
Abstract
The microbial reduction of nitrate, via nitrite into gaseous di-nitrogen (denitrification) plays a major role in nitrogen removal from aquatic ecosystems. Natural abundance stable isotope measurements can reveal insights into the dynamics of production and consumption of nitrite during denitrification. In this study, batch experiments with environmental bacterial communities were used to investigate variations of concentrations and isotope compositions of both nitrite and nitrate under anoxic conditions. To this end, denitrification experiments were carried out with nitrite or nitrate as sole electron acceptors at two substrate levels respectively. For experiments with nitrate as substrate, where the intermediate compound nitrite is both substrate and product of denitrification, calculations of the extent of isotope fractionation were conducted using a non-steady state model capable of tracing chemical and isotope kinetics during denitrification. This study showed that nitrogen isotope fractionation was lower during the use of nitrite as substrate (ε = −4.2 and −4.5‰ for both treatments) as compared to experiments where nitrite was produced as an intermediate during nitrate reduction (ε = −10 and −15‰ for both treatments). This discrepancy might be due to isotopic fractionation within the membrane of denitrifiers. Moreover, our results confirmed previously observed rapid biotic oxygen isotope exchange between nitrite and water.
Similar content being viewed by others

Introduction
The growing world population requires an increased food production, necessitating the intensive use of nitrogen-containing synthetic fertilizers, which has led to nitrate pollution in many surface water and groundwater bodies1,2,3,4,5. Even though essential for plants, dissolved nitrate can be responsible for contamination of drinking water supplies from surface and groundwaters, posing a potential threat to human health6,7,8,9,10. Fortunately, it has been shown that denitrification plays a significant role in the mitigation of nitrate pollution11,12,13. Denitrification is the microbial dissimilatory reduction of NO3− to N2 via several steps, governed by different enzymes14,15,16. During this beneficial removal of NO3−, the intermediates nitrite (NO2−), a toxic component at low concentrations17, and nitrous oxide (N2O), which participates in ozone layer destruction18, are formed. Whereas the production of N2O has received considerable attention due to its role as a potent greenhouse gas, the intermediate compound nitrite (NO2−) has received much less attention since it is often assumed to be thermodynamically unstable, short-lived, and therefore not accumulating in the environment. However, nitrite has been detected at concentrations exceeding the European Water Framework Directive (EU WFD) of 0.009 mg N L−1 in urbanized rivers19,20,21,22,23. Therefore, a better understanding of its dynamics in aquatic systems is highly desirable.
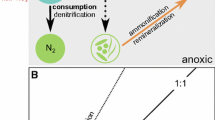
During denitrification, nitrite is both the product of nitrate reduction and the substrate of its own reduction into NO, N2O and N2 (Fig. 1):

Theoretical trends of NO3− and NO2− concentrations during denitrification.
Nitrate reduction initially results in nitrite production. During phase 1, the “net nitrite production” phase, the NO2− concentration rises and reaches a maximum; then, in phase 2, the concentration of nitrite decreases until nitrite is completely consumed. It is noted that during phase 1 production as well as reduction of NO2− can take place simultaneously. The same holds for phase 2, where nitrite is mostly reduced but also still being produced. In addition of being an intermediate during denitrification, nitrite is also an important intermediate at the crossroads of other nitrogen transformation processes, such as codenitrification24,25, dissimilatory nitrate reduction to ammonium (DNRA)26,27,28, anaerobic ammonium oxidation (Anammox)29, nitrifier denitrification30 and nitrification31,32.
Stable isotope techniques can provide unique insights into sources and processes regulating the N cycle in ecosystems. The nitrogen and oxygen isotope ratios of NO3− (δ15N and δ18O) have been used since several decades to better distinguish the origin of NO3− 33,34,35,36. In addition, it provides information regarding the processes involved in its transformation, e.g. denitrification and nitrification in lakes, groundwater, riparian zones, rivers, soils, and marine environments37,38,39,40,41,42,43. Denitrification is accompanied by significant N and O isotope fractionation affecting the remaining NO3− due to reaction kinetics differences between the lighter and the heavier isotopes of N and O (14N vs. 15N and 16O vs. 18O). Enrichment in heavy isotopes of N and O in the remaining nitrate is caused by the fact that 14N and 16O react faster than 15N and 18O during denitrification33,44.
Denitrification is a unidirectional reaction in which nitrate is reduced to gaseous nitrogen (N2) via different intermediates (NO2−, NO, and N2O). The N and O isotope enrichment factors ε15N and ε18O during denitrification are usually determined by measuring variations in concentrations and isotope ratios of the remaining nitrate, while the isotopic compositions of the final product N2 or of the intermediate compounds such as NO2− and N2O are rarely analyzed. In past studies, the concentrations and isotopic compositions of nitrate and nitrite have not always been separately determined due to analytical limitations for the latter. Therefore, the “net ε15N” and “net ε18O” determined for the entire denitrification process assessed solely based on measurements of the progressively disappearing nitrate may not always be accurate.
Only a few studies used nitrite (NO2−) rather than nitrate as source of nitrogen for the determination of ε15N for nitrite reduction. Two of these studies were carried out in soil experiments45,46 another with a microbial culture43. Due to new chemical reduction analytical methods, it is now possible to determine the isotopic composition of NO2− alone, separating the isotopic analyses of NO3− from those of coexisting NO2− 47,48,49,50. Recent studies of the isotopic composition of nitrite focused mainly on nitrification51,52,53,54,55,56. A few studies mention ε15N values for nitrite reduction during denitrification. These studies were conducted using pure cultures of denitrifiers41,42,43, marine incubations42, in-situ marine environments55 or more recently abiotic denitrification57. In order to improve our knowledge on the isotope effects affecting nitrite, we investigated isotope fractionation during denitrification in laboratory experiments with environmental bacterial communities. Nitrite was either applied as a substrate or analyzed as intermediate during nitrate reduction.
Since during denitrification and nitrate consumption nitrite is both substrate and product, it is not possible to use the Rayleigh equation to calculate concentrations and isotope compositions of the intermediate compound nitrite. For this reason, we developed the Isonitrite numerical model that simulates the evolution of oxygen and nitrogen isotope compositions of NO3− and NO2− during simultaneous nitrite production and consumption. The model, constrained by our experimental data, is used to estimate isotope fractionation factors of oxygen and nitrogen (ε15N and ε18O) associated to denitrification. To our knowledge, this study is the first focusing on freshwater benthic denitrifying communities with two distinct sources of dissolved inorganic nitrogen, i.e, nitrite or nitrate.
Results
Isotopic enrichment factors associated with nitrite reduction
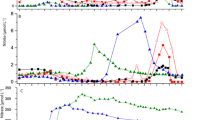
In the low nitrite experiment (101 µM), concentrations decreased with time reaching 9.7 µM after 240 min (Fig. 2a). In the high nitrite experiment (614 µM), concentrations decreased continuously to 0 µM after 390 min (Fig. 2b). Nitrite reduction rates of 119 nmol min−1 g−1 were observed at high initial nitrite concentrations whereas the low initial nitrite concentration resulted in lower nitrite reduction rates of 35 nmol min−1 g−1 under the experimental conditions.

Kinetics of nitrite concentrations (a,b), variations of δ15N (c,d) and calculation of ε15N (e,f) for low and high nitrite treatments respectively.
The decrease in nitrite concentrations was associated with a change in δ15N-NO2− of the remaining NO2− increasing from −18.1‰ to −8.4‰ and from −19.6‰ to −17.6‰ for low and high treatments respectively (Fig. 2c,d). The N isotope enrichment factors associated with the reduction of NO2− was −4.2 and −4.7‰ for low and high nitrite treatments respectively (Fig. 2e,f).
The reduction of nitrite generated constant and low oxygen isotope values in the remaining nitrite (Fig. 3a,b). The oxygen isotope ratios of nitrite remained constant throughout the experiment with mean δ18O-NO2− of −0.6 ± 1.1‰ and −0.9 ± 0.6‰ for low and high nitrite treatments respectively.

Variations of δ18O-NO2− with the decrease of nitrite concentration for low (a) and high (b) nitrite treatments.
Isotopic enrichment factor associated with nitrate reduction
Similar to the experiments carried out with nitrite only, the kinetics of denitrification were investigated with solutions with low (97 µM) and high (508 µM) initial nitrate concentrations (Fig. 4b). Nitrate concentrations decreased from 97 to 1.8 µM and from 508 to 1.2 µM over the 180 and 420 minutes of the experiments. The rates of nitrate reduction were 50 nmol g−1 min−1 and 137 nmol g−1 min−1 for low and high nitrate treatments respectively. The nitrate reduction rates are in the same range as the nitrite reduction rates (35 and 119 nmol g−1 min−1, see previous section).

Variations of nitrite (open symbol) and nitrate (filled symbol) concentrations (a,b), variations of δ15N-NO3− (c,d) and of δ18O-NO3− (e,f) for low and high nitrate treatments respectively.
In both nitrate experiments (low and high), initial NO2− concentrations were less than 1 µM. In the low nitrate treatment, NO2− concentrations increased to 23 µM at 90 minutes and then decreased to 0.2 µM at 105 minutes (Fig. 4a, open symbols). For the high nitrate treatment, NO2− concentrations increased to 105 µM at 300 minutes and subsequently decreased to 0.4 µM at 420 minutes (Fig. 4b, open symbols). As expected, the trends in NO2− concentrations reveal two phases; an increase due to nitrate reduction to nitrite leading to an initial net production of nitrite, followed by a decrease of nitrite concentrations as the newly formed nitrite is further reduced to N2 (see Fig. 1).
In the two experiments with NO3−, the decrease in NO3− concentrations was accompanied by a corresponding enrichment of 15N in the remaining NO3−. The δ15N-NO3− of the remaining nitrate increased from 5.5 to 33.7‰ within 90 minutes and from 7.5 to 47.4‰ within 300 minutes in low and high nitrate treatments (Fig. 4c,d). The decrease in NO3− concentrations due to denitrification was also accompanied by a corresponding enrichment of 18O in the remaining NO3−. δ18O-NO3− increased from 19.8 to 45.6‰ within 90 minutes and from 22.9 to 57.0‰ within 300 minutes in low and high nitrate treatments respectively (Fig. 4e,f). The increase in δ18O-NO3− was initially very slow as long as nitrite was net produced, in all likelihood due to the rapid oxygen isotope exchange between nitrite and water.
The N isotope enrichment factor associated with the reduction of nitrate varied between −21.1 and −24.4‰ (Fig. 5a,b) and the O isotope enrichment factors ranged between −19.6 and −21.9‰ (Fig. 5c,d) for low and high nitrate treatments respectively. Despite different nitrate reduction rates between the two experimental treatments with amended nitrate concentrations, the observed N and O isotopic enrichment factors did not vary by more than 3.3‰.

Calculation of ε15N-NO3− (a,b) and ε18O-NO3− (c,d) for low and high nitrate treatments respectively.
Modeling kinetics and isotopic composition of nitrite and nitrate during denitrification
The Isonitrite model provides the kinetic parameters of the multi-step process of NO3− reduction to N2 via the intermediate NO2−. The model was applied to simulate concentrations and isotope compositions of nitrite and nitrate observed in the batch experiments for the low and high concentration treatments. Overall, the model achieved good simulations of the temporal evolution of NO3− and NO2− concentrations (Fig. 6) and N and O isotope ratios of nitrate and nitrite (Fig. 7).

Measured (red dots) and modeled (blue lines) nitrate (a,b) and nitrite concentrations (c,d) for low and high nitrate treatments respectively.

Measured (red dots) and modeled (blue lines) δ15N-NO3− (a,b) and δ18O-NO3− (c,d) for low and high nitrate treatments respectively.
When simulating the low-nitrate treatment, the model fit of the experimental NO3− and NO2− concentrations was obtained by using kinetic constants of 4.2 × 10−5 min−1 for NO3− reduction (equation 11 in Methods) and 7.2 × 10−5 min−1 for NO2− reduction (Eq. 8), and a reaction order (n in Eqs. 7 and 8) of 0.4. In these experiments, the model constrains N and O isotope enrichment factors (ε15N = −19‰, ε18O = −18‰ for NO3− reduction) that are very close to the values obtained by applying the Rayleigh model to the data (Fig. 5). This indicates that the model is able to correctly reproduce the N and O isotope fractionation of the reduction of NO3− to NO2−. However, in order to obtain a good fit of the model to the concentration and isotope data obtained in the multi-step (NO3− → NO2− → N2) experiment (Fig. 8a), it was necessary to impose a higher ε15Ν for NO2− reduction (−10‰) than the value determined in batch experiments where only NO2− reduction occurred (−4.2‰; Fig. 2).

Determination of ε15N of nitrite during the reduction of nitrate for low (a) and high (b) nitrate treatment.
Similar results were obtained with the model applied to the experiment with high nitrate treatment. However, to obtain a good fit of the NO3− and NO2− concentration trends, a slightly different approach was implemented to simulate the kinetics of NO3− and NO2− reduction. To reproduce the increase in NO3− consumption at 170 minutes observed in this experiment (Fig. 5c), a change in the kinetic constant of NO3 reduction from 1.8 × 10−5 min−1 to 6.9 × 10−5 min−1 at 170 minutes was necessary. Also, the NO2− reduction rate was set to 6 × 10−1 µM min−1 for the first 170 minutes, and to 1.8 × 10−2 µM min−1 for the remaining part of the experimental high nitrate treatment to achieve a satisfactory fit of the observed nitrite concentrations (Fig. 6d). A very good fit of the experimental trends in δ15N-NO3− and δ18O-NO3− was obtained by imposing model N and O isotope enrichment factors (ε15Ν = −19.1‰, ε18O = −19.2‰ for NO3− reduction) that were very close to the values measured in the batch experiments where only NO3− was reduced (Fig. 7). Similar to the low nitrate experiment, it was necessary to use a higher model ε15Ν for NO2− reduction (ε varied between −10 and −15‰) than that determined in batch experiments where only NO2− reduction took place (ε = −4.7‰) to obtain a good model fit for the data of the multi-step NO3− → NO2− → N2 experiment (Fig. 8b). This confirms that the Isonitrite model is capable of accurately predicting the concentrations of nitrate and nitrite, and the isotope composition of the remaining nitrate during denitrification.
Discussion
N isotope fractionation during denitrification and transformation of nitrite and nitrate
Even though initial nitrite concentrations affected the nitrite reduction rates, a similar isotope enrichment factor (ε15N) for nitrite reduction to N2 of −4.5 ± 0.3‰ was determined. This ε15N value for nitrite reduction is on the low end of previously reported values. Chien et al.45 reported ε15N values between −33.2 and −2.9‰ in soil experiments. Martin and Casciotti (2016) calculated ε15N between −22 to −8‰ for reduction of nitrite associated with different marine strains.
During denitrification, a gradual decrease in NO3− concentration is usually accompanied by a significant enrichment of 15N in the remaining nitrate33. The nitrogen isotope fractionation factors calculated in the current study for denitrification of nitrate (low and high nitrate treatments) of −21.1 and −24.4‰ are in good agreement with those found in the literature under similar conditions where denitrification occurs58. During nitrate reduction, the observed 15N enrichment of the remaining nitrate is caused by the preferential reduction of 14N-NO3− and a consequent enrichment of 14N of the product and intermediate compound NO2− during phase 1 of “net NO2− production” as shown in Fig. 1. As a result, NO2− concentrations increase and reach a maximum value during this phase 1 of denitrification (see Fig. 4a,b). During phase 2 or “net NO2− reduction” (Fig. 1), the NO2− concentration progressively decreases, leading to 15N enrichment of remaining NO2− due to preferential reaction of 14N-NO2−. During phase 1, production and reduction of NO2− take place simultaneously, however production occurs at a higher rate leading to an increase in nitrite concentrations. In phase 2, production and reduction of NO2− also take place simultaneously, but since NO2− reduction rates are higher than production rates, nitrite concentrations decrease.
Based on the N isotope enrichment factor (ε15Ν) measured for nitrite reduction alone (ε = −4.2‰) in the experiment with the low nitrite treatment, and combined with the ε15N of nitrate reduction (ε = −21‰), the model did not accurately predict the measured δ15N-NO2− and constantly underestimated the measured δ15N-NO2− value (Fig. 8). When the apparent N isotope enrichment factor (ε15Ν) of NO2− reduction was changed to −10‰ in the model, an almost perfect fit between simulated and observed δ15N-NO2− was achieved (Fig. 8a). A similar approach was used for the experiment with the high nitrate treatment with an N isotope enrichment factor varying between −10 and −15‰ to fit simulated and observed δ15N-NO2− (Fig. 8b). For both low and high nitrate treatments, the apparent N isotopic enrichment factor of nitrite reduction required by the model to achieve a good fit with the observed data (ε15N = −10 and −15‰ respectively) is higher than the ε15N obtained from the experiments with nitrite as sole source of nitrogen (ε15N = −4.2 and −4.7‰ for low and high nitrite treatment respectively).
It is also interesting to note that the calculated rates in the model revealed that the nitrite production rate varies during nitrate reduction, as shown especially for the high nitrate treatment. The nitrogen isotope enrichment factor determined for experiments with nitrite as the sole electron acceptor was lower for both treatments (ε15N varied between −4.7 and −4.2‰) than suggested by the Isonitrite model, independent of the nitrite reduction rates or nitrite concentrations. The apparent nitrogen isotope fractionation of nitrite during nitrate reduction seemed to increase with increasing availability of nitrite. The amount of nitrite available for nitrite reductase during denitrification is variable and lower than in the experiments with nitrite addition. Hence, either the variable or the lower nitrite concentrations may explain the difference in nitrogen isotope enrichment factors compared with those calculated in the nitrite reduction experiment.
Nitrite reduction is mediated by two different types of periplasmic enzymes, the copper containing nitrite reductase (nirK or Cu-NIR) or the cytochrome cd1-containing nitrite reductase (nirS or Fe-NIR)16. During denitrification with nitrite as a sole electron acceptor, nitrite is directly used by one of these enzymes at the outer side of the cell wall. During the experiment with nitrate as electron acceptor, nitrite is produced in the cytoplasm and consumed within the periplasm. Nitrite concentrations as substrate are low compared to those in the case of nitrite addition. When nitrite is produced from the reduction of nitrate, the comparatively low amount of nitrite-nitrogen could be limiting and generate low N isotope fractionation. In addition to lower net concentrations of nitrite, nitrite is subject to transport upon reduction of nitrate, which may affect the extent of isotope fractionation59. Nitrate reduction can occur in the periplasm (via the enzyme Nap), but in most cases nitrate is reduced by the membrane bound nitrate reductase Nar. The membrane bound Nar reduces nitrate to nitrite in the cytoplasm, nitrite is then transported over the cell membrane via a transporter (nark or nasH) back to the periplasm where it is reduced further by either nirS or nirK16. The affinity with the transporter might thus generate nitrogen isotope fractionation. Combined transport and limiting nitrite concentrations could thus be responsible for higher nitrogen isotope fractionation during nitrate reduction as opposed to experiments were nitrite was the sole electron acceptor.
O isotope fractionation during nitrate and nitrite reduction
During denitrification, a gradual decrease in NO3− concentrations is usually accompanied by a significant enrichment of 18O in the remaining nitrate44. The oxygen isotope fractionation factors of −19.6 and −21.9‰ calculated in the current study are on the high end of what has been previously reported in the literature42,44.
In our study, the nitrogen and oxygen isotope fractionation during denitrification were in the same range (ε15N −21.1 to −24.4‰ and ε18O −19.6 to −21.9‰) for low and high nitrate treatments with a ratio of N and O isotope enrichment factors (18ε/15ε) observed in this study of 0.9 for low and high nitrate treatments. This is in agreement with results from a number of recent isotopic denitrification studies where the observed 18ε/15ε ratio was close to 1.050,60,61,62. The role of the different nitrate reductases from various bacterial strains has recently been discussed potentially explaining the large range of ε15N values observed during denitrification in marine sediments42. Moreover, Frey et al.63 showed with pure cultures that the extent of N isotope fractionation during denitrification may depend on the different nitrate reduction enzymes (periplasmic Nap versus membrane bound Nar).
In the experiments with nitrite as sole source of nitrogen, δ18O-NO2− remained constant during the nitrite reduction experiments. This is clear evidence for rapid oxygen isotope exchange between H2O and NO2− under experimental conditions keeping the δ18O-NO2− constant. Therefore, the observed oxygen isotope enrichment for denitrification of −21‰, as revealed in experimental low and high nitrate treatments, appears to be entirely associated with reduction of nitrate to nitrite.
The Isonitrite model was used to obtain a lower limit for the kinetics of oxygen isotope exchange between NO2− and H2O. Kinetics of this exchange were estimated by ensuring that the oxygen isotope fractionation associated with the reduction of NO3− and NO2− was not able to deviate the δ18O-NO2− from its equilibrium value with water having a δ18O-H2O value of −10‰. Repeated model runs varying the kinetic constant of the oxygen isotope exchange between NO2− and H2O (Eq. 21 in Methods) suggest that this kinetic constant must be greater than 5 * 10−10 µM s−1 in order for δ18O-NO2− to not deviate from its equilibrium value with water near 0‰ (Fig. 8). The fact that the δ18O-NO2− remained constant despite that NO2− production and consumption probably fractionate oxygen isotope ratios considerably suggests that the kinetic isotope constant for O isotope exchange between nitrite and water represented in Eq. (21) is greater than 3 * 10−8 µM−1 min−1.
In the absence of published values of the kinetic constant of oxygen isotope exchange k+ (reaction 17), it was compared with the chemical kinetic constant of NO3− reduction kNO3 (Eq. 7). In order to do this comparison, the units of k+ (µM−1 min−1) were converted into the units of kNO3 (min−1) by considering relation 18 and multiplying k+ (µM−1 min−1) by the concentration of H216O (55 × 106 µM). A k+ constant of 1.65 min−1 was obtained which is 5 orders of magnitude higher than the kNO3 (4.2 × 10−5 min−1). It appears thus that oxygen isotope exchange in our experiment is very rapid compared to the microbial mediated reduction of NO3−. Previous studies hypothesized that isotopic exchange can occur between the oxygen of water and the oxygen of nitrite over long time periods41,43,53. Our study confirms rapid oxygen isotope exchange between the intermediate nitrite and water. Due to the fast change of the δ18O-NO2−, it appears that isotope variations are driven not by physical parameters but are caused by bacteria activity.
Conclusions
This study reports the N and O isotope enrichment factors associated with the multi-step biogeochemical process of denitrification. Isotope measurements and calculation of isotope fractionation based on the Isonitrite model provide insights into the apparent isotopic fractionation of nitrite during denitrification. Due to rapid isotopic exchange between the oxygen of nitrite and water, δ18O-NO2− changed immediately and remained stable during nitrate reduction. Model calculations also confirmed rapid oxygen isotope equilibrium exchange between water and nitrite with a kinetic isotope constant for O isotope exchange of >5 * 10−2 kg mol−1 s−1.
Based on N and O isotope enrichment factors obtained from batch experiments, model simulations revealed an apparent nitrogen isotope enrichment factor for nitrite reduction (of −10‰ and −15‰) higher than those calculated for the experiments (−4.1‰ and −4.7‰) with nitrite as initial source of nitrogen with two different concentrations. The reduction of nitrate seems to drive the change of the nitrogen isotope fractionation factor affecting nitrite reduction. We hypothesize that this difference is due to variations of the amount of available nitrite. This suggests that the amount of available nitrite and transport determine the extent of nitrogen isotope fractionation during the process of denitrification.
Methods
Experimental setup and materials
In order to determine the variations in nitrogen and oxygen isotope ratios, N and O isotopic enrichment factors for nitrite reduction were determined under denitrifying conditions (in the absence of oxygen) with NO2− as the sole electron acceptor in a laboratory batch incubation experiment. Subsequently, the same experimental approach was used with NO3− as electron acceptor in order to determinate the N (ε15N) and O (ε18O) isotope enrichment during denitrification of NO3−. For both experiments, two treatments at the low (101 µM for nitrite and 97 µM for nitrate) and high end (614 µM for nitrite and 508 µM for nitrate) of concentrations measured in surface waters were used. Subsequently, a non-steady state model was developed to simulate the changes in concentrations and N and O isotope ratios of nitrate and nitrite tracing kinetics and isotope variations during reduction of nitrate concentrations associated with initial net production of nitrite and subsequent net reduction of nitrite.
Batch experiments
Muddy organic sediments were collected from the Morbras Stream, La Queue en Brie, a tributary of the Marne River in the Seine watershed upstream of Paris (France). Sediments from this stream had been used in a preliminary test and showed significant nitrate reduction potential (data not shown). The land use in the upstream catchment is mainly crops. The sediment was homogenized in the laboratory prior to the experiments. Contents and isotopic compositions ot total organic carbon and total nitrogen were determined by EA-IRMS (Corg = 8.6%; Norg = 0.8%; C/N = 14.9; δ13Corg = −24.6‰; δ15Ntotal = 2.9‰). Thirty grams of sediment were weighed in 300 ml glass bottles and 280 mL of nitrite or nitrate in milliQ water was added to create a sediment slurry. The slurries were flushed with He for twenty minutes to achieve anoxic conditions prior to crimp capping the bottles with a septum and a ring. The solutions in the first set of experiments contained 101 and 614 µM of nitrite derived from KNO2 (low and high nitrite treatments), and the second set of experiments contained 97 and 508 µM of nitrate derived from KNO3 (low and high nitrate treatments). Prior to addition, the salts were dissolved in milliQ water with a δ18O-H2O value of −10‰. Bottles were incubated under continuous agitation in order to maintain sediments in suspension and optimize denitrification (Sebilo et al. 2003) in the dark at 20 °C for 270 and 330 minutes for low and high nitrite treatments and for 180 and 420 minutes for low and high nitrate treatments, respectively. For each treatment, 14 similar slurries were prepared and considered homogeneous at the beginning of the experiment. Every 15 to 30 minutes for each treatment, a bottle was sacrificed and the slurry was centrifuged at 4000 rpm for 15 minutes. The supernatant was then filtered first through a 0.45μm glass microfiber filters (GFF-Whatman) and subsequently through a 0.2 µm nylon membrane (Whatman) to remove all sediment. Solutions were immediately preserved with mercuric chloride (0.3 ml at 5%) to avoid any residual transformation of the dissolved compounds in the solution. An aliquot of this solution was used to determine nitrite and nitrate concentrations. Ammonium concentrations were determined at the beginning and at the end of the experiments to check the ammonium budget. As ammonium consumption due to nitrification can be excluded under anoxic conditions, ammonium production should be only caused by ammonification (degradation of organic matter) or DNRA. Ammonium concentrations were below the limit of detection for all analyzed samples. Another aliquot of the solution was frozen and stored for subsequent N and O isotope analysis on nitrite and nitrate.
Nutrient concentration analyses
Nutrient concentrations were determined by colorimetry. Nitrate concentrations were determined using the colorimetric hydrazine method. Nitrate was converted to nitrite with hydrazine. Then nitrite reacted with sulphanilamide and N-1-naphthylethylenediamine dihydrochloride. Nitrite concentrations were directly measured by colorimetry by using the same reactants. Ammonium reacted with sodium nitroprussiate and sodium hydroxide. The absorbance of coloration was determined at 540 nm and 660 nm (Gallery Water ThermoFisher Scientific) to obtain nitrite or nitrate and ammonium concentrations respectively with a precision of the measurement ±0.3 µM.
Isotope ratio measurements
Nitrogen and oxygen isotope ratios of nitrate and nitrite were determined separately following a modified protocol of McIlvin and Altabet64. Nitrate was reduced to nitrite in an activated column of cadmium. Yields of nitrate reduction were calculated using concentration measurements with a yield of reduction >95%.
When both nitrite and nitrate concentrations were not negligible, the liquid samples were diluted with a salted buffer to obtain a concentration of 20 µM of NO2− at the column outlet, which is composed of reduced NO3− and the initial NO2− (called later NOx−). Subsequently, the NO2− (i.e. NOx− and NO2−) was further reduced to nitrous oxide by sodium azide solution. To achieve this, each sample was diluted with a salt solution (NaCl = 35.5 g l−1) to obtain 1 µM of NO2− concentration in 15 ml and transferred into a 20 ml glass vial. The vials were crimp-sealed with Teflon-backed silicone septa (IVA, 70220804) and aluminum caps. The sodium azide solution, which consists of a 40 ml mixture of 20 ml of azide (NaN3, 2 M) and 20 ml of acetic acid (C2H4O2, 20%), was flushed with He for 30 min to avoid any N2O pollution from the buffer. In order to reduce the NO2− to N2O, 0.8 ml of azide solution was injected in each vial, and vials were placed in a water bath at 30 °C for one hour. The yield of conversion was better than 95%.
The isotope compositions of all N2O samples were measured with an isotope ratio mass spectrometer (IRMS, Delta Vplus, Thermo Scientific, Bremen, Germany) in continuous-flow mode with a purge-and-trap system coupled with a Finnigan GasBench II system (Thermo Scientific, Bremen, Germany). Results are reported in the internationally accepted delta notation in ‰ with respect to the standards air for δ15N and Vienna Standard Mean Ocean Water (V-SMOW) for δ18O, respectively. Nitrate and nitrite reference materials subject to the same analytical procedures were used to calibrate the isotopic composition of N2O (USGS34, δ15N = −1.8‰, δ18O = −27.9‰, USGS35, δ15N = +2.7‰, δ18O = +57.5‰ and USGS32, δ15N = +180‰, δ18O = +25.7‰ for nitrate standards; lab nitrite standards Lb1, δ15N = −63‰ and Lb2, δ15N = +2.7‰ for nitrite standards). The precision was ±0.3‰ for δ15N-NO2− and ±0.5‰ for δ18O-NO2−.
Finally, using the isotopic compositions of NOx− (NO2− + NO3−) and NO2− alone, the δ15N-NO3− and δ18O-NO3− were calculated as follows:
where [NO3−] and [NO2−] are the concentrations of NO3− and NO2−, respectively.
Based on this calculation, the precision for δ15N-NO3− was ±0.5‰ and ±0.8‰ for δ18O-NO3−.
Determination of isotope enrichment factors
The nitrite experiment in a closed system without any renewal of substrate ensured Rayleigh conditions and therefore the N and O isotope enrichment factors (ε) were calculated using the following equations:
and
where δ15N and δ18O-NO2− (t0) are the N and O isotope ratios of the substrate at time zero, and C0 and Ct are the concentrations of nitrite at time zero and t, respectively. This relationship was used to determine the N and O isotope enrichment factor of nitrite reduction by plotting measured values of δ15N(t) and δ18O(t) against ln Ct/C0. The same approach was used to determine the N and O isotope enrichment factor for denitrification for the experiments where nitrate was the sole source of nitrogen.
Isonitrite model
As nitrite is both a substrate and a product during denitrification of nitrate, Rayleigh conditions and equations are not applicable to determine the N and O isotope enrichment factor associated with the reduction of NO2− when these two processes occur concurrently in a batch reactor. Although our batch experiments alone are sufficient to derive enrichment factors (ε) for the separately tested NO3− = >NO2− and NO2− = >N2 steps, a model was developed to calculate the isotope enrichment factors when these processes occur simultaneously in a batch reactor. The model simulates the multi-step denitrification process and considers the presence of the intermediate product (NO2−) as well as the N and O isotopic compositions of nitrite and nitrate. The model is constrained with experimentally determined concentrations and N and O isotopic ratios of nitrite and nitrate. The model was used to estimate instantaneous isotopic enrichment factors associated with NO3− reduction to NO2− and NO2− reduction to N2. Moreover, the model was used to investigate the kinetics of oxygen isotope exchange between water and NO2−.
The model considers that denitrification is a two-stage process:
The rate of these processes is computed as:
and
where NO3− and NO2− concentrations are expressed in µM, Rx (x = NO3− or NO2−) is the nitrite production or reduction (reaction) rate in µM min−1, kx is the kinetic constant in min−1 and nx is the reaction order.
To simulate both N and O isotope ratios simultaneously, three isotopic species must be considered for each chemical species, with the total chemical concentration being equal to the sum of the isotopic species:
With 18O3: 18O16O16O and 18O2: 18O16O.
From these isotopic species, the δ15N and the δ18O values of NO2− and NO3− can be calculated as follows (equations shown for NO3):
where SMOW is the 18O/16O ratio in the SMOW standard and NSTD is the 15N/14N ratio of air.
The rate of reaction for NO3− and NO2− reduction for the different isotope species have to sum up to the rate of the total chemical species calculated with Eqs. 3 and 4. For example, for NO3− reduction:
The expressions for the rates of the isotopic species are obtained by solving a system of three equations comprising equation 14 and equations 12 and 13 written in terms of rates rather than concentrations, and introducing isotopic enrichment factors for N and O isotopes (15εNO3 and 18εNO3, respectively):
Exchange of oxygen isotopes between NO2− and H2O takes place according to the following isotopic exchange reaction:
The forward and backwards rates of this reaction are calculated as:
where N16O2 = 14N16O2 + 15N16O2, the rates are in µM min−1, and the forwards (k+) and backwards (k−) kinetic constants (in µM−1 min−1) are related to the equilibrium constant of the isotope exchange reaction a follows:
where the equilibrium constant for the isotope exchange reaction is obtained from the equilibrium concentrations of the species involved in the oxygen isotope exchange:
where the subscript i refers to the initial, equilibrium concentration of isotopic species.
The concentrations of the six isotopic model species in time are calculated by integrating the following system of six coupled differential equations starting from initial chemical and isotopic equilibrium conditions:
To summarise, known model input parameters are: (1) the simulation time (that corresponds to the duration of the experiment), (2) the initial concentrations of NO3− and NO2−, (3) the oxygen and nitrogen isotope compositions of NO3− and NO2−, and (4) the δ18O of water. Unknown (fitting) parameters are: (1) the kinetic constants for NO2− production, for NO2− consumption and for isotope exchange between NO2− and H2O, and (2) the oxygen and nitrogen isotope fractionation factors in NO2− production and NO2− consumption.
The model was implemented with the software Mathematica version 9 (Wolfram Research). Model outputs are changes of nitrite and nitrate concentrations and nitrogen and oxygen isotope compositions for both NO2− and NO3−.
References
Splading, R. F. & Exner, M. E. Occurrence of nitrate in groundwater - A review. J. Environ. Qual. 22, 392–402 (1993).
Zhang, W. L., Tian, Z. X., Zhang, N. & Li, X. Q. Nitrate pollution of groundwater in northern China. Agr. Ecosys. Environ. 59, 223–231 (1996).
Harter, T., Davis, H., Mathews, M. C. & Meyer, R. D. Shallow groundwater quality on dairy farms with irrigated forage crops. J. Contam. Hydrol. 55, 287–315 (2002).
World Population Prospects: The 2010 Revision vol. I Comprehensive Tables. United Nations Department of Economic and Social Affairs Population Division 2011 pp. 1–481. (New York).
Gu, B. et al. The role of industrial nitrogen in the global nitrogen biogeochemical cycle. Scientific Reports 3, https://doi.org/10.1038/srep02579 (2013a).
Tilman, D. et al. Forecasting agriculturally driven global environmental change. Science 292, 281–284 (2001).
Gulis, G., Czompolyova, M. & Cerhan, J. R. An ecology study of nitrate in municipal drinking water and cancer incidence in Trnava District, Slovakia. Environmental Research 88, 182–187 (2002).
Wolfe, A. H. & Patz, J. A. Reactive nitrogen and human health: acute and long-term implications. Ambio 31, 120–125 (2002).
Townsend, A. R. et al. Human health effects of a changing global nitrogen cycle. Front. Ecol. Environ. 1, 240–246 (2003).
Gu, B., Ge, Y., Chang, S. X., Luo, W. & Chang, J. Nitrate in groundwater of China: sources and driving forces. Global Environ. Change 23, 112–1121 (2013b).
Zayed, G. & Winter, J. Removal of organic pollutants and of nitrate from wastewater from the dairy industry by denitrification. Appl. Microbiol. Biotechnol. 49, 469–474 (1998).
Wang, X. & Wang, J. Removal of nitrate from groundwater by heterotrophic denitrification using the solid carbon source. Sci. China Ser. B-Chem. 52, 236–240 (2009).
Harrison, M. D., Groffman, P. M., Mayer, P. M. & Kaushal, S. S. Nitrate removal in two relict oxbow urban wetlands: a 15N mass-balance approach. Biogeochemistry 111, 647–660 (2012).
Wijler, J. & Delwiche, C. C. Investigations on the denitrifying process in soil. Plant and Soil 5, 155–169 (1954).
Firestone, M. K. & Davidson, E. A. Microbiological basis of NO and N2O production and consumption in soils in Exchanges of Trace Gases Between Terrestrial Ecosystems and the Atmosphere (eds Andreae, M. O. & Schimel, D. S.), 7–21 (Wiley, 1989).
Zumft, W. G. Cell biology and molecular basis of denitrification. Microbiol. Mol. Biol. Rev. 61, 533–616 (1997).
Bancroft, K., Grant, I. F. & Alexander, M. Toxicity of NO2 −: Effect of nitrite on microbial activity on in acid soil. Applied and Environmental Microbiology 38, 940–944 (1979).
Crutzen, P. J. The influence of nitrogen oxides on the atmospheric ozone content. Quart. J. R. Met. Soc. 96, 320–325 (1970).
Helder, W. & De Vries, R. T. P. Estuarine nitrite maxima and nitrifying bacteria (Ems-Dollard estuary). Neth. J. Sea Res. 17, 1–18 (1983).
Morris, A. W., Howland, R. J. M., Woodward, E. M. S., Bale, A. J. & Mantoura, R. F. C. Nitrite and ammonia in the Tamar estuary. Neth. J. Sea Res. 19, 217–222 (1985).
Von der Wiesche, M. & Wetzel, A. Temporal and spatial dynamics of nitrite accumulation in the River Lahn. Water Res. 32, 1653–1661 (1998).
Garnier, J. et al. Nitrous oxide emission in the Seine River estuary (France): comparison with upstream sector of the Seine basin. Biogeochemistry 77, 305–326 (2006).
Raimonet, M., Vilmin, L., Flipo, N., Rocher, V. & Laverman, A. M. Modelling the fate of nitrite in an urbanized river using experimentally obtained nitrifier growth parameters. Water Research 73, 373–387 (2015).
Tanimoto, T., Hatano, K., Kim, D. H., Uchiyama, H. & Shoun, H. Co-denitrification by the denitrifying system of the fungus Fusarium oxysporum. FEMS Microbiology Letters. 93, 177–180 (1992).
Spott, O., Russow, R. & Strange, C. F. Formation of hybrid N2O and hybrid N2 due to codenitrification: First review of a barely considered process of microbially mediated N-nitrosation. Soil Biology and Biochemistry. 43, 1995–2011 (2011).
Payne, W. J. Reduction of nitrogenous oxides by microorganisms. Bacteriological Reviews 37, 409–452 (1973).
Koike, I. & Hattori, A. Denitrification and ammonia formation in anaerobic coastal sediments. Applied Environmental Microbiology 35, 278–282 (1978).
Kelso, B., Smith, R. V., Laughlin, R. & Lennox, D. Dissimilatory nitrate reduction in anaerobic sediments leading to river nitrite accumulation. Applied Environmental Microbiology 63, 4679–4685 (1997).
Jetten, M. S. M. et al. The anaerobic oxidation of ammonium. FEMS Microbiol. Rev. 22, 421–437 (1999).
Poth, M. & Focht, D. D. 15N kinetic analysis of N2O production by Nitrosomonas europaea: an examination of nitrifier denitrification. Applied and Environmental Microbiology 49, 1134–1141 (1985).
Goreau, T. et al. Production of NO2 − and N2O by nitrifying bacteria at reduced concentrations of oxygen. Applied Environmental Microbiology 40, 526–532 (1980).
Wrage, N., Velthof, G. L., van Beusichem, M. L. & Oenema, O. Role of nitrifier denitrification in the production of nitrous oxide. Soil Biology & Biochemistry 33, 1723–1732 (2001).
Mariotti, A., Gerrmon, J. C. & Leclerc, A. Nitrogen isotope fractionation associated with the NO2 − → N2O step of denitrification in soils. Can. J. Soil Sci. 62, 227–241 (1982).
Kendall, C. Tracing nitrogen sources and cycling in catchments in Isotope Tracers in Catchment Hydrology (eds Kendall, C. & McDonnel, J. J.) 521–576 (Elsevier, 1998).
Mattern, S., Sebilo, M. & Vanclooster, M. Identification of the nitrate contamination sources of the Brusselian sands groundwater body (Belgium) using a dual-isotope approach. Isotopes in Environmental and Health Studies 47, 297–315 (2011).
Briand, C. et al. Isotopic (δ15N, δ18O, δ11B) and fecal tracers for the identification of nitrate sources and pathways to rivers. Scientific Reports. 7, 41703, https://doi.org/10.1038/srep41703 (2017).
Brandes, J. A., Devol, A. H., Yoshinari, T., Jayakumar, D. A. & Naqvi, S. W. A. Isotopic composition of nitrate in the central Arabian Sea and eastern tropical North Pacific: a tracer for mixing and nitrogen cycles. Limnol Oceanogr. 43, 1680–1689 (1998).
Voss, M., Dippner, J. W. & Montoya, J. P. Nitrogen isotope patterns in the oxygen-deficient waters of the Eastern Tropical North Pacific Ocean. Deep-Sea Res I 48, 1905–1921 (2001).
Lehmann, M. F., Reichert, P., Bernasconi, S. M., Barbieri, A. & McKenzie, J. A. Modelling nitrogen and oxygen isotope fractionation during denitrification in a lacustrine redox-transition zone. Geochimica et Cosmochim Acta 67, 2529–2542 (2003).
Sebilo, M., Billen, G., Grably, M. & Mariotti, A. Isotopic composition of nitrate-nitrogen as a marker of riparian and benthic denitrification at the scale of the whole Seine River system. Biogeochemistry 63, 35–51 (2003).
Knöller, K., Vogt, C., Haupt, M., Feisthauer, S. & Richnow, H. H. Experimental investigation of nitrogen and oxygen isotope fractionation in nitrate and nitrite during denitrification. Biogeochemistry 103, 371–384 (2011).
Dähnke, K. & Thamdrup, B. Nitrogen isotope dynamics and fractionation during sedimentary denitrification in Boknis Eck Baltic Sea. Biogeosciences 10, 681–709 (2013).
Martin, T. S. & Casciotti, K. Nitrogen and oxygen isotopic fractionation during microbial nitrite reduction. Limnol. Oceanogr, https://doi.org/10.1002/lno.10278 (2016).
Böttcher, J., Strebel, O., Voerkelius, S. & Schmidt, H. L. Using isotope fractionation of nitrate-nitrogen and nitrate-oxygen for evaluation of microbial denitrification in a sandy aquifer. J Hydrol 114, 413–424 (1990).
Chien, S. H., Shearer, G. & Kohl, D. H. The nitrogen isotope effect associated with nitrate and nitrite loss from waterlogged soils. Soil Sci. Soc. Amer. J. 41, 63–69 (1977).
Mariotti, A., Landreau, A. & Simon, B. 15N isotope biogeochemistry and natural denitrification process in groundwater: Application to the chalk aquifer of northern France. Geochimica et Cosmochim Acta 52, 1869–1878 (1988).
McIlvin, M. R. & Altabet, M. A. Chemical conversion of nitrate and nitrite to nitrous oxide for nitrogen and oxygen isotopic analysis in freshwater and seawater. Anal. Chem. 77, 5589–5595 (2005).
Granger, J., Sigman, D. M., Prokopenko, M. G., Lehmann, M. F. & Tortell, P. D. A method for nitrite removal in nitrate N and O isotopes analyses. Limnol Oceanogr. Methods 4, 205–212 (2006).
Böhlke, J. K., Smith, R. L. & Hannon, J. E. Isotopic analysis of N and O in nitrite and nitrate by sequential selective bacterial reduction to N2O. Anal Chem 79, 5888–5895 (2007).
Hu, H., Bourbonnais, A., Larkum, J., Bange, H. W. & Altabet, M. A. Nitrogen cycling in shallow low-oxygen coastal waters off Peru from nitrite and nitrate nitrogen and oxygen isotopes. Biogeosciences 13, 1453–1468 (2016).
Buchwald, C. & Casciotti, K. L. Oxygen isotopic fractionation and exchange during bacterial nitrite oxidation. Limnol Oceanogr. 55, 1064–1074 (2010).
Casciotti, K. L., McIlvin, M. R. & Buchwald, C. Oxygen isotopic exchange and fractionation during bacterial ammonia oxidation. Limnol Oceanogr. 55, 753–62 (2010).
Buchwald, C., Santoro, A. E., McIlvin, M. R. & Casciotti, K. L. Oxygen isotopic composition of nitrate and nitrite produced by nitrifying cocultures and natural assemblages. Limnol Oceanogr. 57, 1361–1375 (2012).
Casciotti, K. L. & Buchwald, C. Insights on the marine microbial nitrogen cycle from isotopic approaches to nitrification. Frontiers in Microbiology 3, 0.3389/fmicb.2012.00356 (2012).
Casciotti, K. L., Buchwald, C., Santoro, A. E. & Frame, C. Assessment of nitrogen and oxygen isotopic fractionation during nitrification and its expression in the marine environment in Research on Nitrification and Related Processes, Part A (ed. Klotz, M. G.) 253–280 (San Diego, CA: Academic, 2011).
Jacob, J., Sanders, T. & Dähnke, K. Nitrification and nitrate isotope fractionation as a case study in a major European river. Biogeosciences 13, 5649–5659 (2016).
Buchwald, C., Grabb, K., Hansel, C. M. & Wankel, S. Constraining the role of iron in environmental nitrogen transformations: Dual isotope systematics of abiotic NO2 − reduction by Fe(II) and its production of N2O. Geochimica et Cosmochimica Acta 186, 1–12 (2016).
Sebilo, M. et al. The use of nitrogen and oxygen natural isotopic composition to characterize and quantify nitrification and denitrification processes in the Seine River and Estuary. Ecosystems 9, 1–16 (2006).
Goddard, A. D. et al. The Paracoccus denitrificans NarK-like nitrate and nitrite transporters-probing nitrate uptake and nitrate/nitrite exchange mechanisms. Molecular Microbiology 103, 117–133 (2017).
Granger, J., Sigman, D. M., Needoba, J. A. & Harrison, P. J. Coupled nitrogen and oxygen isotope fractionation of nitrate during assimilation by cultures of marine phytoplankton. Limnol Oceanogr. 49, 1763–1773 (2004).
Granger, J., Sigman, D.M., Lehmann, M.F. & Tortell, P.D. Nitrogen and oxygen isotope fractionation during dissimilatory nitrate reduction by denitrifying bacteria. Limnol Oceanogr. 53, 2533–2545 (2008).
Kritee, K., Sigman, D.M., Granger, J., Ward, B.B., Jayakumar, A. & Deutsch, C. Reduced isotope fractionation by denitrification under conditions relevant to the ocean. Geochimica et Cosmochimica Acta 92, 243–259 (2012).
Frey, C., Hietanen, S., Jürgens, K., Labrenz, M. & Voss, M. N and O isotope fractionation in nitrate during chemolithoautotrophic denitrification by sulfurimonas gotlandica. Environ. Science and Technol. 48, 13229–13237 (2014).
Semaoune, P., Sebilo, M., Templier, J. & Derenne, S. Is there any isotopic fractionation of nitrate associated with diffusion and advection ? Env. Chem. 9, 158–162 (2012).
Acknowledgements
The contributions of BM to this project were supported by the Natural Sciences and Engineering Research Council of Canada (NSERC) via a discovery grant.
Author information
Authors and Affiliations
Contributions
M.S. wrote the manuscript, M.S., A.M.L., E.P. and A.M. designed the study. E.P., A.M. and V.V. performed the analysis, G.A. and M.S. developed the model. M.S., A.M.L., B.M. and G.A. discussed the results and reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sebilo, M., Aloisi, G., Mayer, B. et al. Controls on the Isotopic Composition of Nitrite (δ15N and δ18O) during Denitrification in Freshwater Sediments. Sci Rep 9, 19206 (2019). https://doi.org/10.1038/s41598-019-54014-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-54014-3
- Springer Nature Limited