proteomic analysis via liquid chromatography-mass spectrometry (LC-MS). Plasma samples (10 µL) were depleted of the most abundant proteins using Proteome Purify 2 Mouse Serum Protein Immunodepletion Resin (R&D Systems, Catalog no. MIDR002-020) according to manufacturer’s protocol. Depleted samples were buffer exchanged into water on a Corning Spin × 5 kDa molecular weight cut off spin column and quantified by Qubit fluorometry (Life Technologies). 50 μg of each sample was reduced with dithiothreitol, alkylated with iodoacetamide and digested overnight with trypsin (Promega). Each digested sample was processed by solid phase extraction and 3 μg of each digest was analyzed by nano LC-MS/MS with a Waters M-Class NanoAcquity HPLC system interfaced to a ThermoFisher Fusion Lumos mass spectrometer for 3 hrs per sample. Raw label-free quantitation (LFQ) protein intensity values were Log2 transformed prior to calculating percent change difference and statistical analysis.
Statistical analysis
Statistical analyses between individual data points and between survival plots was done with student’s two tailed t-test and log-rank (Mantel-Cox) test, respectively, with alpha = 5.000% (GraphPad Prism).
Significance statement
Mycotoxins continue to pose a serious threat as biological weapons due to their high systemic toxicity, environmental stability and ready accessibility. With the ongoing and unpredictable threat from terror groups, there is an acute need for an effective medical countermeasure. This study used extracorporeal blood purification with highly porous polymer beads to demonstrate an effective therapeutic method to reduce toxicity and mortality following administration of a lethal dose of aflatoxin in an animal model. Comprehensive evaluation of potential mechanisms indicates the device works through a combination of direct toxin adsorption and modulation of the levels of reactive proteins in the plasma from the aflatoxin-mediated inflammation and tissue damage.
Ethics statement
All animal work was done in accordance with protocols approved by the Institutional Animal Care and Use Committee at Rutgers University and the US Army Animal Care and Use Review Office.
Data availability
The authors agree to policies on data sharing and materials. The mass spectrometry proteomics data generated during the current study have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE partner repository with the dataset identifier PXD015442.
References
Kosalec, I. & Pepeljnnak, S. Mycotoxigenicity of clinical and environmental Aspergillus fumigatus and A. flavus isolates. Acta Pharm. 55, 365–375 (2005).
Matsumura, M. & Mori, T. Detection of Aflatoxins in Autopsied Materials from a Patient Infected with Aspergillus flavus. Jpn. J. Med. Mycol. 39, 167–171 (1998).
Bedard, L. L. & Massey, T. E. Aflatoxin B1-induced damage and its repair. Cancer Lett. 241, 174–183 (2006).
Bennett, J. W. & Klich, M. Mycotoxins. Clin. Microbiology Rev. 16(3), 497–516 (2003).
Croy, R. G. & Wogan, G. N. Temporal Patterns of Covalent DNA Adducts in Rat Liver after Single and Multiple Doses of Aflatoxin B1. Cancer Research. 41, 197–203 (1981).
Wang, T. V. & Cerutti, P. A. Formation and removal of aflatoxin B1-induced DNA lesions in epithelioid human lung cells. Cancer Res. 39, 5165–5170 A (1979).
Bbosa, G. S. et al. Review of the biological and health effects of aflatoxins on body organs and body systems: Aflatoxins—Recent advances and future prospects. Intechopen Publisher 12, 239–265 (2013).
Woods, J. B. USAMRIID’s Medical Management of Biological Casualties Handbook (6th Ed, April 2005). Fort Detrick, MD. BlueBook 6th Edition - Sep 2006.pdf Downloaded from, http://www.usamriid.army.mil/education/bluebookpdf/USAMRIID (2006).
Boonen, J. et al. Human skin penetration of selected model mycotoxins. Toxicology. 301(1–3), 21–32 (2012).
Wannemacher, R. W. & Wiener, S. L. Trichothecene mycotoxins: in Medical Aspects of Chemical and Biological Warfare. USArmy Medical Department. (1997).
Zilinskas, R. A. Iraq’s Biological Weapons: The Past as Future? J. Am. Med. Assoc. 278(418–424), 60 (1997).
De Pont, A. C. Extracorporeal treatment of intoxications. Curr. Opin. Crit. Care 13, 668–673 (2007).
Lionte, C., Sorodoc, L. & Simionescu, V. Successful treatment of an adult with Amanita phalloides-induced fulminant liver failure with molecular adsorbent recirculating system (MARS). Rom. J. Gastroenterol. 14(3), 267–71 (2005).
Samuel, N. et al. Acute Aflatoxicosis Resulting in Fulminant Hepatic Failure and Rhabdomyolysis. Gastroenterology. Research 2, 48–50 (2009).
Stankewicz, R., Lewandowski, Z., Kotulski, M., Patkowski, W. & Krawczyk, M. Effectiveness of Fractionated Plasma Separation and Absorption as a Treatment for Amanita Phalloides Poisoning. Ann. Transpl. 21, 428–432 (2016).
Gruda, M. C. et al. Broad adsorption of sepsis related PAMP and DAMP molecules, mycotoxins, and cytokines from whole blood using CytoSorb® sorbent porous polymer beads. PLoS One. 13(1), e0191676 (2018).
Hassan, K. et al. CytoSorb Adsorption During Emergency Cardiac Operations in Patients at High Risk of Bleeding. Ann. Thorac. Surg. 108(1), 45–51 (2019).
Donmez, N., Donmez, H., Keskin, E. & Kisadere, I. Effects of Aflatoxin on Some Haematological Paramaters and Protective Effectiveness of Esterified Glucomannan in Merino Rams. Scientific World Journal. Artic. ID 342468, 4 (2012).
Kuntsevich, V. I. et al. In-Vitro Myoglobin Clearance by a Novel Sorbent System. Artif. Cell Blood Substit. Immobil. Biotechnol. 37(1), 45–7 (2009).
Calabro, M. G. et al. Blood Purification with CytoSorb in Critically Ill Patients: Single-Center Preliminary Experience. Artif. Organs 43(2), 189–94 (2019).
Körtge, A., Mitzner, S. & Wasserkort, R. Removal capability of CytoSorb hemoadsortion columns for selected prescription drugs frequently related to drug overdose. 44th ESAO and 7th IFAO Congress. Vienna, Austria (2017).
Schroeder, I., Zoller, M., Angstwurm, M., Kur, F. & Frey, L. Venlafaxine intoxication with development of takotsubo cardiomyopathy: successful use of extracorporeal life support, intravenous lipid emulsion, and CytoSorb. Int. J. Artif. Organs 40(7), 358–360 (2017).
Schnitzler, S., Lange, T. & Nguyen H. Use of CytoSorb in potential intoxication with Psilocybe cubensis. C.O.W, https://literature.cytosorb-therapy.com/ (2019).
Sun, Z. et al. Circulating proteomic panels for diagnosis and risk stratification of acute-on-chronic liver failure in patients with viral hepatitis B. Theranostics 9, 1200–1214 (2019).
Zhou, N. et al. Discovery of a Potential Plasma Protein Biomarker Panel for Acute-on-Chronic Liver Failure Induced by Hepatitis B Virus. Frontiers in Physiology. 8:Article 1009 (2017).
Sun, Y., Wen, J., Chen, R. & Deng, Y. Variable protein homeostasis in housekeeping and nonhousekeeping pathways under mycotoxins stress. Sci. Rep. 9, 7819, https://doi.org/10.1038/s41598-019-44305-0 (2019).
Law, R. H. et al. An overview of the serpin superfamily. Genome Biology. 7(5), 216 (2006).
Janeway, C. A. et al. The complement system and innate immunity. Immunobiology: The Immune System in Health and Disease. 5th Ed, Ch 2. New York: Garland Science (2001).
Singh, K. B., Maurya, B. K. & Trigun, S. K. Activation of oxidative stress and inflammatory factors could account for histopathological progression of aflatoxin B1 induced hepatocarcinogenesis in rat. Mol. Cell Biochem. 401(1–2), 185–196 (2015).
Cancado et al. Glutathione S-transferase and aluminum toxicity in maize. Funct. Plant. Biol. 32, 1045–1055 (2005).
Cotgreave, I. A. & Gerdes, R. G. Recent trends in glutathione biochemistry glutathione-protein interactions: a molecular link between oxidative stress and cell proliferation? Biochem. Biophys. Res. Commun. 242, 1–9 (1998).
Nagai, H., Matsumaru, K., Feng, G. & Kaplowitz, N. Reduced glutathione depletion causes necrosis and sensitization to tumor necrosis factor-α-induced apoptosis in cultured mouse hepatocytes. Hepatology 36, 55–64 (2002).
Sarasin, A., Moule, Y. & Darracq, N. In vivo effect of aflatoxin B1 on protein synthesis in rat liver. FEBS Lett. 29(Issue 3), 329–332 (1973).
Ishikawa, A. T. et al. Impact of a Single Oral Acute Dose of Aflatoxin B1 on Liver Function/Cytokines and the Lymphoproliferative Response in C57Bl/6 Mice. Toxins 9(11), E374, https://doi.org/10.3390/toxins9110374 (2017).
Qian, G. et al. Aflatoxin B1 modulates the expression of phenotypic markers and cytokines by splenic lymphocytes of male F344 rats. J. Appl. Toxicol. 34(3), 241–9 (2014).
Pierce, R. H. et al. Disruption of redox homeostasis in tumor necrosis factor-induced apoptosis in a murine hepatocyte cell line. Am. J. Pathol. 157, 221–236 (2000).
Han, D., Hanawa, N., Saberi, B. & Kaplowitz, N. Hydrogen peroxide and redox modulation sensitize primary mouse hepatocytes to TNF-induced apoptosis. Free. Radic. Biol. Med. 41, 627–639 (2006).
Guengerich, F. P., Arneson, K. O., Williams, K. M., Deng, Z. & Harris, T. M. Reaction of aflatoxin B1 oxidation products with lysine. Chem. Res. Toxicol. 15, 780–792 (2002).
Josse, R., Dumont, J., Fautre, A., Robin, M. & Guillouzo, A. Identification of early target genes of aflatoxin B1 in human hepatocytes, inter-individual variability and comparison with other genotoxic compounds. Toxicol. Appl. Pharmacology 258, 176–187 (2012).
Shen, H. M., Shi, C. Y., Shen, Y. & Ong, C. N. Detection of elevated reactive oxygen species level in cultured rat hepatocytes treated with aflatoxin B1. Free. Radic. Biol. Medicine. 21, 139–146 (1996).
Peskin, A. V. et al. The high reactivity of peroxiredoxin 2 with H2O2 is not reflected in its reaction with other oxidants and thiol reagents. J. Biol. Chem. 282, 11885–11892 (2007).
Berggren et al. Thioredoxin peroxidase-1 (peroxiredoxin-1) is increased in thioredoxin-1 transfected cells and results in enhanced protection against apoptosis caused by hydrogen peroxide but not by other agents including dexamethasone, etoposide, and doxorubicin. Arch. Biochem. Biophys. 392, 103–109 (2001).
Ziglari, T. & Allameh, A. The significance of glutathione conjugation in aflatoxin metabolism. Intech. Chapter 13 (2013).
Marin, D. et al. Changes in performance, blood parameters, humoral and cellular immune responses in weanling piglets exposed to low doses of aflatoxins. J. Anim. Science. 80(5), 1250–7 (2002).
Park, S. et al. Hemoperfusion leads to impairment in hemostasis and coagulation process in patients with acute pesticide intoxication. Sci. Reports. 9, 13325 (2019).
Ankawi, G. et al. What have we learned about the use of CytoSorb adsorption columns? Blood Purif. 48, 196–202 (2019).
Reiter, K. et al. In vitro removal of therapeutic drugs with a novel adsorbent system. Blood Purif. 20(4), 380–8 (2002).
Kumagai, S. et al. The fate and acute toxicity of aflatoxin B1 in the mastomys and rat. Toxicon. 36(1), 179–88 (1997).
Pong, R. S. & Wogan, G. N. Toxicity and Biochemical and Fine Structural Effects of Synthetic Aflatoxins M1 and B1 in Rat Liver. Journal of the National Cancer Institute 47(3) (1971).
Wong, Z. & Hsieh, D. The comparative metabolism and toxicokinetics of aflatoxin B1 in the monkey, rat, and mouse. Toxicol. Appl. Pharmacology. 55, 115–125 (1980).
Acknowledgements
This work was funded by the U.S. Joint Program Executive Office for Chemical and Biological Defense (JPEO-CBD) under contract W911QY-17-C-0007.
Author information
Authors and Affiliations
Contributions
K.G.R., P.O.S. and T.J.K. carried out the experiments and analysis of the results. K.D. performed statistical analysis. V.J.C., P.P.C. and T.D.G. participated in conception and coordination of the study. K.G.R. and M.C.G. designed the study, analyzed the results and wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
K.G.R., P.O.S., T.J.K., K.D., V.J.C., P.P.C., T.D.G. and M.C.G. are all employed by CytoSorbents Medical which produces the CytoSorb device and receive salary and stock options.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ruggeberg, KG., O’Sullivan, P., Kovacs, T.J. et al. Hemoadsorption Improves Survival of Rats Exposed to an Acutely Lethal Dose of Aflatoxin B1. Sci Rep 10, 799 (2020). https://doi.org/10.1038/s41598-020-57727-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-57727-y
- Springer Nature Limited